

Abstract
The type 2 secretion system (T2SS), a multi-protein machinery that spans both the inner and the outer membranes of Gram-negative bacteria, is used for the secretion of several critically important proteins across the outer membrane. Here we report the crystal structure of the N-terminal cytoplasmic domain of EpsF, an inner membrane spanning T2SS protein from Vibrio cholerae. This domain consists of a bundle of six anti-parallel helices and adopts a fold that has not been described before. The long C-terminal helix α6 protrudes from the body of the domain and most likely continues as the first transmembrane helix of EpsF. Two N-terminal EpsF domains form a tight dimer with a conserved interface, suggesting that the observed dimer occurs in the T2SS of many bacteria. Two calcium binding sites are present in the dimer interface with ligands provided for each site by both subunits. Based on this new structure, sequence comparisons of EpsF homologs and localization studies of GFP fused with EpsF, we propose that the second cytoplasmic domain of EpsF adopts a similar fold as the first cytoplasmic domain and that full-length EpsF, and its T2SS homologs, have a three-transmembrane helix topology.
Keywords: General Secretory Pathway, Type 4 Pilin Biogenesis, GspF, XcpS, PilG
1. Introduction
In many pathogenic as well as non-pathogenic Gram-negative bacteria a diversity of unrelated proteins is secreted from the periplasm across the outer membrane into the extracellular milieu by the type 2 secretion system (T2SS) (Cianciotto, 2005; Evans et al., 2008; Sandkvist, 2001b). The T2SS is also called the terminal branch of the general secretory pathway (Gsp) (Pugsley, 1993) and, in Vibrio species, the extracellular protein secretion (Eps) apparatus (Sandkvist et al., 1997). This sophisticated machinery spans both the inner and the outer membrane and contains 11 to 15 different proteins, with many, if not all, of these proteins present in multiple copies (Filloux, 2004; Johnson et al., 2006). The nomenclature of the T2SS proteins is quite complex. In this paper, proteins from the Eps system from Vibrio species are referred to as “Eps” followed by a capital letter, while the non-Vibrio T2SS homologs will be called “Gsp” followed by the same capital letter. Hence, the T2SS homologs of EpsF are called GspF proteins. GspF will also be used as the name of the family of T2SS inner membrane proteins to which EpsF belongs (See Supplementary Figure 1 for a GspF family sequence alignment).
In V. cholerae, the major virulence factor cholera toxin and several other proteins including a soluble colonization factor, hemagglutinin-protease, lipase and chitinase are secreted by the T2SS across the outer membrane (Hirst and Holmgren, 1987; Kirn et al., 2005; Sandkvist, 2001b; Sikora et al., 2007). In enterotoxigenic Escherichia coli (ETEC), the T2SS is responsible for secretion of heat-labile enterotoxin (Tauschek et al., 2002), a close homolog of cholera toxin (Merritt and Hol, 1995; O'Neal et al., 2004; Sixma et al., 1991). Other bacteria secreting a variety of proteins using the T2SS include important human pathogens such as Pseudomonas aeruginosa (Bally et al., 1992), Klebsiella spp. (d'Enfert et al., 1987; Uweh, 2006), Legionella pneumophila (Söderberg et al., 2004), Yersinia pestis (Yen et al., 2008), enteropathogenic E. coli (EPEC) and enterohemorrhagic E. coli (EHEC) (Schmidt et al., 1997). Also several plant pathogens, like Erwinia chrysanthemi and E. carotovora, Xanthomonas campestris and X. fastidiosa (Bouley et al., 2001; Cianciotto, 2005; Sandkvist, 2001b), contain a T2SS system. Among the different types of protein secretion systems identified so far in Gram-negative bacteria, the T2SS is remarkable in that proteins are secreted by the T2SS across the outer membrane in a folded conformation (Bortoli-German et al., 1994; Hardie et al., 1995; Hirst and Holmgren, 1987; Pugsley, 1992; Sandkvist, 2001b).
It has become evident that several components of the T2SS are related to components of the type 4 pilin biogenesis (T4PB) system (Bally et al., 1992; Craig and Li, 2008; Filloux, 2004; Hobbs and Mattick, 1993; Nunn, 1999; Peabody et al., 2003). Type 4 pili are thin, strong filaments extending from a wide variety of human bacterial pathogens (Craig et al., 2004; Hansen and Forest, 2006). The T4PB system is responsible for a diversity of functions including pilus assembly and disassembly, protein export, DNA import and phage entry (Burrows, 2005; Mattick, 2002; Nudleman and Kaiser, 2004). Studies of the T2SS proteins are therefore important for increasing our understanding of critical membrane transport phenomena in many bacterial species, several of which are of great medical importance.
The T2SS can be envisioned as consisting of three major subassemblies (Filloux, 2004; Johnson et al., 2006; Peabody et al., 2003; Py et al., 2001; Sandkvist, 2001a; Sauvonnet et al., 2000): (i) the outer membrane complex, comprising mainly the crucial multi-subunit secretin EpsD, which is thought to be the pore that opens and closes to allow passage of the secreted proteins; (ii) the pseudopilus, which consists of one major and several minor pseudopilins that may form a retractable plug or piston; and, (iii) an inner membrane platform, containing the cytoplasmic “secretion ATPase” EpsE and the membrane proteins EpsL, EpsM, EpsC and EpsF. The central protein of the current paper is the polytopic inner membrane protein EpsF, which previously was shown to be indispensable for secretion in V. cholerae (Sandkvist et al., 1997).
GspF interacts with other proteins from the inner membrane platform and is thought to be a key player in the T2SS and T4PB systems (Crowther et al., 2004; Py et al., 2001). Bioinformatics analysis of the amino acid sequence and BlaM-fusion experiments of GspF from E. caratovora indicated that this T2SS EpsF homolog crosses the inner membrane three times (Thomas et al., 1997). These authors also concluded that the N-terminus of the GspF protein in E. caratovora is cytoplasmic and the C-terminus periplasmic. This topology was confirmed by alkaline phosphatase fusion experiments of the T2SS GspF homolog in P. aeruginosa (Arts et al., 2007). Peabody et al. analyzed a large number of sequences from members of the superfamily of bacterial inner membrane proteins to which EpsF belongs (Peabody et al., 2003), hereafter called the “GspF/PilG/BfpE superfamily”. The GspF family represents members of the T2SS machinery, whereas the PilG and BfpE families consist of homologs from the T4PB system. Members from the entire superfamily display an internal sequence repeat such that the first cytoplasmic domain is related to the second half of the protein (Peabody et al., 2003). In spite of considerable attention for these important bacterial inner membrane proteins, no high resolution structural information has been reported to date for full length proteins or domains of members from this superfamily.
In continuation of our studies aimed to unravel the architecture of the T2SS and its components in human pathogens and close relatives (Abendroth et al., 2004a; Abendroth et al., 2004b; Abendroth et al., 2005; Korotkov et al., 2006; Korotkov and Hol, 2008; Korotkov et al., 2009; Robien et al., 2003; Yanez et al., 2008a; Yanez et al., 2008b), we report here the crystal structure of a truncated form of the first N-terminal cytoplasmic domain of V. cholerae EpsF. This truncated domain (hereafter also called “cyto1-EpsF56-171”) appears to adopt a novel fold, is entirely helical and forms a tight dimer with the residues of the dimer interface well conserved. Two symmetry-related calcium binding sites occupy a dimer with well conserved ligand residues across the GspF family. Sequence analysis of the GspF family suggests that the second cytoplasmic domain of members of this family adopts the same conformation as the first cytoplasmic domain. A tentative model of the cytoplasmic domains of a dimer of full-length EpsF is proposed.
2. Materials and methods
2.1 Protein expression and purification
DNA encoding for V. cholerae cyto1-EpsF56-171 was amplified by PCR and ligated into a pACYC-CT vector using NcoI and NheI restriction sites. The pACYC-CT vector encodes a C-terminal TEV-cleavable His6-tag and provides chloramphenicol resistance. BL21(DE3) E. coli cells were used for protein expression. For expression of native protein, the main culture was grown in LB medium at 37°C, and induced for 3 hrs with 0.5 mM IPTG. For expression of Se-Met labeled protein, the main culture was grown in M9 medium. 30 minutes before induction with 0.5 mM IPTG, an amino acid mixture for suppression of methionine biosynthesis was added essentially as described elsewhere (van Duyne et al., 1993). Cells were resuspended in buffer A (50 mM Tris-HCl, pH 8, 300 mM NaCl) and lysed with lysozyme and by sonication. The cell debris was pelleted by centrifugation at 20,000 g for 20 minutes. 3 ml of Ni-NTA (Qiagen) were incubated for 30 minutes with the clarified lysate from 1l culture, washed with 30 ml buffer B (20 mM Tris-HCl, pH 8, 300 mM NaCl, 1 mM TCEP), a second time with 30 ml buffer B plus 20 mM imidazole, and then eluted with 20 ml buffer B plus 200 mM imidazole. The Ni-NTA elution fractions were incubated with TEV protease over night at 4°C while dialyzing against 10-fold the volume of buffer A. The sample was then passed over 1ml Ni-NTA resin equilibrated with buffer A and washed with another 10ml buffer A. Flowthrough and wash were combined and concentrated to 5ml. The concentrated sample was further purified in buffer B by gel permeation chromatography using Superdex S200 pre-grade resin (Amersham-Pharmacia). Typically, 50 mg protein could be purified per liter culture. The protein was concentrated to 10 mg/ml for crystallization experiments.
2.2 Crystallization and data collection
Crystallization conditions were screened for using the commercial screens ProComplex (Qiagen); Index, PEG-Ion, SaltRX (Hampton Research); and Wizard (Emerald BioSystems). Various hits were found, each of them included PEG (400, 3000, 3350, 8000), a divalent cation such as Ca2+ or Mg2+, and a buffer pH 6.0-8.0. Conditions were optimized to 12.5% PEG 400, 200 mM CaOAc2, 100 mM MES pH 7.0.
For cryo-cooling, the crystals were sequentially transferred in buffers containing 200 mM CaOAc2, 100 mM MES pH 7.0, plus increasing concentrations of PEG 400 and NaCl in the following ratios: 20%/300 mM, 20%/600 mM, 25%/600 mM, 30%/600 mM. For the NaI soak, NaCl was gradually replaced by NaI. The crystals were frozen in the cryostream and screened for diffraction. Two cycles of cryo-annealing by blocking the cryostream could reduce the mosaicity of the crystals considerably.
