

Abstract
Ionotropic glutamate receptor (iGluR) desensitization can be modulated by
mutations that change the stability of a dimer formed by the agonist binding
domain. Desensitization of
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors can be
blocked by a single point mutation (e.g., GluR2 L483Y) that stabilizes this
dimer in an active conformation. In contrast, desensitization of kainate
receptors can be slowed, but not blocked, by similar dimer interface
mutations. Only covalent cross-linking via introduced disulfides has been
previously shown to block kainate receptor desensitization completely. We have
now identified an apparently nondesensitizing GluR6 point mutant (D776K)
located at the apex of the ligand binding (S1S2) domain dimer interface.
Asp776 is one of a cluster of four charged residues in this region that
together mediate direct dimer interactions and contribute to the binding sites
for one chloride and two sodium ions. Despite the localized +4 change in the
net charge of the S1S2 dimer, the D776K mutation actually increased the
thermodynamic stability of the dimer. Unlike GluR6 wild type, the D776K mutant
is insensitive to external cations but retains sensitivity to external anions.
We therefore hypothesize that the unexpected phenotype of this charge reversal
mutation results from the substitution of the sodium ions bound within the
dimer interface by the introduced lysine
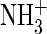 groups. The nondesensitizing D776K
mutant provides insights into kainate receptor gating and represents a
potentially useful new tool for dissecting kainate receptor function.
groups. The nondesensitizing D776K
mutant provides insights into kainate receptor gating and represents a
potentially useful new tool for dissecting kainate receptor function.
Desensitization, receptor inactivation in the continued presence of agonist, has a profound influence on the magnitude and duration of kainate- and AMPA-selective iGluR responses. After activation by the endogenous agonist Glu, desensitization is generally rapid (time constants typically ∼1-10 ms) and substantially complete (96-99.7%) (Ozawa et al., 1998; Dingledine et al., 1999). Desensitization is an intrinsic property of the receptor subunits themselves and is therefore an integral part of the overall gating mechanism of these receptors.
Viewed from a functional perspective, desensitization occurs independently of channel opening. As tetramers, iGluRs can bind up to four agonist molecules; agonist binding to at least two subunits is required to drive channel gating (Heckmann et al., 1996; Clements et al., 1998; Rosenmund et al., 1998), but agonist binding to one or more subunits is sufficient for entry into desensitized states (Heckmann et al., 1996; Robert and Howe, 2003). Concentrations of agonist too low to activate responses are therefore able to cause desensitization. The process of desensitization is reversible, agonist responses recovering completely once agonist is removed. The rate of this recovery from desensitization varies significantly between iGluR subtypes; multiple overlapping time-constants reveal the existence of a number of distinct desensitized states (Robert and Howe, 2003).
From a structural standpoint, our view of the desensitization process comes largely from crystallographic studies of the isolated agonist-binding or “S1S2” domain. Structures of the S1S2 domain have been determined for both AMPA and kainate receptor subunits, revealing a conserved bilobate structure (Gouaux, 2004; Mayer, 2006). In both subtypes, S1S2 domains have been observed to form 2-fold symmetrical, back-to-back dimers (Armstrong and Gouaux, 2000; Nanao et al., 2005). The stability of the S1S2 dimer has been found to correlate inversely with desensitization rates. In particular, a leucine-to-tyrosine AMPA receptor point mutant, which completely blocks desensitization (Stern-Bach et al., 1998), stabilizes the S1S2 dimer by strengthening intersubunit contacts (Sun et al., 2002). These observations suggest that dimer rearrangement is required for desensitization, an interpretation subsequently corroborated for both AMPA- and kainate-receptor subtypes (Horning and Mayer, 2004; Priel et al., 2006; Weston et al., 2006; Zhang et al., 2006).
Despite the common role of the S1S2 dimer interface, there are also differences between AMPA and kainate receptor desensitization. For one thing, it has proven difficult to suppress desensitization in the kainate receptors by stabilizing purely noncovalent interactions. In addition, in kainate receptors, response amplitudes and desensitization rates are dependent on external ions (Bowie, 2002; Paternain et al., 2003). Binding sites have been identified at the apex of the S1S2 dimer for both anions and cations (Plested and Mayer, 2007; Plested et al., 2008), consistent with studies in which mutations in this region affected desensitization rates (Fleck et al., 2003; Wong et al., 2006, 2007). In this study, we have characterized the electrophysiological and biochemical consequences of additional apical mutations; in particular, the effects on receptor responses of charge-reversal mutations. Their effects highlight the key role of this region as a regulator of receptor desensitization.
Materials and Methods
Mutagenesis. All mutagenesis was carried out on a rat GluR6(Q) cDNA clone. Residue numbering is from the start methionine (subtract 31 for GluR6 and 30 for GluR5 to obtain numbering based on predicted mature polypeptide). Mutants were generated using the QuikChange protocol and Pfu turbo polymerase (Stratagene, La Jolla, CA), essentially as described in Zhang et al. (2006). All mutants were confirmed either by sequencing of the entire open reading frame, or by sequencing after subcloning of the 1.65-kilobase XagI-Eco47III fragment. Crystal structures for GluR6 S1S2-Dom (Protein Data Bank code 1yae) (Nanao et al., 2005) and GluR5 S1S2-KA (Protein Data Bank code 3c32) (Plested et al., 2008) were used to generate figures using MacPyMOL (http://pymol.sourceforge.net/).
Cell Culture, Electrophysiology, and Data Analysis. HEK-293 cell culture, whole-cell patch clamp, and data analysis were carried out as described previously (Zhang et al., 2008). Whole-cell recordings were made 48 to 72 h after transfection at a holding potential of -70 mV using an amplifier (EPC 10; HEKA Elektronic, Lambrecht/Pfalz, Germany). The electrode solution contained 110 mM CsF, 30 mM CsCl, 4 mM NaCl, 0.5 mM CaCl2, 10 mM HEPES, and 5 mM EGTA (adjusted to pH 7.3 with CsOH). The external bath solution contained 150 mM NaCl, 2.8 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, and 10 mM HEPES (adjusted to pH 7.3 with NaOH). Rapid agonist application was achieved using a piezo-based perfusion system (Burleigh LSS-3200; Scientifica, Harpenden, UK). The rate of solution exchange in this system (determined by open-tip junction currents) was <250 μs, and 20-to-80% rise-times for control (GluR6 WT) responses were <1 ms. Recordings were performed on small-diameter cells (20 μm) lifted into the perfusion stream to ensure rapid solution exchange (Zhang et al., 2008). Time constants were determined using single-exponential fits in PulseFit (HEKA). For GluR6 WT, steady-state decay rates were determined from responses with clear steady states (those fit ranged between 15 and 75 pA from peak responses of 3-15 nA).
