

Abstract
A family of 20 tris-azaaromatic quaternary ammonium (AQA) compounds were tested for their inhibition of α7 nicotinic acetylcholine receptors (nAChRs) expressed in Xenopus laevis oocytes. The potency of inhibitory activity was related to the hydrophobic character of the tris head groups. Two tris-AQA compounds were studied in detail: the highly effective inhibitor 1,3,5-tri-[5-(1-quinolinum)-pent-1-yn-1-yl]-benzene tribromide (tPyQB) and the less potent antagonist 1,3,5,-tri-{5-[1-(2-picolinium)]-pent-1-yn-1-yl}benzene tribromide (tPy2PiB). In addition, we evaluated 1,2,4,5-tetra-{5-[1-(3-benzyl)pyridinium]pent-1-yl}benzene tetrabromide (tkP3BzPB), a tetrakis-AQA with very hydrophobic headgroups. We compared the activity of the AQA compounds to the frequently used α7-antagonist methyllycaconitine (MLA). Both tPyQB and tkP3BzPB were selective antagonists of α7. However, although inhibition by tPyQB was reversible within 5 min, the recovery time constant for tkP3BzPB inhibition was 26.6 ± 0.8 min, so that the equilibrium inhibition in the prolonged presence of nanomolar concentrations of tkP3BzPB was nearly 100%. The potency, selectivity, and slow reversibility of tkP3BzPB were comparable with or greater than that of MLA. The inhibitory actions of tPyQB, tPy2PiB, and tkP3BzPB were evaluated on the acetylcholine (ACh)-evoked responses of native nAChRs in rat brain slices. The α7-mediated responses of hippocampal interneurons were effectively reduced by 1 μM tPyQB and tkP3BzPB but not tPy2PiB. In rat medial septum, tkP3BzPB produced a greater inhibition of ACh-evoked responses of cells with fast inward currents (type I) than of cells with predominantly slow kinetics (type II), suggesting that tkP3BzPB can block α7 yet preserve the responsiveness of non-α7 receptors. These agents might be helpful in elucidating complex receptor responses in brain regions with mixed populations of nAChRs.
Nicotinic acetylcholine receptors (nAChRs) are distributed throughout the central and peripheral nervous systems (Role and Berg, 1996; Wonnacott, 1997). Nine neuronal α subunits (α2-α10) and three neuronal β subunits (β2-β4) have thus far been identified and cloned in vertebrate systems. One type of neuronal nAChR is formed by the assembly of α and β subunits, with functional properties depending on both α and β subunits within the receptor complex (Buisson and Bertrand, 2002). In Xenopus laevis oocytes, pairwise combinations of some neuronal α and β subunits form functional receptors. However, the existence of complex subtypes consisting of more than two different subunits has been documented in native brain regions. In addition to the heteromeric receptors, α7, α8, or α9 nAChR subunits can form functional α-bungarotoxin-sensitive homopentamers (Couturier et al., 1990; Séguéla et al., 1993; Elgoyhen et al., 1994; Peng et al., 1994). The two major subtypes in the central nervous system (CNS) are α4β2* (asterisk denotes the possibility of additional subunits) and α7 nAChRs (Flores et al., 1992; Lindstrom et al., 1996). The majority of the α7 nAChRs in the brain are believed to be homopentameric receptors; however, recent data suggest the existence of putatively heteromeric α7β2 nAChRs on the medial septum/diagonal band (MS/DB) neurons (Liu et al., 2009).