Diffraction data of the iodide soak and the high resolution crystal were collected in house using a MicroMax-007 HF rotating anode (Rigaku) equipped with VariMax HF (Osmic) monochromator and a Saturn 994 (Rigaku) CCD detector. Diffraction data of the native crystal were collected at beamline 9.2 at SSRL on a Mar 325 CCD detector. Further details are provided in Table 1.
Table 1. Crystallographic data and refinement statistics.
| NaI soak SeMet protein | Native Met protein | High resolution SeMet protein | |
|---|---|---|---|
| Wavelength | 1.5418 Å | 2.0663 Å | 1.5418 Å |
| Scan angle | 0.5° | 1.0° | 0.5° |
| # of images | 720 | 360 | 620 |
| Cell dimensions: | 48.82 | 49.20 | 46.99 |
| a, b, c (in Å) | 54.37 | 53.83 | 52.12 |
| 88.59 | 89.07 | 89.47 | |
| Resolution (in Å) | 50 – 1.90 | 50 – 1.95 | 45 – 1.70 |
| (highest shell) | (1.97 – 1.90) | (2.02 – 1.95) | (1.76 – 1.70) |
| Reflections, total | 225,415 | 211,998 | 181,046 |
| Reflections, unique | 18,609 | 16,365 | 23,908 |
| Redundancy | 12.1 (5.4) | 13.0 (8.4) | 7.6 (3.9) |
| Rsym | 9.3% (32.6%) | 4.6% (14.4%) | 8.0% (27.7%) |
| I/sigma | 16.1 (3.9) | 36.0 (10.2) | 17.8 (4.8) |
| Rwork | 15.6% (18.9%) | ||
| Rfree | 20.7% (30.3%) | ||
| RMSD bonds | 0.012 Å | ||
| RMSD angles | 1.274 ° | ||
| RMSD chirals | 0.072 Å3 | ||
| Ramachandran plot -most favorable | 210 (96.6%) | ||
| additionally allowed | 7 (3.4%) | ||
2.3 Data processing and structure solution
Generally, the diffraction images were indexed, integrated and scaled using the d*TREK suite through the Crystal Clear interface (Pflugrath, 1999). For the iodide soak and the native data set, the strength of the anomalous signal was estimated with SHELXC. The positions of the anomalous atoms were found with SHELXD (Schneider and Sheldrick, 2002). Sites were refined and phases were calculated with SHARP (Bricogne et al., 2003). Phases were improved within SHARP using SOLOMON (Abrahams et al., 1996) and DM (Cowtan and Zhang, 1999). Non-crystallographic symmetry was not applied for initial phasing. ARP/wARP (Perrakis et al., 1999) was used for automatic model building. The structures were manually inspected and completed using COOT (Emsley and Cowtan, 2004) and refined with REFMAC5 (Murshudov et al., 1997) using TLS groups defined by the TLSMD server (Painter and Merritt, 2006). All crystallographic calculations were done within the CCP4 suite (Bailey, 1994).
Specific procedures and considerations for both data sets for which experimental phase information was derived, were as follows:
(i) NaI soak: Data were collected at λ=1.5418Å. The following atoms contribute to the anomalous scattering of the crystal: 12× I- (f″=6.75 e-), 2× Ca2+ (f″=1.28 e-), 10× Se (f″=1.14 e-), and 2× S (f″=0.56 e-). ShelxC indicated a strong anomalous signal over the entire resolution range. ShelxD (Schneider and Sheldrick, 2002) could find 16 sites, which in hindsight could be identified as iodide and calcium sites. After refinement and phasing with Sharp (Bricogne et al., 2003), and density modification with Solomon/DM (Abrahams et al., 1996; Cowtan and Zhang, 1999) within Sharp, ARP/wARP (Perrakis et al., 1999) could build 232 out of 246 residues of the asymmetric unit and assign all of them to the sequence.
(ii) Native protein: Data were collected at λ=2.06633 Å to 1.95 Å resolution. ShelxC indicated a significant anomalous signal up to 2.2 Å resolution. ShelxD could find two strong sites (Ca2+, f″=2.13 e-), and 13 weak sites (S from Met and Cys, f″=0.95 e-). After refinement of the positions and phasing with Sharp, and density modification with Solomon/DM within Sharp, ARP/wARP could build 228 out of 246 residues.
2.4 Functional Studies
Plasmid construction
DNA encoding EpsF 1-174 (hereafter also called “cyto1-EpsF”) was amplified by PCR and ligated into the pQE30 vector and then moved into the pMMB67 vector using EcoRI and SalI restriction enzymes. The resulting construct was named pMMB-cyto1-EpsF. For plasmid GFP-EpsF, primers CGGATCCGCCGCGTTTGAATACA and GCTGCAGACTATGTCGTTTTCGCC were used to amplify epsF flanked by BamHI and PstI restriction sites. After digestion of pMMB66-gfp with BamHI and PstI, gfp-epsF was constructed. Plasmid EpsF-GFP was constructed first using primers GAATTCGATGCGGGTGACTAAG and GGATCCACGACTCATTAAGTTATTC to amplify epsF flanked by EcoRI and BamHI restriction sites and then moved into pMMB66-Cterminal-gfp following digestion with EcoRI and BamHI.
Bacterial strains and growth conditions
Wild type and epsF mutant strains of V. cholerae were grown to stationary phase at 37°C in LB (Luria broth) supplemented with thymine and 200μg/ml of carbenicillin or in M9 growth medium supplemented with 4% casamino acids and 0.4% glucose. When indicated, over-expression of cyto1-EpsF was induced with isopropyl β-D-thiogalactopyranoside (IPTG).
Inhibition of secretion in wild type V. cholerae cells
The cyto1-EpsF construct (pMMB-cyto1-EpsF) was expressed in wild type V. cholerae with and without induction by IPTG, and tested for extracellular protease secretion using a fluorescence based assay (Johnson et al., 2007). Briefly, supernatants from overnight cultures grown in LB were assayed in 5 mM HEPES pH 7.5 and 0.05 mM N-tert-butoxy-carbonyl-Gln-Ala-Arg-7-amido-4-methyl-coumarin (Sigma-Aldrich, St. Louis MO) for 10 min at 37° C using the excitation and emission wavelengths 385 nm and 440 nm, respectively.
In vivo crosslinking
Cells of wt and epsF mutant strains of V. cholerae containing either pMMB-cyto1-EpsF or control plasmid pMMB67 were induced with IPTG (100 μM final concentration). Following centrifugation, cells were resuspended in phosphate buffered saline to OD600 of 6 and subjected to crosslinking. Disthiobis succinimidyl propionate (DSP) (Pierce, Rockford, IL) was added to a final concentration of 0, 250 or 400 μM and the cells were incubated at room temperature for 60 minutes. The crosslinker was then quenched by adding Tris-HCl, pH 8 to a final concentration of 50 mM. Cells were centrifuged and resuspended in 500 μl 50 mM Tris-HCl, pH 8 with 100 μg lysozyme and 10 μg DNase. After incubation for 10 minutes at room temperature, the suspensions were sonicated (1 sec pulse followed by 1 sec rest for 10 sec) and analyzed by SDS-PAGE with no reducing agent and immunoblotting with anti-EpsF antibodies as described previously (Johnson et al., 2007).
Co-purification of histidine-tagged cyto1-EpsF and EpsL
Cell extracts were incubated with 10 μl Cobalt-Immobilized Metal Affinity Chromotography Resin (IMAC beads; BD Biosciences, San Jose CA) in binding buffer (50mM Tris-HCl, pH 8.0, 1% Triton X-100) for 2 hours with rocking at 4°C. After incubation, samples were centrifuged, the supernatant removed and beads were washed three times with binding buffer and once with 50mM Tris-HCl, pH 8.0. 20μl of SDS-PAGE sample buffer containing DTT were added to the beads and boiled for 10 minutes prior to centrifugation. Supernatants were subjected to SDS-PAGE and immunoblotting with anti-EpsL antibody.
Expression of GFP fusion proteins
GFP-EpsF and EpsF-GFP fusions were expressed from plasmid pMMB67 in the V. cholerae epsF mutant strain with and without induction by IPTG (20 μM final concentration). Cells were washed and assayed for fluorescence with a BioTek microplate reader (BioTek Instruments, Winooski VT) using the excitation and emission wavelengths 380 nm and 440 nm, respectively.
2.5 Preparation of figures
For Figures 1 and 7a, sequences were aligned with T-coffee (Notredame et al., 2000). Rendering was done with ESPRIPT (Gouet et al., 1999). Figures 2 and 6 were prepared with PyMol (DeLano, 2002).
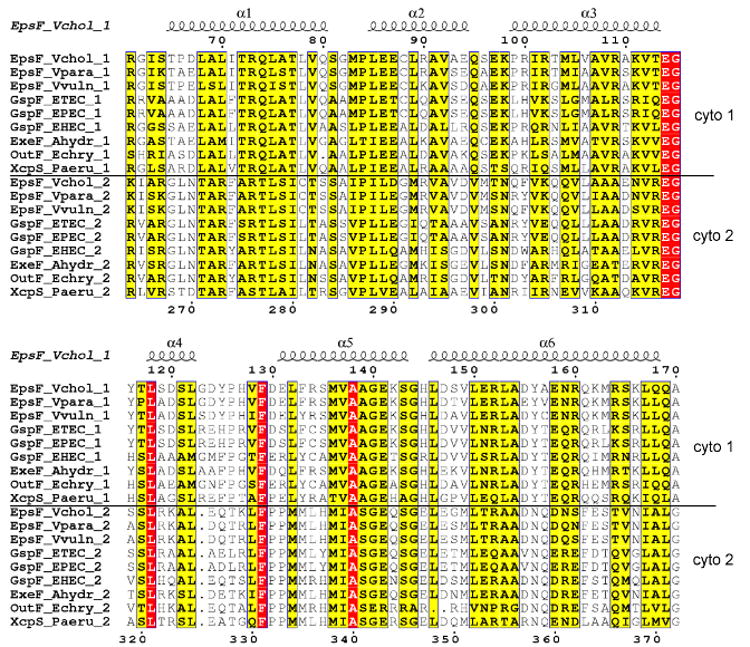
Figure 1. Family sequence alignment of the first cytoplasmic domain of EpsF and selected T2SS homologues.