Oocytes were injected with cRNA (∼50 nl; 0.8 mg/ml) transcribed from the GluR6 WT and mutant cDNAs (in pcDNA3.1; linearized with XbaI) using the mMessage mMachine T7 kit (Ambion, Warrington, UK). Current recordings were made 48 to 56 h after injection using the setup described in Thompson and Lummis (2008). The control external medium (96 mM NaCl, 1.1 mM KCl, 1.8 mM BaCl2, 5 mM HEPES, adjusted to pH 7.5 with NaOH) was modified by substitution of NaCl with NaF, NaNO3, CsCl, or RbCl as appropriate. Where the cation was exchanged, the pH was adjusted using CsOH instead of NaOH. Recordings were made at a holding potential of -60 mV; agonist (10 mM Glu) was applied for 10 s at a flow rate of 10 ml/min. Oocytes expressing GluR6 WT were pretreated with concanavalin A (0.3 mg/ml, 0.2-μm filter; incubated for 5-10 min) before recording. The program WCP (J. Dempster, University of Strathclyde, UK) was used for oocyte data recording and analysis.
All data are presented as mean ± S.E.M. Statistical analyses were carried out using Prism (ver. 5; GraphPad Software, San Diego, CA). Significant differences compared with the GluR6 WT control were identified by unpaired t test for single comparisons and using oneway ANOVA for multiple comparisons. In the latter case, significance was tested using either Dunnett's or Dunn's multiple comparison tests depending on data variances.
S1S2 Domain Expression and Characterization. Overlap-extension polymerase chain reaction was used to generate a GluR6 S1S2 domain construct in the pET21 bacterial expression vector (Novagen, Nottingham, UK). The construct was based on the domain boundaries described by Mayer (2005) and included GluR6 residues Ser429 to Lys544 and Pro667 to Glu806, joined by a GlyThr linker sequence and ending with a six-histidine tag. This construct was recombinantly expressed (induction with 1 mM isopropyl β-d-thiogalactoside; 4 h, 23°C) in BL21(DE3) cells (Bioline, London, UK). Constructs were purified to homogeneity after chemical lysis (Cellytic-B; Sigma-Aldrich, Poole, UK) in three chromatography steps, using a base buffer containing 150 mM NaCl, 25 mM HEPES, 5% (v/v) glycerol, and 1 to 5 mM glutamate. The steps were His-select nickel-affinity (pH 7.5, elution with 400 mM imidazole; Sigma), HiTrapQ anion exchange (pH 8.0, sample in flow-through; GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK), and Superdex-75 gel filtration (pH 7.5; GE Healthcare). The final yield ranged between 0.2 and 0.5 mg/liter culture.
Purified S1S2 constructs were concentrated by ultrafiltration as required. Blue Native polyacrylamide gel electrophoresis (BN-PAGE) was carried out using Novex 4-to-16% Native PAGE gels and associated buffers (Invitrogen, Paisley, UK) according to the manufacturer's instructions. Protein bands were visualized either directly with Coomassie Blue R-250 or silver staining or after electroblotting onto Immobilon P (Millipore, Watford, UK) and detection with an anti-His tag monoclonal antibody (1:1000 dilution; Sigma).
For size-exclusion chromatography and ultracentrifugation, samples were initially concentrated to an A280 of ∼0.4 to 0.6 and then dialyzed into 25 mM HEPES, pH 7.5, 150 mM NaCl, 5% (w/v) glycerol, 1 mM sodium glutamate, and 0.02% (w/v) NaN3. Size-exclusion chromatography was performed on a Superdex-75 10/30 column. The WT and D776K proteins each eluted as single peaks, and corresponding ln(mol. wt.) and Stokes radius estimates were obtained by linear regression analysis of globular standards.
Ultracentrifugation experiments were performed at 20°C in a centrifuge
(ProteomeLab XL-A; Beckman Coulter, Fullerton, CA) equipped with an AN-60
rotor and absorbance optics. Sedimentation velocity experiments were performed
at a rotor speed of 3.5 × 104 rpm. Absorbance scans were
taken at 280 nm at ∼1.25-min intervals, with a 0.003-cm radial scan step.
Sedimentation equilibrium data were recorded for 10 h each at speeds of 7, 10,
14, 20, and 28 × 103 rpm. Scans were taken at 1-h intervals
with a 0.001-cm step size along the radial axis and five replicates per data
point. Attainment of sedimentation equilibrium was verified using the program
WinMATCH (D. A. Yphantis and J. W. Lary;
http://www.biotech.uconn.edu/auf/ftp/WINMATCH.ZIP)
Six-sector cells were loaded with 1×, 2×, and 4× dilutions
of ∼10 μM stock solutions. Curves collected at all five speeds for the
two channels with the highest concentrations were globally fit. Protein
partial specific volume (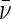 and buffer
density and viscosity (ρ, η) were calculated using the program
SEDNTERP (J. Philo, D. Hayes, and T. Laue)
(Laue et al., 1992).
Sedimentation coefficient distributions were calculated using the program
SEDFIT87 (Dam and Schuck,
2004). Sedimentation equilibrium data were analyzed using the
program SEDANAL (Stafford and Sherwood,
2004) under both single-species and oligomerization models.
and buffer
density and viscosity (ρ, η) were calculated using the program
SEDNTERP (J. Philo, D. Hayes, and T. Laue)
(Laue et al., 1992).
Sedimentation coefficient distributions were calculated using the program
SEDFIT87 (Dam and Schuck,
2004). Sedimentation equilibrium data were analyzed using the
program SEDANAL (Stafford and Sherwood,
2004) under both single-species and oligomerization models.
Biotinylation, Deglycosylation, and Radioligand Binding. Cell surface expression was determined after biotinylation as described previously (Zhang et al., 2006) and quantified using KODAK 1D Image Analysis software (Carestream Health, Rochester, NY). Deglycosylation experiments using Endo H and PNGase F (New England Biolabs, Hitchin, UK) were performed essentially as described by Priel et al. (2006). In brief, membranes were prepared from cells 55 to 60 h after transient transfection with full-length GluR6 constructs, and membrane aliquots containing 20 μg of total protein were incubated with 1 μl of the enzymes for 2 h at 37°C in the supplied buffers. Immunoblots for both surface labeling and deglycosylation experiments were carried out using polyclonal GluR6/7 antisera (1:1000 dilution; Millipore, Watford, UK).