Identical residues are in red, similar residues according to the Risler matrix are in yellow. The predicted transmembrane helix (Met 171 – Val 192) is indicated with a green bar. The extent of the construct is marked with the black arrows above the sequences. The blue bar below the sequences indicates the solvent accessibility of each residue: dark blue is solvent exposed, white is buried. Residues involved in Ca2+-binding are marked with red triangles. Residues which are part of the dimer interface are marked with blue stars. Alpha-helical secondary structure symbols above the cyto1-EpsF sequence represent the observed structure in our experimental model.
Figure 7. The second cytoplasmic domain of EpsF and a model of full-length EpsF dimer.

(A) Alignment of the two cytoplasmic domains of T2SS homologs of EpsF. The two cytoplasmic domains from the same species as in Figure 1 were aligned. The upper block of sequences are sequences of the first cytoplasmic domain, the lower block of sequences represents the second cytoplasmic domain. Residue numbers above the sequences refer to the first cytoplasmic domain of EpsF from V. cholerae, while residue numbers below the sequences refer to the second cytoplasmic domain of EpsF from V. cholerae. Secondary structure assignment refers to that of cyto1-EpsF56-171. Identical residues are in red, similar residues according to the Risler matrix are in yellow.
(B) Global model of a dimer formed by the cytoplasmic parts of two EpsF chains. The central part is identical to the cyto1-EpsF56-171 dimer (Figure 2). A second cytoplasmic domain is added with the same fold as the first domain on the basis of the high degree of sequence identity between the first and second EpsF domains (see text and Figure 7a). The orientation of the second domains with respect to the membrane is the same as of the first domains. The orientation of the second domains with respect to the first domains is arbitrary, but the two second domains are related by the same twofold axis relating the first cytoplasmic domains. Note that the N-terminal 70 residues, the two anti-parallel transmembrane helices TMH1 and TMH2, the 31-residue loop in the periplasm connecting these helices, and the C-terminal TMH3 plus the 17-residue tail in the periplasm are not included. The overall dimensions of this dimer are 81 by 55 Å parallel to the membrane, and 54 Å perpendicular to the inner membrane.
Figure 2. The dimer of cyto1-EpsF56-171 from V. cholerae.
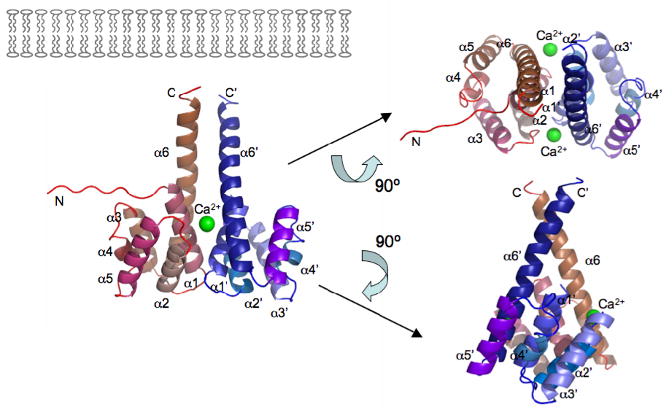
(A) Each cyto1-EpsF56-171 subunit consists of a six-helix bundle. The N-terminal residues have no secondary structure; the second half of the C-terminal helix protrudes away from the body of the domain and may extend towards the inner membrane as sketched in the left upper corner. Green spheres indicate calcium ions in the dimer interface. The dimer is shown in three different orientations related by 90° rotations about two different axes.
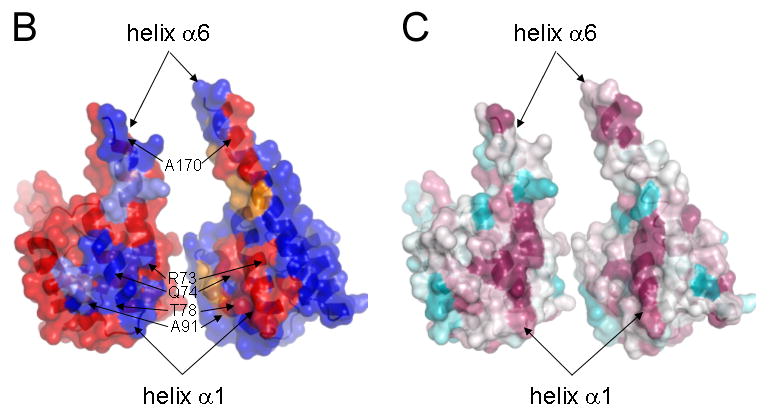
(B) The interface of the cyto1-EpsF56-171 dimer. In this butterfly figure, the surfaces of the two cyto1-EpsF56-171 domains per dimer are colored in red and blue, respectively. Residues involved in dimer interactions are colored with the same color as the domain they contact. Light colors of the “imprint” indicate residues involved in hydrogen bonds, dark colors indicate residues involved in hydrogen bonds and hydrophobic interactions. Twelve hydrogen bonds in the dimer interface involve the following residues: (i) Glu 74 and Thr 77 provided by helix α1; (ii) Glu 95 from the loop between α2 and α3; and, (iii) Gln 161, Lys 162, Ser 165, Lys 166 and Glu 172, all belonging to the C-terminal part of helix α6. The following residues are involved in hydrophobic interactions: Leu 70, Arg 73, Gln 74, Thr 77, Leu 78, Gln 80 and Ser 81 provided by helix α1; Ala 91 from helix α2; and Lys 166, Gln 169, Ala 170, Leu 174 from helix α6. Clearly, most of the dimer interactions occur between equivalent helices α1 and α1′ and between α6 and α6′.
(C) Conservation of interface residues according to the Consurf server (Landau et al., 2005)based on the sequence alignment of T2SS homologs of EpsF as shown in Figure 1. Comparison of Figures 2b and 2c shows that most dimer interactions involve residues from helices α1 and α6 and that interface residues are conserved. (Red: well conserved; blue less conserved).
Figure 6. Calcium binding site in the dimer interface of V. cholerae cyto1-EpsF56-171.

Calcium is coordinated as an approximate tetragonal bipyramid by Glu 97 from chain A, and Glu 151 and Asp 155 from chain B, and three water molecules. Asp 155 is binding the calcium ion in a bidentate manner. Arg 73 forms a salt bridge with both Glu 151 and Asp 155. Glu 151 has an additional hydrogen bond with Gln 80. The calcium ion is surrounded by anomalous Fourier electron density contoured at 7.5 sigma colored in gold.
3. Results
3.1 Structure Determination
An extensive search was required in order to obtain a GspF construct that could be crystallized. Initially, full length EpsF proteins from V. cholerae, V. vulnificus, V. parahaemolyticus, and GspF from ETEC, were expressed and purified in the presence of various detergents. Despite good expression and homogeneous preparations, as determined by SDS-PAGE and size exclusion chromatography, no crystals were obtained. The first cytoplasmic domain of EpsF from all four species (spanning residues 1-171 in V. cholerae) could not be crystallized either. As the degree of sequence homology between different species drops significantly between residues ∼ 45 and ∼ 60 (Figure 1), constructs with several N-terminal truncations were made and tested for crystal growth. Eventually, an N-terminally truncated version of V. cholerae EpsF containing residues 56-171 yielded diffraction quality crystals, but solely in the presence of calcium ions, whereas the presence of magnesium gave crystals of poor quality.
Crystals of Se-Met cyto1-EpsF56-171 diffracted very well and an initial 1.7 Å resolution data set could be collected in house. In order to obtain experimental phase information to determine the structure, anomalous scattering differences were measured at long wavelengths to obtain phases by two independent approaches. Since it was unknown beforehand which phasing method would work, the following two additional data sets were collected. First, a crystal of Se-Met protein was transferred to an iodide-containing buffer and diffraction data to 1.9 Å resolution were collected on a rotating anode generator with λ=1.5418 Å (Table 1). A sufficiently large signal was obtained from the anomalous scatterers iodide and calcium to allow an initial structure determination as described in Materials and Methods. Second, a crystal of native protein was exposed at the SSRL synchrotron using a wavelength of 2.0663 Å, and a dataset up to 1.95 Å resolution was collected (Table 1). Here, calcium and sulphur provided a sufficiently strong anomalous signal such that useful phase information could be extracted (Materials and Methods). Refinement against the 1.7 Å resolution data set yielded a final structure with an Rwork of 15.6 %, an Rfree of 20.7 % and good geometry (Table 1). The crystal structure for cyto1-EpsF56-171 reported in this paper consists of consecutive residues Phe 57 to Ser 171 for chain A, and residues Ser 62 to Gln 177 for chain B. Residues Ser 171 to Gln 177 originate from the vector sequence of the C-terminal TEV-cleavable His6-tag.
3.2 Tertiary and quaternary structure of cyto1-EpsF56-171
The first domain of V. cholerae EpsF adopts an all-helical fold (Figure 2) with its six helices forming two layers. One layer is made up by helices α1, α2 and α6, and the other by α3, α4 and α5, with adjacent helices running anti-parallel to each other. Four of the six helices have a rather similar length of 10 to 15 residues, helix α4 consists of only five residues, whereas the long C-terminal helix α6 consists of 23 and 30 residues in chains A and B, respectively. Starting from residue Lys162, the C-terminal part of helix α6 protrudes from the body of the domain (Figure 2). The high helix content of EpsF is in agreement with the result of CD studies (Collins et al., 2007) on the homolog PilG. Neither of the common structural similarity search programs SSM (Krissinel and Henrick, 2004) or DALI (Holm and Sander, 1993) reported proteins with significant structural homology. Therefore, we assume that cyto1-EpsF56-171 has a novel fold.
The cyto1-EpsF56-171 crystals contain two types of dimers with a substantial interface. One is an arrangement in which two α6 helices from different subunits run anti-parallel to each other, making interactions along their entire length (not shown). It is unlikely that this is an arrangement of physiological relevance because of the different directions of the two α6 helices in this dimer. The α6 helices are each continued by a transmembrane helix (TM helix) which starts 2 to 3 residues after the end of helix α6 (see discussion below). If one α6 helix would continue as a TM helix into the inner membrane, the end of the other α6 helix in this dimer would be more than 50 Å away from the inner membrane surface.