For radioligand binding assays, membrane preparations and [3H]kainate saturation and displacement assays were performed as described previously (Zhang et al., 2006), using membrane preparations from HEK cells stably expressing GluR6 D776K (after G418 selection). Data were fitted by nonlinear regression using built-in functions in Prism.
Results
We had previously used a combination of site-directed mutagenesis and whole-cell patch clamp in HEK-293 cells to identify GluR6 S1S2 domain mutations that slowed desensitization kinetics (Zhang et al., 2006). The residues in that study were clustered around Tyr521, the homolog of the nondesensitizing GluR2 L483Y mutant. Intersubunit interactions in this region play an important role in determining dimer stability and therefore desensitization rates. A second point of contact in S1S2 dimers is evident in crystal structures (Fig. 1A) (Armstrong and Gouaux, 2000; Nanao et al., 2005). This comprises either a pair (in AMPA receptor subunits) or quartet (in kainate receptor subunits) of charged residues capable of forming intersubunit salt bridges at the apex of the dimer. In GluR6, contacts are formed between Glu524 and Lys531, the equivalent of which is also found in AMPA receptor subunits, and between Arg775 and Asp776 (Fig. 1B), an interaction found only in kainate receptor subunits (Nanao et al., 2005). This region is also where a single chloride and two sodium ions bind in kainate receptor subunits (Fig. 1C) (Plested and Mayer, 2007; Plested et al., 2008), complicating any interpretation of how the various charged amino acids interact. Indeed, the GluR5 homolog of Arg775 (Arg790) shows conformational flexibility in the presence of different cations, such that an intersubunit contact is not always formed (Plested et al., 2008).
Fig. 1.

Ionic interactions at the apex of kainate S1S2 domain dimers. A, the GluR6 S1S2-domoate dimer interface (Nanao et al., 2005) viewed from the side. Protomers B and D (gold and blue, respectively, cartoon representation) are shown with domoate (black) and the apical residues Glu524 (yellow), Lys531 (gray), Arg775 (green), and Asp776 (magenta), all in space-fill representation. Nitrogens and oxygens are colored blue and red, respectively. B, closer view of apical cluster, from the top (left) and side (right; looking through protomer B) of the S1S2 dimer. Residues are colored as in A. For clarity, only selected parts of the protomer B main chain (partially transparent cartoon) are shown on the right. Polar contacts are indicated (dotted gray lines). C, equivalent views of the GluR5 S1S2-KA complex (Plested et al., 2008), viewed as in B (protomer A, pink; protomer B, light green). The GluR5 homologs of Glu524, Lys531, Arg775, and Asp776 (Glu539, Lys546, Arg790, and Asp791) are shown, as are the chloride (green) and sodium (blue) ions.
Switching Charges at the S1S2 Apex Has Varied Effects on Desensitization Kinetics. To better understand the role of these residues in receptor function, we determined the effect of single and double charge exchanges at these four sites. These experiments revealed a spectrum of effects consistent with perturbations of the S1S2 dimer interaction, ranging from loss of response to almost complete block of desensitization (Fig. 2, Table 1). We first assessed the effect of double mutations at the two charge pairs Glu524-Lys531 and Arg775-Asp776, in which the respective charged amino acids were exchanged. Mutants were transiently transfected into HEK cells, and responses to applications of Glu (3 mM) and KA (1 mM) were determined by whole-cell voltage-clamp recordings. As their overall effect was charge-neutral, these mutants should reveal the extent to which geometry at these sites, as opposed simply to charge, is important in receptor function. The E524K-K531E mutant was still functional but with responses to Glu and KA that desensitized 3- to 4-fold faster than those of GluR6 WT (Fig. 2A, Table 1). For the R775D-D776R mutant, in contrast, no responses were observed with either agonist (Table 1). This indicates that the environment in the vicinity of Arg775 and Asp776 is more sensitive to changes in charge distribution than that around Glu524 and Lys531.
Fig. 2.

Varied effects of charge exchanges to apical GluR6 residues. A, representative current responses recorded from HEK cells transiently transfected with the E524K-K531E double mutant and the R775D and K531E single mutants. Applications (100 ms) of Glu (3 mM, black line above trace) and KA (1 mM, gray line above trace) are shown, along with normalized GluR6 WT traces (gray dotted; top and bottom mutants only), to allow comparison of desensitization rates. B, representative responses to applications of Glu and KA recorded from cells expressing GluR6 D776K. Traces from 4 s agonist applications are shown to illustrate the almost complete lack of desensitization. C, traces from brief (1 ms) agonist applications to illustrate peak decay rates from D776K (black traces), compared with GluR6 WT (normalized traces; gray, dotted).
TABLE 1.
Desensitization kinetics of GluR6 mutants
Presented are peak currents (Ipeak), desensitization time-constant (τDes), desensitization (Des), and relative KA efficacy (IKA/Glu) (mean ± S.E.M. for n determinations), recorded from transiently-transfected HEK cells. Kinetics were determined from 100-ms agonist applications except as indicated in parentheses. Boldface type indicates significant differences comparing τDes, %des and IKA/Glu, values with those of GluR6 WT (not tested for E524K-K531E or R775D mutants owing to small current sizes);
|
Mutant
|
Glu
|
KA
|
||||||
|---|---|---|---|---|---|---|---|---|
| Ipeak | τDes | Des | n | IKA/Glu | τDes | Des | n | |
| nA | ms | % | ms | % | ||||
| Wild type | 6.8 ± 0.9 | 5.0 ± 0.2 | 99.7 ± 0.1 | 21 | 0.62 ± 0.05 | 3.9 ± 0.2 | 99.4 ± 0.1 | 12 |
| E524K | N.R. | 10 | N.R. | 10 | ||||
| K531E | 5.6 ± 0.7 | 9.1 ± 0.6*** | 99.8 ± 0.1 | 11 | 1.03 ± 0.03*** | 34.5 ± 3.9*** | 98.4 ± 0.7 | 13 |
| (1 s) | (1 s) | |||||||
| E524KK531E | 0.03 ± 0.01 | 1.3 ± 0.2a | 100 | 11 | N.D.b | 1.4 ± 0.1c | 100 | 11 |
| R775D | 0.01 ± 0.003 | N.D. | N.D. | 11 | N.D.d | N.D. | N.D. | 11 |
| D776K | 0.83 ± 0.3 | N.D. | 9.5 ± 1.6***e | 30 | 0.86 ± 0.03** | N.D. | 10.3 ± 1.2**f | 24 |
| (4 s) | (4 s) | |||||||
| R775DD776R | N.R. | 7 | N.R. | 7 | ||||
N.R., no response; N.D., not determined.
P < 0.01.