Two symmetry-related cyto1-EpsF56-171 chains form another dimer with approximate dimensions of 55 by 55 by 35 Å (Figure 2a). There are in total 14 residues involved in intra-dimer contacts, engaging in a mixture of hydrogen bonds and hydrophobic interactions (Figures 1 and 2). The interface between the two molecules in the dimer buries 1750 Å2 solvent accessible surface area. This is in the range often observed for physiological dimers (Jones and Thornton, 1996). A gap volume index of 2.26 Å, when compared to values for physiological relevant interfaces (Jones and Thornton, 1996), indicates excellent complementarity of the interface surfaces. Although gel permeation chromatography indicates that cyto1-EpsF56-171 is monomeric in solution (not shown), the high protein concentration in the crystal likely promotes a concentration-dependent oligomerization of cyto1-EpsF56-171 that may be reflective of its oligomerization within the T2SS in vivo.
Support for intracellular oligomerization of cyto1-EpsF comes from in vivo cross-linking experiments (Figure 3). When intact V. cholerae cells that express cyto1-EpsF were incubated with the membrane-permeable amine-reactive cross-linker dithiobis succinimidyl propionate (DSP) and then subjected to SDS-PAGE and immunoblotting with anti-EpsF antibodies, cyto1-EpsF was found to migrate as a dimer of approximately 40 kDa (Figure 3). Additionally, several higher molecular mass species were detected suggesting that cyto1-EpsF is capable of oligomerization. This is in agreement with the finding that the N-terminal cytoplasmic domain of BfpE, a cyto1-EpsF homolog, forms dimers in the yeast two-hybrid system (Crowther et al., 2004). Although we were able to detect oligomers of cyto1-EpsF, the cross-linking was inefficient and only a fraction of cyto1-EpsF was oligomeric. This is common for in vivo cross-linking and may reflect low cell envelope permeability of the cross-linker and/or lack of free amines that are in close proximity to the subunit interface and available for cross-linking. The low cross-linking efficiency and the small amount of full length EpsF produced in these cells likely prevented the detection of cross-linked EpsF species.
Figure 3. In vivo oligomerization of cyto1-EpsF.
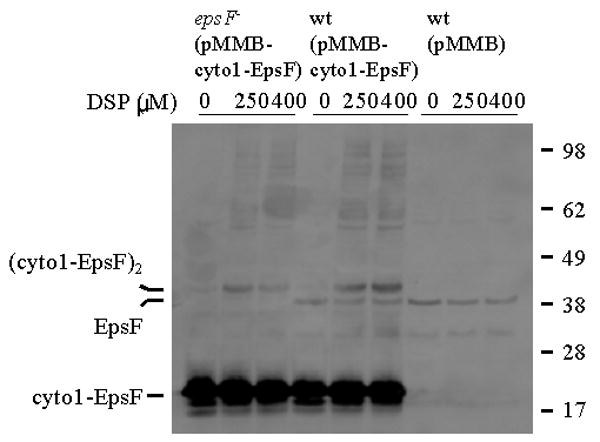
Wild type and epsF mutant strains of V. cholerae containing either pMMB67 or pMMB-cyto1-EpsF were grown in LB in the presence of IPTG to induce expression of cyto1-EpsF. Following growth, intact cells were incubated with increasing concentrations of DSP for 60 min. Then, the cells were centrifuged and subjected to SDS-PAGE and immunoblot analysis using anti-EpsF antibodies. The position of native EpsF and monomeric and cross-linked dimeric forms of cyto1-EpsF is indicated on the left. Molecular mass markers are shown on the right.
We do believe that in vivo oligomerization of cyto1-EpsF does not represent an artifact due to misfolding as cyto1-EpsF is produced in a soluble form (Section 2.1) and is capable of interacting with the remainder of the T2SS as indicated by the following experiments. We found that when cyto1-EpsF was expressed at increasing concentrations in wild type V. cholerae (Figure 4a), it inhibits secretion in a concentration-dependent manner (Figure 4b). This may occur through the formation of mixed non-active oligomers of cyto1-EpsF and full length EpsF. Alternatively, cyto1-EpsF may compete with native EpsF for interaction with EpsE and/or EpsL. To test this hypothesis, we examined the ability of cyto1-EpsF to interact with EpsE and EpsL using metal affinity chromatography. Cell extracts of V. cholerae expressing histidine-tagged cyto1-EpsF were incubated with Cobalt-Immobilized Metal Affinity Chromotography Resin (IMAC beads). Following washing steps, bound proteins were eluted from the IMAC beads and subjected to SDS-PAGE followed by immunoblotting with antibodies directed against either EpsL or EpsE to detect proteins capable of binding to cyto1-EpsFHis6. We reasoned that histidine-tagged cyto1-EpsF would bind to the metal affinity resin and that proteins capable of interacting with cyto1-EpsFHis6 would also be recovered. EpsL was co-purified with cyto1-EpsFHis6 when cyto1-EpsFHis6 was expressed in both epsF mutant and wild type cells (Figure 5, lanes 7 and 8), indicating that EpsL and cyto1-EpsF are capable of interaction in vivo. EpsL alone did not react with IMAC beads, and was therefore not detected without cyto1-EpsFHis6 expression (Figure 5, lanes 5 and 6). No detectable interaction between cyto1-EpsFHis6 and EpsE was observed (data not shown).
Figure 4. Cyto1-EpsF inhibits secretion of protease via the T2SS in V. cholerae.
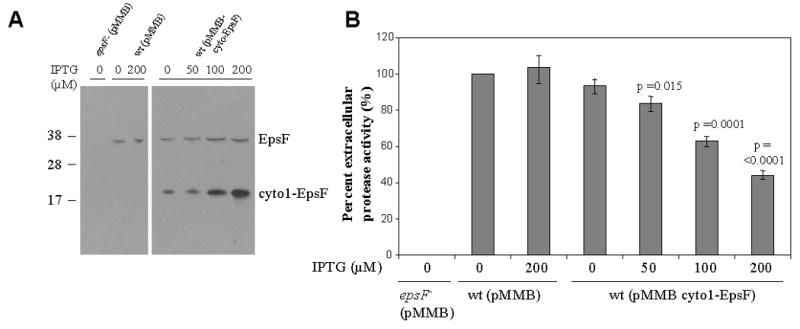
Wild type V. cholerae containing either pMMB67 as negative control or pMMB-cyto1-EpsF was grown in LB in the absence or presence of increasing concentrations of IPTG to induce the expression of cyto1-EpsF. Culture supernatants and cells were separated by centrifugation. (A) Cells were disrupted and subjected to SDS-PAGE and immunoblotting with anti-EpsF antibodies to determine the relative level of cyto1-EpsF expression at different IPTG concentrations. The position of molecular mass markers is shown on the left and native EpsF and cyto1-EpsF are indicated on the right. (B) Culture supernatants were tested for the presence of extracellular protease. The rate of hydrolysis was obtained from three independent experiments and the results are presented as percent extracellular activity ± standard error (SEM). The extracellular activity in the supernatant from the negative control was set to 100%. The epsF mutant, which is unable to secrete the protease, is shown as positive control.
Figure 5. Interaction of cyto1-EpsF and EpsL.
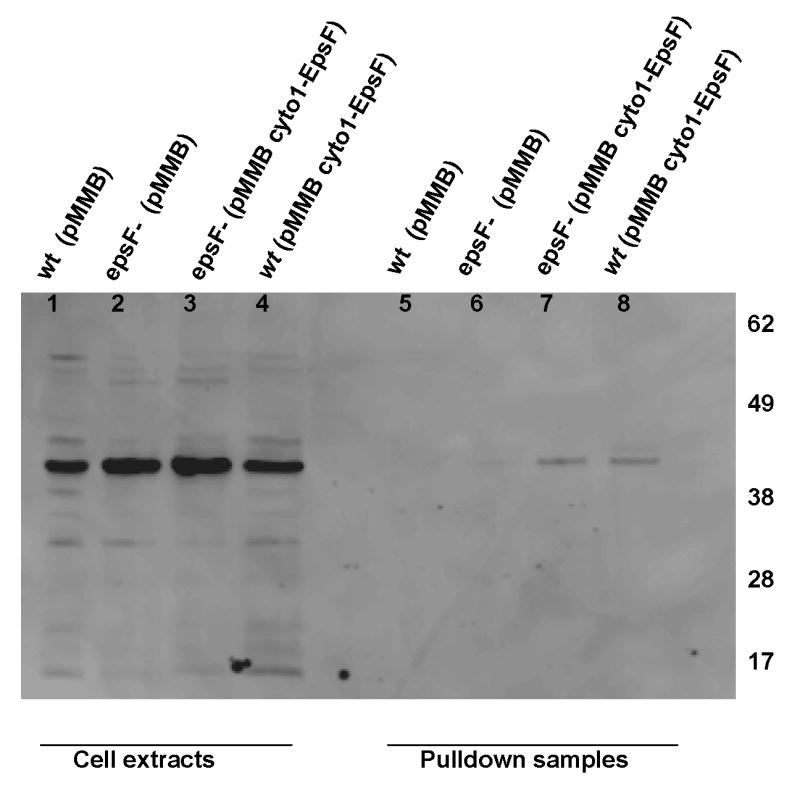
EpsL was co-purified with cyto1-EpsFhis6 from V. cholerae cell extracts by metal-affinity chromatography and subjected to SDS-PAGE and immunoblotting with anti-EpsL antibodies. Lanes 1-4 show the level of EpsL detected in cell extracts prior to purification, and lanes 5-8 represent EpsL proteins that bound to hexahistidine-tagged cyto1-EpsF. No EpsL was purified when cyto1-EpsF was not expressed (lanes 5 and 6). Positions of molecular weight markers are shown.
3.3. The metal binding sites
In the interface between the two subunits in the crystal structure, extra density is observed which indicates the presence of two metal ions which are 21.5 Å apart and follow the non-crystallographic twofold of the cyto1-EpsF56-171 dimer (Figure 2a). We assume that these ions are calcium ions because of: (i) the anomalous difference maps in the λ=1.5418 Å data reveal 12.2 to 12.8 sigma peaks at the metal binding sites which is slightly higher than the 10.2 sigma peak at the Se positions with the highest anomalous peaks, which agrees with the f″=1.28 electrons for Ca2+ and the f″=1.14 electrons for selenium at this wavelength; (ii) the anomalous difference maps in the λ=2.0663 Å data set show 34.1 and 33.5 sigma peaks at the two metal binding sites, compared to the 14.2 sigma peak at the highest sulphur position, which agrees with the f″=2.13 electrons for Ca2+ and the f″=0.95 electrons for sulphur ions at this wavelength; (iii) refinement as calcium ions at full occupancy yields temperature factors for the cations of 11.4 Å2 in chain A and 13.6 Å2 in chain B which are close to the average temperature factors of 11-18 Å2 and 14-24 Å2, respectively, for the liganding atoms.
The conclusion that the metal ions are calcium is further supported by the environment of the sites. Since these are so similar in the two cases (Table 2), we will only describe here site A, shown in Figure 6. The protein metal ligands are one carboxylate oxygen from Glu 151 and two carboxylate oxygen atoms provided by Asp 155 from one subunit, with Glu 97 from the second subunit providing a fourth carboxylate oxygen as ligand. Three water molecules complete the coordination sphere. The ligands of each Ca2+ ion form a distorted tetragonal bipyramid, involving seven atoms since Asp 155 is ligating in a bidentate manner. The ligands are all oxygen atoms, with the three protein ligands being negatively charged, as is often the case in calcium binding sites in proteins. The distances between Ca2+ and its ligands (Table 2) are very close to values reported in the literature (Harding, 2006), which is in good agreement with all other data mentioned above indicating that two calcium ions are bound per cyto1-EpsF56-171 dimer at symmetry-related sites. The guanidinium group of Arg 73 interacts with the metal-coordinating carboxylates of both Glu 151 and Asp 155, and appears to have a “ligand-positioning” function (Figure 6).
Table 2. Calcium to Ligand distances in the dimer of the first cytoplasmic domain of V. cholerae EpsF.
| Ligand atom | Ca 1 | Ca 2 | Literature value |
|---|---|---|---|
| Glu 97 Oε2 | 2.32 | 2.30 | 2.36 |
| Glu 151 Oε2 | 2.27 | 2.27 | 2.36 |
| Asp 155 Oδ1 | 2.54 | 2.54 | < 2.8 * |
| Asp 155 Oδ2 | 2.52 | 2.51 | < 2.8 * |
| Wat 1 O | 2.38 | 2.42 | 2.39 |
| Wat 2 O | 2.46 | 2.33 | 2.39 |
| Wat 3 O | 2.49 | 2.38 | 2.39 |
Inter-atomic distances are given in Å for both Ca2+-ions in the dimer. For comparison, recently revised literature values are included in the last column (Harding, 2006). (*) These literature values (Harding, 2006) refer to a bidentate ligand.
3.4 Conservation of the cyto1-EpsF dimer and the metal binding site in the EpsF family
Sequences of the first cytoplasmic domains of the GspF family are well conserved (Figure 1; Table 3). When comparing V. cholerae cyto1-EpsF56-171 with T2SS homologs, the amino acid sequence identity for the 116 residues in the truncated domain for which the structure was solved, ranges from 86 % between V. cholerae and V. vulnificus to 46 % when comparing V. cholerae EpsF and P. aeruginosa GspF (Table 3). When considering the 14 residues involved in the interactions across the interface then these residues from V. cholerae share 100 % identity with the homologs from two other Vibrio species, and an identity of 64 % or higher with GspF homologs. Therefore, taking also into account the extensive and well-packed nature of the interface observed in the cyto1-EpsF56-171 dimer (Figure 2), it is likely that all N-terminal domains of GspF homologs use a similar interface to form dimers.
Table 3. Conservation of overall conservation versus contact residues in the first cytoplasmic domains in the T2SS EpsF family.
| T2SS homolog of EpsF in the following species: | % identity of EpsF domain 1 (residues 55-168) vs. V. cholerae | % identity of 14 dimer contact residues vs. V. cholerae |
|---|---|---|
| Vibrio cholerae | 100 | 100 |
| Vibrio parahaemolyticus | 86 | 100 |
| Vibrio vulnificus | 84 | 100 |
| ETEC | 64 | 86 |
| EPEC | 65 | 86 |
| EHEC | 57 | 64 |
| Aeromonas hydrophila | 69 | 64 |
| Erwinia chrysanthemi | 56 | 64 |
| Pseudomonas aeruginosa | 46 | 71 |
The three residues involved in Ca2+-binding by the first domain, and the “ligand-positioning” Arg 73, are conserved well in the GspF family (Figure 1). Of particular interest is that the bidentate ligand Asp 155 of V. cholerae EpsF (Figure 6) is only once replaced by a Glu, suggesting that the side chain at this position coordinates a metal ion in all GspF family members in a bidentate manner. The second calcium ligand, Glu 97, is only once substituted by another amino acid: a Thr in the GspF homolog of P. aeruginosa. This still would allow the side chain oxygen atom from this Thr to coordinate a calcium ion. The third calcium-coordinating residue, Glu 151, is frequently replaced by an Asn, and once by a Ser, i.e. each time by a residue which still could provide an oxygen atom as ligand to the metal binding site. Interestingly, in each member of the GspF family, two or three negatively charged carboxylates are available to coordinate divalent cations at this site. The side chains of the coordinating Glu 151 and Asp 155 interact with Arg 73, which is an invariant residue. Hence, replacing the Asp 155 by a Glu, or Glu 151 by an Asn or Ser, most likely allows the geometry of this Arg-stabilized calcium site to be essentially maintained, since the arginine's guanidinium group interacts with the very oxygen atom which is liganding the metal ion (Figure 6). This would explain why arginine is consistently present at this position across the GspF family.
3.5 Similarity between the second and the first cytoplasmic domains of GspF
A significant degree of amino acid sequence similarity exists between the first and the second cytoplasmic domains of V. cholerae EpsF (Table 4), as has been noted previously (Peabody et al., 2003) for members of the GspF/PilG/BfpE superfamily. When comparing residues 65-162 from the solved cyto1-EpsF56-171 structure with residues 268-364 of the second domain of V. cholerae EpsF (hereafter also called “cyto2-EpsF”), 28 % of the residues are identical. Importantly, the levels of identity between the corresponding sequences in the first and second domains in GspF homologs are similar or higher (Figure 7a; Table 4). The highest identity, 34 %, occurs between the first and second domain of the GspF homolog in P. aeruginosa. Interestingly, the first domain of V. cholerae EpsF displays a sequence identity with the second domains of the GspF family ranging from 28 to 39 %. Because of this clearly detectable homology between the cyto1-GspF and cyto2-GspF domains throughout the GspF family, it seems reasonable to assume that across the family both cytoplasmic domains adopt the same all-helical cyto1-EpsF fold.
Table 4. Comparison of the amino acid sequence in the first versus the second cytoplasmic domains in the T2SS GspF family. (For alignment see Figure 6a).
| Species | % identity of Domain 1 vs. Domain 2 within each species at left | % identity of V. cholerae EpsF Domain 1 (Residues 65-162) vs. species Domain 2 |
|---|---|---|
| Vibrio cholerae | 28 | 28 |
| Vibrio parahaemolyticus | 29 | 29 |
| Vibrio vulnificus | 28 | 29 |
| ETEC | 31 | 31 |
| EPEC | 30 | 32 |
| EHEC | 33 | 34 |
| Aeromonas hydrophila | 28 | 36 |
| Erwinia chrysanthemi | 31 | 35 |
| Pseudomonas aeruginosa | 34 | 39 |
The three residues which provide the ligands for Ca2+-binding sites, Glu 97, Glu 151 and Asp 155, plus the invariant Arg 73 that interacts with both Glu 151 and Asp155, are only poorly conserved in the second domain of T2SS EpsF homologs (Figure 7a). Of the 14 residues involved in the cyto1-EpsF56-171 interface only 1 or 2 are conserved in the second cytoplasmic domain of the homologs. Since the metal binding sites occur in the dimer interface of the cyto1-EpsF dimer (Figure 2), it is unlikely that cyto2-EpsF, while adopting most likely a similar fold as cyto1-EpsF56-171, forms the same type of dimers as cyto1-EpsF56-171.
3.6 Localization of fusion proteins of EpsF with GFP
Membrane topology prediction of EpsF with the TMHMM program (Krogh et al., 2001) identified three putative transmembrane helices and suggested a cytoplasmic location for the N-terminus and a periplasmic location for the C-terminus of EpsF. In order to test the predicted topology of EpsF, we constructed Green Fluorescent Protein (GFP) fusions to both the N-terminus and the C-terminus of EpsF. Both fusion proteins were detected by immunoblotting with anti-EpsF antibodies (data not shown) when expression of the fusion proteins was induced in V. cholerae. When the level of fluorescence was measured in these strains, we found GFP fused to the N-terminus of EpsF (GFP-EpsF) was fluorescent, whereas fusion of GFP to the C-terminus (EpsF-GFP) resulted in a non-fluorescent protein (Figure 8). This latter finding suggests that the C-terminus of EpsF is likely located in the periplasmic space, as GFP is unable to fold in the periplasm due to the reducing environment of this compartment (Feilmeier et al., 2000). Although non-fluorescent, EpsF-GFP was able to complement the secretion defect in the epsF mutant strain, suggesting that the protein is inserted in the membrane similarly to non-tagged EpsF (data not shown).
Figure 8. Fusion of GFP to the EpsF C-terminus results in a non-fluorescent protein.