P < 0.001.
n = 4.
Mean response to KA was 0.12 ± 0.05 nA.
n = 5.
Mean response to KA 0.01 ± 0.004 nA.
n = 21.
n = 20.
We next mutated these four residues singly to reverse the charge of their side chains. Given their location and the effect of the double mutants, these single mutants would be expected to destabilize the dimer and therefore attenuate responses. For the E524K and R775D mutations, this was indeed the case, with responses either entirely absent (E524K) or extremely small (R775D) (Fig. 2A, Table 1). This was in agreement with the reported effects of other changes to these sites in AMPA and kainate receptor subunits (Fleck et al., 2003; Horning and Mayer, 2004; Plested and Mayer, 2007; Wong et al., 2007). With the K531E mutation, in contrast, desensitization in response to both Glu and KA was actually slowed (Fig. 2A, Table 1). Although unexpected, this was again consistent with published data, at least for kainate receptors. In GluR6, the K531G mutation has been found to slow desensitization, particularly in response to KA (Fleck et al., 2003). Similar mutations to the homologous site in GluR2 (i.e., K493A and Met) accelerated desensitization, as would be predicted (Horning and Mayer, 2004). Our K531E mutation does not resolve the question of why changes to this site have unexpected effects on kainate receptor responses, but it does appear to rule out simple steric effects (Horning and Mayer, 2004).
Replacing Asp776 with a Lysine Blocks Receptor Desensitization. The last of the four charge-reversal mutations we tested, Asp776 to lysine (D776K), would again be expected to severely attenuate or even eliminate receptor responses. This was the effect of other reported mutations at this site in GluR6. Mutation of Asp776 to asparagine, glutamate or glycine-accelerated desensitization, whereas mutation to threonine abolished responses (Fleck et al., 2003; Plested and Mayer, 2007). The D776K mutant had a very different phenotype. Although most cells showed no agonist-activated responses, currents were observed in a minority (30/116). These responses were essentially nondesensitizing to agonist applications up to 4 s, desensitizing by approximately 10% with both Glu and KA (Fig. 2B and Table 1). Current amplitudes varied greatly for responses to both Glu (range, 11 pA to 5.5 nA; mean, 830 pA; median, 262 pA) and KA (range, 9 pA to 4.7 nA; mean, 810 pA; median, 270 pA). Consistent with the absence of desensitization, repeated 100-ms applications in a paired-pulse protocol showed no sign of desensitization or appreciable run-down (n = 6; data not shown). Whereas only 25% of transiently transfected cells gave currants for D776K, later tests on HEK cells stably transfected with D776K showed smaller but more reliable responses to Glu (22/52 responded; range, 13-620 pA; mean, 156 pA; median, 61 pA). Unless otherwise indicated, the characterization of D776K described below was carried out using transiently transfected cells.
Comparing agonist efficacy between GluR6 WT and D776K, the overall rank order of efficacy for Glu, KA, and domoate (another partial agonist) was maintained (Glu > KA > domoate). The relative efficacy of KA was slightly higher in D776K (Table 1), but there was no difference in the relative efficacy of domoate (100 μM) between GluR6 WT (0.33 ± 0.03, n = 6) and D776K (0.35 ± 0.03, n = 14). Comparing D776K currents in response to Glu and KA, their rise times were similar at ∼4 and ∼3 ms, respectively (compared with <1 ms for WT and other constructs), as were their desensitization kinetics (for the ∼10% of the D776K response that desensitized, the kinetics were poorly fitted by exponential decays, but time-constants were approximately 1-2 s).
The responses to the two agonists were different in their decay rates from peak responses (Fig. 2C). The decay rates from brief (1 ms) agonist applications were determined for GluR6 WT and D776K. Deactivation rates from peak responses to Glu were unaffected by the mutation, whereas deactivation rates from peak responses to KA were slowed more than 10-fold in D776K compared with GluR6 WT (Fig. 2C and Table 2). A similar agonist-selective effect on decay was not apparent with decay rates from steady-state responses (τDec-SS). For both agonists, τDec-SS was higher than τDec-1ms, but this was true for GluR6 WT as well as D776K (Table 2). In the nondesensitizing GluR1 mutant L497Y, the deactivation rate from peak responses is slowed, whereas the modulator cyclothiazide blocks desensitization without affecting deactivation rates (Mitchell and Fleck, 2007). Therefore, at least in terms of deactivation rates, KA-induced D776K responses behave similarly to those of GluR1 L497Y (slower deactivation), whereas Glu-induced D776K responses are more akin to cyclothiazide-treated GluR1 (normal deactivation). In very general terms, this implies differences in the stability of receptor states when Glu and KA are bound (see Discussion).
TABLE 2.
Deactivation kinetics of GluR6 mutants
τDec-1 ms and τDec-SS are the deactivation rate from peak and from steady state, respectively, determined as described under Materials and Methods. Values are presented as mean ± S.E.M. for n determinations. Boldface type indicates significant differences comparing τDec-1 ms or τDec-SS values with those of GluR6 wild type. “Quad” is the GluR6 K525E/K696R/I780L/Q784K mutant characterized in Zhang et al. (2006).
|
Mutant
|
Glu
|
KA
|
||||||
|---|---|---|---|---|---|---|---|---|
| τDec-1 ms | n | τDec-SS | n | τDec-1 ms | n | τDec-SS | n | |
| Wild type | 3.9 ± 0.2 | 17 | 10.8 ± 2.4 | 6 | 4.4 ± 0.2 | 17 | 127 ± 32 | 5 |
| “Quad” | 37 ± 2.2*** | 4 | 93 ± 16*** | 12 | N.D. | 1050 ± 170 | 12 | |
| K531E | 2.5 ± 0.3 | 10 | N.D. | 14.2 ± 1.0*** | 10 | N.D. | ||
| D776K | 3.9 ± 0.3 | 18 | 5.7 ± 0.4* | 25 | 47 ± 2.3*** | 18 | 57 ± 3.9 | 21 |
N.D., not determined.
P < 0.05.
P < 0.001.
Increased Dimer Stability in GluR6 D776K. It is clear from studies in GluR2 that AMPA-receptor desensitization is inversely correlated with the stability of the dimer formed between S1S2 domains. Although the S1S2 domain of GluR2 WT shows only a very weak tendency to dimerize (Kd ≈ 6 mM), the GluR2 L483Y mutant dimerizes with a dissociation constant of 30 nM (Sun et al., 2002). We therefore investigated the tendency of the D776K S1S2 domain to dimerize in comparison with GluR6 WT. S1S2 domains were recombinantly expressed and purified as described under Materials and Methods, and their aggregation state was assessed by gel filtration, BN-PAGE, and analytical ultracentrifugation.
Gel filtration experiments indicated a higher apparent molecular weight, with a clear difference in retention time for D776K compared with GluR6 WT (Fig. 3A). When the retention times were calibrated against globular standards, the relative molecular mass (Mr) of the WT protein was estimated as 31,000, compared with 50,000 for the D776K mutant. The calculated value for both proteins is 32,000. To test the possibility that the altered mutant retention time was caused by a change in hydrodynamic radius (e.g., elongation), sedimentation coefficients were determined for both wild-type and mutant proteins in velocity sedimentation experiments, and the gel-filtration data were re-calibrated in terms of Stokes radius. The shape-independent Mr values obtained from the Svedberg equation were 30,000 for WT and 56,000 for the D776K mutant, confirming the increase in average molar mass of the mutant versus WT S1S2 domains. The Mr values estimated for the D776K mutant are intermediate between the calculated values for monomeric and dimeric species, consistent with the possibility of an equilibrium among oligomeric species, with an exchange rate faster than the time-scale (hours) of the gel-filtration and sedimentation experiments. This possibility was confirmed by BN-PAGE. Although the GluR6 WT was largely (81 ± 7%, n = 3) monomeric at the concentrations used (∼30 μM protein stock), a significant proportion (47 ± 6%, n = 3) of the D776K mutant ran as a dimer (Fig. 3A). There were also higher-order associations evident for D776K (representing 5 ± 2%, n = 3); there is, however, no structural or biochemical evidence that S1S2 domains associate as trimers or tetramers, so it is unlikely that these larger species have a physiological significance.
Fig. 3.

D776K oligomerization, surface expression and trafficking. A, oligomerization differences between GluR6 WT and D776K S1S2 domains. Recombinant S1S2 domains were separated by gel filtration and the protein visualized by Coomassie Blue staining after SDS-PAGE (top). The D776K mutant resolved in earlier fractions, indicating a higher apparent molecular weight. Protein-containing fractions were pooled, concentrated, and separated by Blue-Native PAGE stain using silver stain (bottom). Monomers (open arrow) and dimers (gray arrow) were apparent in both samples, whereas higher-order multimers (black arrow) were also present in D776K S1S2. B, quantification of the GluR6 WT and D776K S1S2 domain monomer/dimer equilibrium by sedimentation equilibrium. Absorbance data collected at 14,000 rpm (○, top) are shown for one channel for the WT (left) and D776K mutant (right), together with global fits obtained using fixed-molecular-weight single-species (dashed line) or monomer/dimer equilibrium (solid line, mutant only) models. The residuals (observed-calculated) associated with each fit are shown at the bottom for the single-species (○) and monomer/dimer equilibrium (▪). The data indicate that there is a significant dimer component for D776K but not GluR6 WT, under the conditions tested. C, immunoblots (with anti-GluR6/7) showing relative total (Tot) and surface (Sur) expression of GluR6 WT, E524K, and D776K determined by surface biotinylation (see Materials and Methods). Surface labeling of D776K was extremely weak or absent (n = 3). The positions of molecular weight markers are indicated. D, immunoblot showing the results of enzymatic deglycosylation of GluR6 WT and D776K using Endo H and PNGase F, carried out as described under Materials and Methods. Control samples were treated as for PNGase F but without enzyme. The sizes of immature (Endo H-sensitive; black arrow), mature (Endo H-insensitive; gray arrow), and unglycosylated (open arrow) GluR6 are indicated (right), as are the positions of molecular weight markers (left). No mature glycosylation was evident in D776K samples (n = 3).
To further test this model and to quantify the affinity of potential oligomerization interactions, we performed equilibrium analytical ultracentrifugation experiments (Fig. 3B). The absorbance curves obtained for the GluR6 WT S1S2 could be globally well fit as a single species, with a molecular weight fixed at the calculated value of 32,214. A representative curve obtained at 14,000 rpm is shown with the associated fit and residuals in Fig. 3B (left curves). If a monomer/dimer equilibrium was fitted instead, error analysis showed that the dimerization affinity could not be distinguished from Keq = 0 at the 95% confidence level.
In contrast to the WT protein, a very poor fit was obtained when the equivalent set of D776K S1S2 absorbance curves were fitted with a fixed-molecular-weight, single-species model (Fig. 3B, right curves, dashed line, and ○ residuals). However, if a monomer/dimer equilibrium was fitted, a significant improvement was obtained in the quality of fit (Fig. 3B, right curves, solid line, and ▪ residuals). The Kd of dimerization refined to a value of 0.9 μM (95% confidence interval, 0.4-1.4 μM). The most parsimonious explanation for the hydrodynamic data are that the WT GluR6 S1S2 domain dimerizes only weakly, consistent with previously reported estimates of >8 mM for the WT Kd (Chaudhry et al., 2009), and that the D776K mutation stabilizes the dimerization interaction by more than 3 orders of magnitude.
Desensitization block and dimer stability have also been shown to affect receptor maturation and trafficking (Priel et al., 2006; Penn et al., 2008). The relatively high nonresponse rate for HEK cells transiently transfected with D776K implies that expression and/or trafficking are affected by this mutation. This was confirmed by biotin labeling and immunoblotting (Fig. 3C), which showed very low total and surface expression of D776K compared with GluR6 WT and other constructs, including mutants with very small responses (e.g., E524K in Fig. 3C and R775D, data not shown). Quantification of these immunoblots showed that total levels of both E524K and D776K were approximately a quarter of GluR6 WT levels (24 ± 7% and 23 ± 6% respectively). As a fraction of total levels, surface expression of E524K was not significantly different from GluR6 WT. The fraction of total D776K expressed at the surface, however, was less than half that of GluR6 WT (0.44 ± 0.19; n = 5, P < 0.05), suggesting a specific trafficking defect associated with the D776K mutation. We also determined ligand affinity (by radioligand binding assay) as a proxy for correct folding. Specific binding of [3H]kainate in transiently transfected HEK cells was too low to allow determination of kainate and glutamate binding affinities. HEK cells stably expressing D776K were therefore used to determine affinity constants. The affinity of kainate for D776K was not significantly different from that for GluR6 WT (Kd = 12.9 ± 1.7 nM, n = 4 and 9.0 ± 1.9 nM, n = 3, respectively; P = 0.18). The apparent affinity of Glu was also unchanged in D776K (Ki = 390 ± 68 nM, n = 3) compared with GluR6 WT (Ki = 280 ± 7 nM, n = 3; P = 0.21).