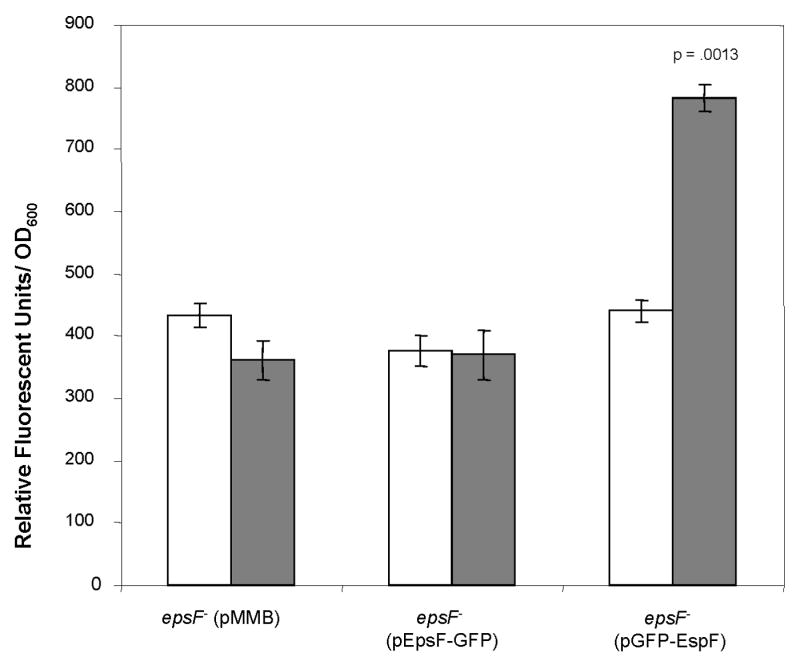
V. cholerae epsF mutant strains containing pMMB alone, or expressing either GFP-EpsF or EpsF-GFP were grown in M9 growth medium and carbenicillin without (white bars) and with (grey bars) IPTG at a final concentration of 20 μM to induce expression of the fusion proteins. Following growth, fluorescence was measured using the excitation and emisssion wavelengths 380 nm and 440 nm, respectively. Results from three independent experiments are presented as average relative fluorescence units ± standard error.
4. Discussion
4.1 Phasing at long wavelengths
We have solved the V. cholerae cyto1-EpsF56-171 crystal structure along two different routes using anomalous scattering effects at long X-ray wavelengths. Both approaches, one a halide soak with a seleno-Met EpsF crystal and using CuKα radiation, and the other sulphur-cum-calcium phasing with X-rays of wavelengths larger than 2 Ångströms with a sulphur-Met crystal (see Materials and Methods), were successful. The structure solution of cyto1-EpsF56-171 is therefore another encouraging example how relatively long wavelength radiation can lead to de novo crystal structure determinations (for other examples, see (Dauter et al., 2000; Dauter et al., 1999; Liu et al., 2000; Weiss et al., 2001a; Weiss et al., 2001b; Xu et al., 2005; Yang et al., 2003). The availability of data sets with different wavelengths was, serendipitously, very useful in verifying the nature of the metal bound as described above.
4.2 The structure of cyto1-EpsF and implications for its orientation with respect to the inner membrane
The topology with a double layer of three anti-parallel helices that is formed by cyto1-EpsF56-171 has not been observed previously. Two cyto1-EpsF56-171 domains form a tight dimer and contain two symmetry-equivalent calcium sites (Figure 2). This suggests that a tight interaction between the first cytoplasmic domains very likely also occurs in full length EpsF, and possibly also in the assembled T2SS. Support for this suggestion comes from analysis of cyto1-EpsF in V. cholerae. When cyto1-EpsF was expressed in an otherwise wild type V. cholerae strain, the amount of protease secreted was reduced as the expression of cyto1-EpsF was increased (Figure 4). This dominant negative effect on secretion indicates that cyto1-EpsF interacts with the rest of the T2SS and provides information that cyto1-EpsF is likely folded into a conformation that is very similar to the first cytoplasmic domain in full length EpsF. One possible mechanism for the negative dominance is that cyto1-EpsF interacts with full length EpsF thus preventing the formation of functional EpsF oligomers. Although we were unable to demonstrate an interaction between cyto1-EpsF and EpsF in vivo, likely due to low cross-linking efficiency, we did detect cross-linked dimers and larger oligomers of cyto1-EpsF (Figure 3) suggesting that cyto1-EpsF has the potential to form mixed inactive oligomers with EpsF. An alternative explanation for the dominant negative effect is that cyto1-EpsF competes with EpsF for interaction with EpsL as cyto1-EpsF and EpsL were shown to co-purify from V. cholerae cell extract (Figure 5). Either way, the functional data suggest that the fold of cyto1-EpsF56-171 as determined in our crystal structure is likely identical or close to that of the first domain of EpsF in the native T2SS.
The long protruding C-terminal helix α6 (Figure 2a) is followed by approximately 4 residues connecting α6 with the predicted TMH1 (Figure 1). Interestingly, helices α6 and α6′ from both molecules in the cyto1-EpsF56-171 dimer point in a similar direction and likely both continue into the membrane. Helix α6 and α6′ make an angle of about 45 degrees with each other and if continuing in the same direction the predicted TMH1 helices from the two subunits would be far apart in the lipid bilayer. However, Pro 174 near the N-terminus of TMH1 could introduce a kink in this transmembrane helix that might allow interactions between the two TMH1 helices within the membrane. Alternatively, the TMH1 helices may interact with other transmembrane helices of EpsF subunits in the dimer, or with transmembrane helices from other T2SS proteins. Whatever the actual case will be, the location and extended nature of the C-terminal α-helices of the cyto1-EpsF56-171 dimer suggest an approximate orientation with respect to the inner membrane as sketched in Figure 2a, where in both subunits the loops between helices α2 and α3, and between α4 and α5, are nearest to the membrane while the α1-α2 and α3-α4 loops face the cytoplasm.
The N-terminal nine residues 56-64 of cyto1-EpsF56-171 in our structure adopt an extended conformation and are only observed in one molecule of the asymmetric unit. This region corresponds to a drop in sequence homology in the T2SS EpsF subfamily (Figure 1). The DISOPRED server (Ward et al., 2004) indicates disorder only for residues Val 43 to Gly 62 (data not shown), whereas residues 1–45 are well conserved in the EpsF family and could possibly form a well-folded subdomain connected via a flexible linker, comprising residues ∼45 – ∼60, to the globular cyto1-EpsF56-171 domain. Truncating the N-terminal subdomain from residue 1 to 55 appeared to be crucial for crystallization. This is similar to the requirement for crystallization of another T2SS protein, the secretion ATPase EpsE from V. cholerae (Robien et al., 2003). There, the N1-domain of ∼ 90 residues needed to be removed in order to obtain crystals. Yet, the N1-domain of EpsE has a critically important function: binding to the cytoplasmic domain of EpsL, as observed in the crystal structure of the N1-domain of EpsE in complex with the cytoplasmic domain of the inner membrane T2SS protein EpsL. This leads to membrane association of EpsE and stimulation of its ATPase activity (Abendroth et al., 2005; Camberg et al., 2007; Sandkvist et al., 1995). Similarly, the N-terminal subdomain of EpsF might be involved in critical interactions and motions during T2SS assembly or function.
4.3 Metal binding sites
Two symmetry-equivalent metal binding sites are present in the interface of the cyto1-EpsF56-171 dimer (Figure 2). Strong anomalous scattering, a calcium-dependent crystallization and the geometry of the binding site identified the metals as calcium ions. Notably, the calcium binding residues are conserved within the GspF family (Figure 1). Whereas cyto1-EpsF56-171 could only be crystallized in the presence of rather high concentrations (200mM) of externally added Ca2+, both protein and crystals were extremely sensitive to the nature of divalent ion during crystallization experiments. When calcium salts were replaced even with small amounts of strontium, barium, or lanthanide salts, protein in crystallization drops precipitated immediately. Moreover, crystals grown in the presence of calcium disintegrated when calcium was replaced with strontium, barium or lanthanides. Crystals obtained from Mg2+-containing solutions were far inferior in quality to those obtained from Ca2+-containing buffer. Undoubtedly, the presence of calcium ions has been critical for crystal stability. The significant degree of conservation of the ligands of the calcium site and of the invariant “Ca-ligand positioning” Arg 73, (Figures 1 and 6) suggests that calcium-binding by EpsF and T2SS homologs may be important during one or more of the many steps in the assembly or functioning of the T2SS system. Obviously, the precise nature of the metal bound under physiological conditions at the putative metal binding sites of EpsF remains to be established.
4.4. Two similar domains and membrane topology of EpsF and its T2SS homologs
The second half of full length V. cholerae EpsF displays a significant degree of sequence homology with the first cytoplasmic domain with a sequence identity of 28 % for 97 residues. This degree of sequence identity is similar or higher across the GspF family of EpsF homologs (Figure 7a, Table 4) suggesting that full length EpsF and its T2SS homologs contain two cytoplasmic domains of similar fold. This is in agreement with the analysis of Peabody et al., who reported that in the GspF/PilG/BfpE superfamily of membrane proteins a homology exists between the N-terminal and C-terminal half of members in this large superfamily of bacterial inner membrane proteins (Peabody et al., 2003).
A transmembrane helix prediction with the program TMHMM (Krogh et al., 2001) for EpsF from V. cholerae indicates three transmembrane helices, with residues 171-193 forming TMH1, residues 220-242 TMH2 and residues 370-392 TMH3 (not shown). This is perfectly compatible with helix α6, observed in the crystal structure of the first cytoplasmic domain of EpsF, ending at approximately position 168, i.e. just prior to TMH1, and residues ∼260 to ∼370 forming the second cytoplasmic domain, i.e. situated between TMH2 and TMH3. Given the significant sequence identities between EpsF and its T2SS homologs (Figure 1, Tables 3 and 4), and the very similar pattern of transmembrane helix prediction across the GspF family (data not shown), it is likely that the entire GspF family has the same topology with two similarly folded cytoplasmic domains and three transmembrane helices. This conclusion is in agreement with the three-transmembrane membrane topology arrived at for E. carotovora GspF (Thomas et al., 1997) and for P. aeruginosa GspF (Arts et al., 2007) using fusion protein techniques, and with the GFP fusion studies shown in Figure 8.
4.5 Topology of non-T2SS EpsF homologs
The GspF/PilG/BfpE superfamily has a diversity of members with weak sequence homology between the three families, although with detectable similarity between the first and second halves of these proteins (Peabody et al., 2003). For BfpE from Enteropathogenic E. coli, a four-transmembrane helix topology has been proposed in the basis of biochemical studies (Blank and Donnenberg, 2001), which is clearly different from the evidence for a three-transmembrane helix topology for the T2SS GspF family, based on: (i) GspF-enzyme fusion studies (Arts et al., 2007; Thomas et al., 1997) (ii) in vivo fluorescence studies on EpsF fusions with GFP (Figure 8); (iii) the topology of the first domain of EpsF (Figure 1) coupled with the significant degree of sequence identity between the first and second domains of T2SS family members (Table 4).