In a final assay, we used the maturation state of receptor glycosylation as an indicator of intracellular localization (Greger et al., 2003; Priel et al., 2006). Membranes isolated from cells transfected with GluR6 WT and D776K were digested using Endo H and PNGase F (Fig. 3D). Endo H only cleaves immature glycosylation added in the endoplasmic reticulum, whereas PNGase F also removes sugar moieties after maturation in the Golgi. Although most GluR6 WT glycosylation was resistant to Endo H treatment (78.4 ± 4.1%, n = 4), this percentage was much lower for D776K (6.6 ± 1.5%, n = 4). No mature glycosylation was detectable with D776K. It is possible that constitutive activation of D776K leads to cytotoxicity, selecting against cells expressing high receptor levels. To counter this, we added the non-selective iGluR antagonist CNQX (10 μM) to the culture medium, and characterized the resulting responses and glycosylation patterns of D776K. The proportion of nonresponding cells treated with CNQX (19/27) was not significantly lower than for nontreated cells (67/89; P = 0.62, χ2 test). There were also no significant differences in the response sizes, relative agonist efficacies, or response kinetics for the two groups. The addition of CNQX to the culture media also had no effect on the glycosylation pattern observed for D776K. In particular, the percentage of receptors resistant to Endo H treatment was unchanged (7.6 ± 1.4%, n = 4).
The Effect of External Cations Is Abolished by D776K Mutation.
Residue Asp776 sits between the anion and cation binding sites located within
the dimer interface (Fig. 1C).
We therefore investigated whether the D776K mutant affected the sensitivity of
the receptor to ions. Current responses of GluR6 WT to glutamate are
attenuated, and desensitization rates are increased if sodium or chloride ions
in the external solution are replaced by other monovalent ions
(Bowie, 2002;
Paternain et al., 2003). We
determined the response amplitude of D776K expressed in Xenopus
laevis oocytes in various external solutions, replacing sodium ions with
either rubidium or cesium ions and chloride ions with either nitrate or
fluoride ions. When the external anion was replaced with either
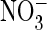 or F-, D776K responses
were significantly attenuated (to 1-2% of control values;
Fig. 4). In contrast,
exchanging the external cation for either Rb+ or Cs+
resulted in responses that were more than 30% larger than those in the NaCl
control (Fig. 4).
or F-, D776K responses
were significantly attenuated (to 1-2% of control values;
Fig. 4). In contrast,
exchanging the external cation for either Rb+ or Cs+
resulted in responses that were more than 30% larger than those in the NaCl
control (Fig. 4).
Fig. 4.

Anion and cation sensitivity of D776K. Scatter-plot showing responses to Glu (10 mM) from X. laevis oocytes expressing the GluR6 D776K mutant, recorded in buffers where either the cation or anion was exchanged. Responses were normalized to control responses (in NaCl), recorded before and after each test response. Scatter for control NaCl responses reflects the variability observed in individual oocytes. Data were collected from five oocytes. Dotted lines show the means for each group, with S.E.M. in gray. Responses in all four test buffers differed significantly from the NaCl control (P < 0.001). For comparison, normalized response levels from ConA-treated oocytes expressing GluR6 WT are indicated by arrows for each ion condition.
Direct comparison with GluR6 WT responses was not possible in oocytes, but
we tested responses after treatment with concanavalin A (ConA) to increase the
size of steady-state responses (Fig.
4). As expected, exchange of either cations or anions attenuated
GluR6 WT responses in ConA-treated oocytes (responses relative to NaCl control
were: Rb+, 17 ± 2%; Cs+, 17 ± 7%;
 , 3.4 ± 0.2%; F-,
1.0 ± 0.7%; n = 3-5). The cation effects were similar to those
observed in HEK cells (Rb+, 29%; Cs+, 6%)
(Plested et al., 2008). The
anion effects were larger than those reported in HEK cells
(
, 3.4 ± 0.2%; F-,
1.0 ± 0.7%; n = 3-5). The cation effects were similar to those
observed in HEK cells (Rb+, 29%; Cs+, 6%)
(Plested et al., 2008). The
anion effects were larger than those reported in HEK cells
(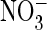 , 75%; F-, 14%)
(Plested and Mayer, 2007),
which may be a consequence of ConA treatment. As noted above, D776K responses
were significantly larger than control in both rubidium and cesium
(Fig. 4), but the permeability
of both ions in GluR6 has been reported to be ∼25% higher than that of
sodium (Jatzke et al., 2002).
We confirmed that this was the most likely explanation for the larger currents
by determining slope-conductance responses at positive potentials. Outward
currents were not significantly higher in the presence of external cesium or
rubidium ions compared with the sodium ion control (data not shown).
, 75%; F-, 14%)
(Plested and Mayer, 2007),
which may be a consequence of ConA treatment. As noted above, D776K responses
were significantly larger than control in both rubidium and cesium
(Fig. 4), but the permeability
of both ions in GluR6 has been reported to be ∼25% higher than that of
sodium (Jatzke et al., 2002).
We confirmed that this was the most likely explanation for the larger currents
by determining slope-conductance responses at positive potentials. Outward
currents were not significantly higher in the presence of external cesium or
rubidium ions compared with the sodium ion control (data not shown).
Discussion
We investigated the effects on agonist responses of reversing the charges at four sites in the apex of the GluR6 S1S2 agonist binding domain. There are several key intersubunit contacts in this region, including salt bridges and binding sites for an anion and two cations (Fig. 1). The ion binding sites and one of the two charge pairs are unique to kainate-selective receptors. Given the proposed importance of dimer interface stability to activation and desensitization, changes that perturb either the charge distribution or the net charge balance in this region would be expected to significantly accelerate desensitization. Among the single mutants at these four sites, E524K and R775D both attenuated responses as expected (Fig. 2A, Table 1). The other two charge-reversal mutations, K531E and D776K, both had unexpected phenotypes, with desensitization respectively slowed and blocked (Fig. 2, A and B). In the case of K531E, the effect was relatively small, with an increase in τDes of less than 2-fold for responses to Glu and ∼9-fold for responses to KA (Table 1). It was not entirely unexpected, however, in that mutation of Lys531 to glycine also slows desensitization of GluR6 (Fleck et al., 2003). For neither of these mutants did the location of Lys531 in the dimer interface, or the interactions it makes, provide an easy explanation for these phenotypes. This is especially true for K531E, which resulted in a change in net charge of minus 4 at the dimer apex relative to GluR6 WT. An explanation for these phenotypes will clearly require further investigation of the conformations adopted by the S1S2 domain dimer during both receptor activation and desensitization.
The second single mutant with an unexpected phenotype, D776K, resulted in apparently nondesensitizing responses. This was in marked contrast to the effects of other published mutations at this site, with conservative changes to asparagine and glutamate accelerating desensitization as would be expected (Plested and Mayer, 2007). Elevated steady-state iGluR responses can result either from changes in the relative stability or kinetic accessibility of the desensitized state (e.g., GluR2 L483Y; Sun et al., 2002), or from changes to the relative contributions of different open states (e.g., Concanavalin A; Bowie et al., 2003). Although we cannot directly distinguish between these possibilities, we believe the nondesensitizing phenotype of D776K can be explained primarily in terms of S1S2 dimer stability and therefore desensitization block. First, our biophysical data confirm that, at least in the isolated S1S2 domain, the dimer is clearly stabilized by the D776K mutation. Although native gel electrophoresis showed that the GluR6 WT S1S2 domain does associate to a limited extent as a dimer, analytical ultracentrifugation data indicated that the affinity is very low, consistent with previous reports (Weston et al., 2006; Chaudhry et al., 2009). In contrast, the apparent affinity of the S1S2 dimer interaction was increased >1000-fold by the D776K mutation (Fig. 3B).
A recent study identified a number of GluR6 S1S2 mutations that stabilized the dimer interface but reduced the extent of desensitization by less than 20% (Chaudhry et al., 2009). It was therefore proposed that the desensitized state is intrinsically more stable in GluR6 than in GluR2. However, the mutants described in that study all exhibited dimerization constants above 20 μM, weaker than the 7 μM value observed for a GluR2 mutant that reduced desensitization by only 10% (Sun et al., 2002). For GluR2, substantial block of desensitization is observed only for mutants with stronger dimerization affinities of ∼1 to 5 μM (26-52% desensitization) and 30 nM for L483Y (8% desensitization). The S1S2 dimer affinity we observed with D776K (0.9 μM), although ∼30-fold lower than that of GluR2 L483Y, falls within the range of affinities observed for strongly nondesensitizing GluR2 mutants. Although our data cannot formally exclude a contribution from destabilization of the desensitized state, the effect of the GluR6 D776K mutation suggests that desensitization in AMPA and kainate receptors is similarly sensitive to the strength of the dimer interface.
These experiments demonstrated that the D776K mutant stabilized the dimer, but they still leave the question of how this occurs. The most likely explanation comes from the location of the sodium binding sites in the dimer interface. It is possible to model a lysine in place of Asp776 without clashes. The Cβ atom of the two Asp776 residues is 6-6.2 Å from the nearest sodium ion, and the amine group of each of the lysines can be positioned close to the center of the opposing sodium binding site using a common rotamer (data not shown). In contrast to the chloride binding site, where residues from both subunits interact with the ion, the two sodium binding pockets are largely contained within single subunits, with only limited interactions across the dimer interface (Plested et al., 2008). Therefore, were the lysine side chains in D776K to extend to and effectively occupy the opposing cation binding sites as surrogate cations, this would represent additional intersubunit interactions. These two new contacts could then serve to lock the conformation of the dimer, preventing desensitization. One consequence would be that the lysines would be expected to displace the sodium ions but not necessarily affect the chloride ion, fully consistent with the observed anion and cation effects of D776K (Fig. 4). The D776K mutant was still sensitive to changes in external anions, unlike the nondesensitizing cystine cross-link mutant (Y521C-L783C), which was insensitive to changes in both external cations and anions (Plested and Mayer, 2007; Plested et al., 2008).
We observed effects of D776K on expression levels and on deactivation kinetics as well. The surface expression and maturation of D776K in HEK cells seemed attenuated. We cannot rule out cytotoxic effects from constitutive leak currents in D776K-expressing cells, but, with that important caveat, the observed low surface expression of D776K is consistent with other reports. The trafficking efficiency of AMPA receptor subunits has been found to correlate closely with their desensitization properties (Greger et al., 2003; Penn et al., 2008), whereas it has been proposed that the ability to desensitize is a key checkpoint in kainate receptor trafficking (Priel et al., 2006). The other interesting effect of D776K was on deactivation kinetics. For the nondesensitizing AMPA receptors, there is a concomitant slowing of the rate of deactivation (Sun et al., 2002). The GluR2 L483Y phenotype was initially proposed to result from both stabilization of the open state and destabilization of the desensitized state (see Fig. 5 in Sun et al., 2002). The former was confirmed by determining the rate of channel closure for GluR1 L497Y (Pei et al., 2007), whereas kinetic modeling of the same mutant suggested that both entry into the desensitized state and the rate of ligand dissociation were decreased relative to GluR1 WT (Mitchell and Fleck, 2007). Although this modeling study did not incorporate slower channel closure, it seems probable that a combination of these three factors blocks desensitization and slows deactivation in the AMPA receptor leucine to tyrosine mutants. It is therefore interesting that peak responses to Glu (but not to KA) deactivate as rapidly as GluR6 WT responses (Fig. 2C), in contrast to the slow deactivation of other GluR6 mutants with attenuated desensitization rates (Zhang et al., 2006; Chaudhry et al., 2009). We have previously found that deactivation rates in GluR6 seem dominated by ligand dissociation (Zhang et al., 2008), which implies that Glu, but not KA, dissociates from D776K as quickly as it does from GluR6 WT. This emphasizes the fact that slower deactivation is not always associated with desensitization block, as with cyclothiazide action at AMPA receptors (Mitchell and Fleck, 2007), but also highlights an interesting ligand-specific difference in the phenotype of D776K.
Chaudhry et al. (2009) rightly underscored the difficulty of improving dimer packing by rational design; as with the original GluR3 L507Y mutant, our discovery of D776K was entirely serendipitous. If anything, the D776K mutation would have been expected to be destabilizing. Over the past 10 years, nondesensitizing AMPA receptor mutants have provided a wealth of insights into the conformational requirements of both the desensitization and trafficking of AMPA receptors. As the first example of a kainate receptor subunit that is nondesensitizing in the absence of covalent cross-linkages, GluR6-D776K is closer in character to these AMPA receptor mutants. It is to be hoped that, low expression notwithstanding, D776K may have the potential to fulfill an equivalent role for kainate receptors in the future.
Acknowledgments
We thank Kate Davis for technical assistance with mutagenesis and cell culture, and Sarah Lummis, Kerry Price, and Andrew Thompson (University of Cambridge, UK) for providing facilities for and assistance with the oocyte recordings. We are also grateful to Steve Heinemann (Salk Institute, La Jolla, CA) for the gift of the rat GluR6(Q) cDNA and David Wyllie (University of Edinburgh, Edinburgh, UK) for help with the fast perfusion system.