It might be possible that members of the superfamily do have weak sequence identity but different membrane topologies. After all, among many similarities, substantial differences between the T2SS and the T4PB exist, in particular regarding the composition of the Inner Membrane Complex. In adition, while both PilG and EpsF are required for the function of the T4P and T2S systems, respectively, they may support these systems with slighty different mechanisms. For example, it has been proposed that in N. menigitidis the GspF homolog PilG counteracts pilT-mediated pilus retraction (Carbonnelle et al., 2006), yet, retraction of T2S pseudopilus has not been demonstrated, nor does T2SS contain a PilT paralog. Due to these differences it seems best to refrain here from extrapolating the GspF three-transmembrane helix topology to other members of the GspF/PilG/BfpE superfamily. New structural data will be required to clarify the intriguing results obtained by different studies so far.
4.6 Full-length EpsF dimers
In order to arrive at a global model of a full-length EpsF dimer with the same conserved interface between the cyto1-EpsF56-171 domain as observed in our structure (Figure 2), we need to add to each of the subunits: (i) a second domain of very similar topology, connected by two anti-parallel transmembrane helices, TMH1 and TMH2, to the first domain, and (ii) a transmembrane helix, TMH3, to the C-terminus of cyto2-EpsF. The orientation of the second domains with respect to the membrane is likely to be roughly the same as that of the first cytoplasmic EpsF domains since the number of residues between the putative α6 and TMH3 is similar to the number of residues between the end of helix α1 of cyto1-EpsF and the start of the predicted TMH1. The mutual orientation of the first and second domains remains to be established, but the requirements of a compact dimer of full length EpsF subunits makes a general shape as depicted in Figure 7b a possibility.
It is of interest to compare the approximate model of the V. cholerae EpsF dimer (Figure 7b) with the 22 Å-resolution structure of the N. meningitidis PilG multimer (Collins et al., 2007) obtained from cryo-electron microscopy studies. The dimensions of our global model of the cytoplasmic part of EpsF dimer are, in the plane parallel to the membrane, approximately 70 by 80 Å when adding an estimate of the space required for the N-terminal domains absent from our structure. The model contains four quite similar major domains which are at low resolution essentially indistinguishable and therefore the model is best compared with the C4 electron microscopy reconstruction (Collins et al., 2007). The dimensions parallel to the membrane of the electron microscopy reconstruction of PilG are ∼80 × 80 Å which agrees reasonably well with our model. The “height” of the EpsF model perpendicular to the membrane extending into the cytoplasm is approximately 54 Å, which is slightly less than the 70 Å of the PilG reconstruction. Nevertheless, the EpsF model does suggest that the quaternary structure of the EpsF-PilG inner membrane protein families is more likely a dimer than a tetramer, options which were both considered possible (Collins et al., 2007).
5. Protein Data Bank accession codes
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank and are available under accession code 3C1Q, 2VMA, and 2VMB.
Supplementary Material
Acknowledgments
We acknowledge Stewart Turley for expert help with data collection. We thank the staff of beamline 9.2 at the Stanford Synchrotron Radiation Lightsource for support during data collection. This research was supported by NIH grant AI34501 to W.G.J.H. from the National Institute of Allergy and Infectious Diseases (NIAID) and by the Howard Hughes Medical Institute (HHMI) and by grant AI49294 from NIAID to M.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the HHMI, NIAID or the National Institutes of Health.
Appendix A. Supplementary Data
Supplementary data associated with this article can be found in the online version.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abendroth J, Bagdasarian M, Sandkvist M, Hol WGJ. The structure of the cytoplasmic domain of EpsL, an inner membrane component of the type II secretion system of Vibrio cholerae: An unusual member of the actin-like ATPase superfamily. Journal of Molecular Biology. 2004a;344:619–633. doi: 10.1016/j.jmb.2004.09.062. [DOI] [PubMed] [Google Scholar]
- Abendroth J, Rice AE, McLuskey K, Bagdasarian M, Hol WGJ. The crystal structure of the periplasmic domain of the type II secretion system protein EpsM from Vibrio cholerae: The simplest version of the ferredoxin fold. Journal of Molecular Biology. 2004b;338:585–596. doi: 10.1016/j.jmb.2004.01.064. [DOI] [PubMed] [Google Scholar]
- Abendroth J, Murphy P, Sandkvist M, Bagdasarian M, Hol WGJ. The X-ray structure of the type II secretion system complex formed by the N-terminal domain of EpsE and the cytoplasmic domain of EpsL of Vibrio cholerae. Journal of Molecular Biology. 2005;348:845–855. doi: 10.1016/j.jmb.2005.02.061. [DOI] [PubMed] [Google Scholar]
- Abrahams JP, Buchanan SK, Van Raaij MJ, Fearnley IM, Leslie AG, Walker JE. The structure of bovine F1-ATPase complexed with the peptide antibiotic efrapeptin. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9420–9424. doi: 10.1073/pnas.93.18.9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts J, de Groot A, Ball G, Durand E, El Khattabi M, Filloux A, Tommassen J, Koster M. Interaction domains in the Pseudomonas aeruginosa type II secretory apparatus component XcpS (GspF) Microbiology-Uk. 2007;153:1582–1592. doi: 10.1099/mic.0.2006/002840-0. [DOI] [PubMed] [Google Scholar]
- Bailey S. The CCP4 Suite - Programs for Protein Crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Bally M, Filloux A, Akrim M, Ball G, Lazdunski A, Tommassen J. Protein secretion in Pseudomonas aeruginosa: characterization of seven xcp genes and processing of secretory apparatus components by prepilin peptidase. Mol Microbiol. 1992;6:1121–1131. doi: 10.1111/j.1365-2958.1992.tb01550.x. [DOI] [PubMed] [Google Scholar]
- Blank TE, Donnenberg MS. Novel topology of BfpE, a cytoplasmic membrane protein required for type IV fimbrial biogenesis in enteropathogenic Escherichia coli. J Bacteriol. 2001;183:4435–4450. doi: 10.1128/JB.183.15.4435-4450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortoli-German I, Brun E, Py B, Chippaux M, Barras F. Periplasmic disulphide bond formation is essential for cellulase secretion by the plant pathogen Erwinia chrysanthemi. Mol Microbiol. 1994;11:545–553. doi: 10.1111/j.1365-2958.1994.tb00335.x. [DOI] [PubMed] [Google Scholar]
- Bouley J, Condemine G, Shevchik VE. The PDZ domain of OutC and the N-terminal region of OutD determine the secretion specificity of the type II out pathway of Erwinia chrysanthemi. J Mol Biol. 2001;308:205–219. doi: 10.1006/jmbi.2001.4594. [DOI] [PubMed] [Google Scholar]
- Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr, Sect D: Biol Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- Burrows LL. Weapons of mass retraction. Mol Microbiol. 2005;57:878–888. doi: 10.1111/j.1365-2958.2005.04703.x. [DOI] [PubMed] [Google Scholar]
- Camberg JL, Johnson TL, Patrick M, Abendroth J, Hol WGJ, Sandkvist M. Synergistic stimulation of EpsE ATP hydrolysis by EpsL and acidic phospholipids. EMBO Journal. 2007;26:19–27. doi: 10.1038/sj.emboj.7601481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonnelle E, Helaine S, Nassif X, Pelicic V. A systematic genetic analysis in Neisseria meningitidis defines the Pil proteins required for assembly, functionality, stabilization and export of type IV pili. Mol Microbiol. 2006;61:1510–22. doi: 10.1111/j.1365-2958.2006.05341.x. [DOI] [PubMed] [Google Scholar]
- Cianciotto NP. Type II secretion: a protein secretion system for all seasons. Trends Microbiol. 2005;13:581–588. doi: 10.1016/j.tim.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Collins RF, Saleem M, Derrick JP. Purification and 3-D electron microscopy structure of the Neisseria meningitidis type IV pilus biogenesis protein PilG. J Bacteriol. 2007;189:6389–6396. doi: 10.1128/JB.00648-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan KD, Zhang KY. Density modification for macromolecular phase improvement. Progress in Biophysics & Molecular Biology. 1999;72:245–270. doi: 10.1016/s0079-6107(99)00008-5. [DOI] [PubMed] [Google Scholar]
- Craig L, Li J. Type IV pili: paradoxes in form and function. Curr Opin Struct Biol. 2008;18:267–77. doi: 10.1016/j.sbi.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig L, Pique ME, Tainer JA. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol. 2004;2:363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- Crowther LJ, Anantha RP, Donnenberg MS. The inner membrane subassembly of the enteropathogenic Escherichia coli bundle-forming pilus machine. Mol Microbiol. 2004;52:67–79. doi: 10.1111/j.1365-2958.2003.03963.x. [DOI] [PubMed] [Google Scholar]
- d'Enfert C, Ryter A, Pugsley AP. Cloning and expression in Escherichia coli of the Klebsiella pneumoniae genes for production, surface localization and secretion of the lipoprotein pullulanase. EMBO J. 1987;6:3531–3538. doi: 10.1002/j.1460-2075.1987.tb02679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauter Z, Dauter M, Rajashankar KR. Novel approach to phasing proteins: derivatization by short cryo-soaking with halides. Acta Crystallogr, Sect D: Biol Crystallogr. 2000;56:232–237. doi: 10.1107/s0907444999016352. [DOI] [PubMed] [Google Scholar]
- Dauter Z, Dauter M, de La Fortelle E, Bricogne G, Sheldrick GM. Can anomalous signal of sulfur become a tool for solving protein crystal structures? J Mol Biol. 1999;289:83–92. doi: 10.1006/jmbi.1999.2743. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMol Molecular Graphics System. 2002 on World Wide Web, http://www.pymol.org.