This work was supported by the UK Medical Research Council [Grant G0200084] and by the National Institutes of Health National Institute of General Medical Sciences [Grant T32-GM008704].
N.N. and Y.Z. contributed equally to this work.
ABBREVIATIONS: AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; iGluR, ionotropic glutamate receptor; HEK, human embryonic kidney; WT, wild type; GluR, glutamate receptor; KA, kainate; PAGE, polyacrylamide gel electrophoresis; BN, Blue-Native; Endo H, endoglycosidase H; PNGase F, peptide-N--(N-acetyl-β-glucosaminyl)asparagine amidase; CNQX, 6-cyano-2,3-dihydroxy-7-nitroquinoxaline; ConA, concanavalin A.
References
- Armstrong N and Gouaux E (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28 165-181. [DOI] [PubMed] [Google Scholar]
- Bowie D (2002) External anions and cations distinguish between AMPA and kainate receptor gating mechanisms. J Physiol 539 725-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Garcia EP, Marshall J, Traynelis SF, and Lange GD (2003) Allosteric regulation and spatial distribution of kainate receptors bound to ancillary proteins. J Physiol 547 373-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry C, Weston MC, Schuck P, Rosenmund C, and Mayer ML (2009) Stability of ligand-binding domain dimer assembly controls kainate receptor desensitization. EMBO J 28 1518-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Feltz A, Sahara Y, and Westbrook GL (1998) Activation kinetics of AMPA receptor channels reveal the number of functional agonist binding sites. J Neurosci 18 119-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam J and Schuck P (2004) Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol 384 185-212. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, and Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51 7-61. [PubMed] [Google Scholar]
- Fleck MW, Cornell E, and Mah SJ (2003) Amino-acid residues involved in glutamate receptor 6 kainate receptor gating and desensitization. J Neurosci 23 1219-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouaux E (2004) Structure and function of AMPA receptors. J Physiol 554 249-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, and Ziff EB (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40 763-774. [DOI] [PubMed] [Google Scholar]
- Heckmann M, Bufler J, Franke C, and Dudel J (1996) Kinetics of homomeric GluR6 glutamate receptor channels. Biophys J 71 1743-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horning MS and Mayer ML (2004) Regulation of AMPA receptor gating by ligand binding core dimers. Neuron 41 379-388. [DOI] [PubMed] [Google Scholar]
- Jatzke C, Watanabe J, and Wollmuth LP (2002) Voltage and concentration dependence of Ca2+ permeability in recombinant glutamate receptor subtypes. J Physiol 538 25-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM and Pelletier SL (1992) Computer-aided interpretation of analytical sedimentation data for proteins, in Analytical Ultracentrifugation in Biochemistry and Polymer Sciences (Harding SE, Rowe AJ and Horton JC eds) pp 90-125, Royal Society for Chemistry, Cambridge, UK.
- Mayer ML (2005) Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron 45 539-552. [DOI] [PubMed] [Google Scholar]
- Mayer ML (2006) Glutamate receptors at atomic resolution. Nature 440 456-462. [DOI] [PubMed] [Google Scholar]
- Mitchell NA and Fleck MW (2007) Targeting AMPA receptor gating processes with allosteric modulators and mutations. Biophys J 92 2392-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanao MH, Green T, Stern-Bach Y, Heinemann SF, and Choe S (2005) Structure of the kainate receptor subunit GluR6 agonist-binding domain complexed with domoic acid. Proc Natl Acad Sci U S A 102 1708-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa S, Kamiya H, and Tsuzuki K (1998) Glutamate receptors in the mammalian central nervous system. Prog Neurobiol 54 581-618. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Cohen A, Stern-Bach Y, and Lerma J (2003) A role for extracellular Na+ in the channel gating of native and recombinant kainate receptors. J Neurosci 23 8641-8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W, Ritz M, McCarthy M, Huang Z, and Niu L (2007) Receptor occupancy and channel-opening kinetics: a study of GLUR1 L497Y AMPA receptor. J Biol Chem 282 22731-22736. [DOI] [PubMed] [Google Scholar]
- Penn AC, Williams SR, and Greger IH (2008) Gating motions underlie AMPA receptor secretion from the endoplasmic reticulum. EMBO J 27 3056-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plested AJ and Mayer ML (2007) Structure and mechanism of kainate receptor modulation by anions. Neuron 53 829-841. [DOI] [PubMed] [Google Scholar]
- Plested AJ, Vijayan R, Biggin PC, and Mayer ML (2008) Molecular basis of kainate receptor modulation by sodium. Neuron 58 720-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A, Selak S, Lerma J, and Stern-Bach Y (2006) Block of kainate receptor desensitization uncovers a key trafficking checkpoint. Neuron 52 1037-1046. [DOI] [PubMed] [Google Scholar]
- Robert A and Howe JR (2003) How AMPA receptor desensitization depends on receptor occupancy. J Neurosci 23 847-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, and Stevens CF (1998) The tetrameric structure of a glutamate receptor channel. Science 280 1596-1599. [DOI] [PubMed] [Google Scholar]
- Stafford WF and Sherwood PJ (2004) Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys Chem 108 231-243. [DOI] [PubMed] [Google Scholar]
- Stern-Bach Y, Russo S, Neuman M, and Rosenmund C (1998) A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron 21 907-918. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, and Gouaux E (2002) Mechanism of glutamate receptor desensitization. Nature 417 245-253. [DOI] [PubMed] [Google Scholar]
- Thompson AJ and Lummis SC (2008) Antimalarial drugs inhibit human 5-HT(3) and GABA(A) but not GABA(C) receptors. Br J Pharmacol 153 1686-1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MC, Schuck P, Ghosal A, Rosenmund C, and Mayer ML (2006) Conformational restriction blocks glutamate receptor desensitization. Nat Struct Mol Biol 13 1120-1127. [DOI] [PubMed] [Google Scholar]
- Wong AY, Fay AM, and Bowie D (2006) External ions are coactivators of kainate receptors. J Neurosci 26 5750-5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, MacLean DM, and Bowie D (2007) Na+/Cl- dipole couples agonist binding to kainate receptor activation. J Neurosci 27 6800-6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nayeem N, and Green T (2008) Mutations to the kainate receptor subunit GluR6 binding pocket that selectively affect domoate binding. Mol Pharmacol 74 1163-1169. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Nayeem N, Nanao MH, and Green T (2006) Interface interactions modulating desensitization of the kainate-selective ionotropic glutamate receptor subunit GluR6. J Neurosci 26 10033-10042. [DOI] [PMC free article] [PubMed] [Google Scholar]