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans FF, Egan S, Kjelleberg S. Ecology of type II secretion in marine gammaproteobacteria. Environ Microbiol. 2008;10:1101–7. doi: 10.1111/j.1462-2920.2007.01545.x. [DOI] [PubMed] [Google Scholar]
- Feilmeier BJ, Iseminger G, Schroeder D, Webber H, Phillips GJ. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol. 2000;182:4068–76. doi: 10.1128/jb.182.14.4068-4076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filloux A. The underlying mechanisms of type II protein secretion. Biochim Biophys Acta. 2004;1694:163–179. doi: 10.1016/j.bbamcr.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- Hansen JK, Forest KT. Type IV pilin structures: insights on shared architecture, fiber assembly, receptor binding and type II secretion. J Mol Microbiol Biotechnol. 2006;11:192–207. doi: 10.1159/000094054. [DOI] [PubMed] [Google Scholar]
- Hardie KR, Schulze A, Parker MW, Buckley JT. Vibrio spp. secrete proaerolysin as a folded dimer without the need for disulphide bond formation. Mol Microbiol. 1995;17:1035–1044. doi: 10.1111/j.1365-2958.1995.mmi_17061035.x. [DOI] [PubMed] [Google Scholar]
- Harding MM. Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:678–682. doi: 10.1107/S0907444906014594. [DOI] [PubMed] [Google Scholar]
- Hirst TR, Holmgren J. Conformation of protein secreted across bacterial outer membranes: a study of enterotoxin translocation from Vibrio cholerae. Proc Natl Acad Sci U S A. 1987;84:7418–7422. doi: 10.1073/pnas.84.21.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs M, Mattick JS. Common components in the assembly of type 4 fimbriae, DNA transfer systems, filamentous phage and protein-secretion apparatus: a general system for the formation of surface-associated protein complexes. Mol Microbiol. 1993;10:233–243. doi: 10.1111/j.1365-2958.1993.tb01949.x. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. Journal of Molecular Biology. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Johnson TL, Scott ME, Sandkvist M. Mapping critical interactive sites within the periplasmic domain of the Vibrio cholerae type II secretion protein EpsM. J Bacteriol. 2007;189:9082–9. doi: 10.1128/JB.01256-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TL, Abendroth J, Hol WGJ, Sandkvist M. Type II secretion: from structure to function. FEMS Microbiol Lett. 2006;255:175–186. doi: 10.1111/j.1574-6968.2006.00102.x. [DOI] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Principles of protein-protein interactions. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn TJ, Jude BA, Taylor RK. A colonization factor links Vibrio cholerae environmental survival and human infection. Nature. 2005;438:863–6. doi: 10.1038/nature04249. [DOI] [PubMed] [Google Scholar]
- Korotkov KV, Krumm BE, Bagdasarian M, Hol WGJ. Structural and Functional Studies of EpsC, a Crucial Component of the Type 2 Secretion System from Vibrio cholerae. J Mol Biol. 2006;363:311–321. doi: 10.1016/j.jmb.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Korotkov KV, Hol WGJ. Structure of the GspK-GspI-GspJ complex from the enterotoxigenic Escherichia coli type 2 secretion system. Nat Struct Mol Biol. 2008;15:462–468. doi: 10.1038/nsmb.1426. [DOI] [PubMed] [Google Scholar]
- Korotkov KV, Pardon E, Steyaert J, Hol WGJ. Crystal Structure of the N-Terminal Domain of the Secretin GspD from ETEC Determined with the Assistance of a Nanobody. Structure. 2009;17:255–265. doi: 10.1016/j.str.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZJ, Vysotski ES, Chen CJ, Rose JP, Lee J, Wang BC. Structure of the Ca2+-regulated photoprotein obelin at 1.7 A resolution determined directly from its sulfur substructure. Protein Sci. 2000;9:2085–2093. doi: 10.1110/ps.9.11.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS. Type IV pili and twitching motility. Annu Rev Microbiol. 2002;56:289–314. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- Merritt EA, Hol WGJ. AB5 toxins. Curr Opin Struct Biol. 1995;5:165–171. doi: 10.1016/0959-440x(95)80071-9. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Nudleman E, Kaiser D. Pulling together with type IV pili. J Mol Microbiol Biotechnol. 2004;7:52–62. doi: 10.1159/000077869. [DOI] [PubMed] [Google Scholar]
- Nunn D. Bacterial type II protein export and pilus biogenesis: more than just homologies? Trends Cell Biol. 1999;9:402–408. doi: 10.1016/s0962-8924(99)01634-7. [DOI] [PubMed] [Google Scholar]
- O'Neal CJ, Amaya EI, Jobling MG, Holmes RK, Hol WGJ. Crystal structures of an intrinsically active cholera toxin mutant yield insight into the toxin activation mechanism. Biochemistry. 2004;43:3772–3782. doi: 10.1021/bi0360152. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Peabody CR, Chung YJ, Yen MR, Vidal-Ingigliardi D, Pugsley AP, Saier MH., Jr Type II protein secretion and its relationship to bacterial type IV pili and archaeal flagella. Microbiology. 2003;149:3051–3072. doi: 10.1099/mic.0.26364-0. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nature Structural Biology. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr, Sect D: Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- Pugsley AP. Translocation of a folded protein across the outer membrane in Escherichia coli. Proc Natl Acad Sci U S A. 1992;89:12058–12062. doi: 10.1073/pnas.89.24.12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugsley AP. The complete general secretory pathway in gram-negative bacteria. Microbiol Rev. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py B, Loiseau L, Barras F. An inner membrane platform in the type II secretion machinery of Gram-negative bacteria. EMBO Reports. 2001;2:244–248. doi: 10.1093/embo-reports/kve042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robien MA, Krumm BE, Sandkvist M, Hol WGJ. Crystal structure of the extracellular protein secretion NTPase EpsE of Vibrio cholerae. Journal of Molecular Biology. 2003;333:657–674. doi: 10.1016/j.jmb.2003.07.015. [DOI] [PubMed] [Google Scholar]
- Sandkvist M. Biology of type II secretion. Molecular Microbiology. 2001a;40:271–283. doi: 10.1046/j.1365-2958.2001.02403.x. [DOI] [PubMed] [Google Scholar]
- Sandkvist M. Type II secretion and pathogenesis. Infection & Immunity. 2001b;69:3523–3535. doi: 10.1128/IAI.69.6.3523-3535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkvist M, Bagdasarian M, Howard SP, DiRita VJ. Interaction between the autokinase EpsE and EpsL in the cytoplasmic membrane is required for extracellular secretion in Vibrio cholerae. EMBO J. 1995;14:1664–73. doi: 10.1002/j.1460-2075.1995.tb07155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkvist M, Michel LO, Hough LP, Morales VM, Bagdasarian M, Koomey M, DiRita VJ. General secretion pathway (eps) genes required for toxin secretion and outer membrane biogenesis in Vibrio cholerae. J Bacteriol. 1997;179:6994–7003. doi: 10.1128/jb.179.22.6994-7003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvonnet N, Vignon G, Pugsley AP, Gounon P. Pilus formation and protein secretion by the same machinery in Escherichia coli. EMBO J. 2000;19:2221–2228. doi: 10.1093/emboj/19.10.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Henkel B, Karch H. A gene cluster closely related to type II secretion pathway operons of gram-negative bacteria is located on the large plasmid of enterohemorrhagic Escherichia coli O157 strains. FEMS Microbiol Lett. 1997;148:265–272. doi: 10.1111/j.1574-6968.1997.tb10299.x. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Sikora AE, Lybarger SR, Sandkvist M. Compromised outer membrane integrity in Vibrio cholerae Type II secretion mutants. J Bacteriol. 2007;189:8484–95. doi: 10.1128/JB.00583-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixma TK, Pronk SE, Kalk KH, Wartna ES, van Zanten BAM, Witholt B, Hol WGJ. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli. Nature. 1991;351:371–377. doi: 10.1038/351371a0. [DOI] [PubMed] [Google Scholar]
- Söderberg MA, Rossier O, Cianciotto NP. The type II protein secretion system of Legionella pneumophila promotes growth at low temperatures. J Bacteriol. 2004;186:3712–3720. doi: 10.1128/JB.186.12.3712-3720.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauschek M, Gorrell R, Strugnell R, Robins-Browne R. Identification of a protein secretory pathway for the secretion of heat-labile enterotoxin by an enterotoxigenic strain of Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:7066–7071. doi: 10.1073/pnas.092152899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Reeves PJ, Salmond GPC. The general secretion pathway of Erwinia carotovora subsp carotovora: Analysis of the membrane topology of OutC and OutF. Microbiology-Uk. 1997;143:713–720. doi: 10.1099/00221287-143-3-713. [DOI] [PubMed] [Google Scholar]
- Uweh O. Klebsiella Infections. 2006 http://www.emedicine.com/MED/topic1237.htm.
- van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- Weiss MS, Sicker T, Hilgenfeld R. Soft X-rays, high redundancy, and proper scaling: A new procedure for automated protein structure determination via SAS. Structure. 2001a;9:771–777. doi: 10.1016/s0969-2126(01)00647-5. [DOI] [PubMed] [Google Scholar]
- Weiss MS, Sicker T, Djinovic-Carugo K, Hilgenfeld R. On the routine use of soft X-rays in macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2001b;57:689–695. doi: 10.1107/s0907444901003572. [DOI] [PubMed] [Google Scholar]
- Xu H, Yang C, Chen LR, Kataeva IA, Tempel W, Lee D, Habel JE, Nguyen D, Pflugrath JW, Ferrara JD, Arendall WB, Richardson JS, Richardson DC, Liu ZJ, Newton MG, Rose JP, Wang BC. Away from the edge II: in-house Se-SAS phasing with chromium radiation. Acta Crystallogr, Sect D: Biol Crystallogr. 2005;61:960–966. doi: 10.1107/S0907444905010644. [DOI] [PubMed] [Google Scholar]
- Yanez ME, Korotkov KV, Abendroth J, Hol WGJ. Structure of the minor pseudopilin EpsH from the Type 2 Secretion system of Vibrio cholerae. J Mol Biol. 2008a;37:91–103. doi: 10.1016/j.jmb.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez ME, Korotkov KV, Abendroth J, Hol WGJ. The crystal structure of a binary complex of two pseudopilins: EpsI and EpsJ from the type 2 secretion system of Vibrio vulnificus. J Mol Biol. 2008b;375:471–486. doi: 10.1016/j.jmb.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Pflugrath JW, Courville DA, Stence CN, Ferrara JD. Away from the edge: SAD phasing from the sulfur anomalous signal measured in-house with chromium radiation. Acta Crystallogr, Sect D: Biol Crystallogr. 2003;59:1943–1957. doi: 10.1107/s0907444903018547. [DOI] [PubMed] [Google Scholar]
- Yen YT, Bhattacharya M, Stathopoulos C. Genome-wide in silico mapping of the secretome in pathogenic Yersinia pestis KIM. FEMS Microbiol Lett. 2008;279:56–63. doi: 10.1111/j.1574-6968.2007.01008.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.