Although the functional diversity of brain nAChRs has been widely documented, the structural composition of many receptor subtypes remains to be elucidated. The therapeutic targeting of isolated neuronal nAChRs is challenged by the diversity in their composition, distribution, and pharmacological properties. Single neurons frequently express multiple nAChR subtypes (Papke, 1993; Henderson et al., 2005; Thinschmidt et al., 2005a). The pharmacological isolation of nicotinic components is possible with the use of subtype-selective ligands. In particular, several “classic” antagonists that have been either obtained from natural sources or synthesized have been used to identify particular nicotinic receptor subtypes. Methyllycaconitine (MLA) is a toxin derived from the seeds of Delphinium brownii that has been reported to be an α7-selective antagonist at low concentrations (Aiyar et al., 1979; Alkondon et al., 1992). However, at higher concentrations, MLA has also been shown to block α4β2 receptors expressed in HEK cells (Buisson et al., 1996). In addition, MLA was shown to inhibit other nAChR subtypes on dopamine neurons from rat striatum at concentrations commonly used to “selectively” block α7-mediated responses (Mogg et al., 2002). Therefore, there is still a need for better ligands to pharmacologically isolate neuronal nAChR subtypes.
In the present work, we evaluated a family of novel azaaromatic quaternary ammonium (AQA) analogs for their ability to inhibit α7 nAChR-mediated responses in X. laevis oocytes. We studied two tris-AQA compounds in detail, 1,3,5,-tri-{5-[1-(2-picolinium)]-pent-1-yn-1-yl}benzene tribromide (tPy2PiB) and 1,3,5-tri-[5-(1-quinolinum)-pent-1-yn-1-yl]-benzene tribromide (tPyQB) (Fig. 1A). Because the activity profile of the tris compounds indicated that potent inhibition of α7 was associated with the presence of multiple hydrophobic head groups, we also tested a tetrakis analog with four hydrophobic head groups, 1,2,4,5-tetra-{5-[1-(3-benzyl)pyridinium]pent-1-yl}benzene tetrabromide (tkP3BzPB, Fig. 1B). All three AQA analogs showed a higher selectivity for α7 than for α4β2 or α3β4 nAChRs expressed in X. laevis oocytes. We evaluated the activity of the AQA analogs both on stratum radiatum interneurons in area CA1 of rat hippocampus, which predominantly express α7 receptors, and on neurons from the medial septum, which express both α7 and α4β2* receptors. Although the more hydrophobic tris analog was most potent in coapplication experiments, the hydrophobic tetrakis analog produced inhibition that was only slowly reversible and was therefore very effective if preapplied to cells or tissue at low concentrations. Our data support the hypothesis that, like MLA, tkP3BzPB can block α7-mediated responses and preserve the responsiveness of non-α7 receptors in neurons with a mixed receptor phenotype. However, whereas MLA is a competitive antagonist, tkP3BzPB inhibition is noncompetitive and is both use- and voltage-independent. The unique mechanism of tkP3BzPB inhibition may be useful to increase our understanding of receptor-mediated signaling and whether ligand binding may have effects that are independent of ion conduction.
Fig. 1.

A, tris-AQA analogs. Shown are structures of 20 tris-AQA molecules tested for their inhibitory effects on α7 nAChR. B, the structure of tkP3BzPB.
Materials and Methods
Chemicals. Tris- and tetrakis-AQA analogs were prepared as described previously (Zheng et al., 2007; Dwoskin et al., 2008). PNU-120596 was purchased from Tocris (Ellisville, MO). All other chemicals for electrophysiology were obtained from Sigma Chemical Co. (St. Louis, MO).
nAChR Expression in X. laevis Oocytes. For recombinant nAChR studies, mature (>9 cm) female X. laevis African frogs (Nasco, Ft. Atkinson, WI) were used as a source of oocytes. Before surgery, frogs were anesthetized by placing the animal in a 1.5 g/l solution of MS222 (3-aminobenzoic acid ethyl ester) for 30 min. Oocytes were removed from an incision made in the abdomen. All procedures involving frogs were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC).
To remove the follicular cell layer, harvested oocytes were treated with 1.25 mg/ml type 1 collagenase (Worthington Biochemicals, Freehold, NJ) for 2 h at room temperature in calcium-free Barth's solution with a composition of 88 mM NaCl, 1 mM KCl, 0.8 mM MgSO4, 2.4 mM NaHCO3, 15 mM HEPES, pH 7.6, and 12 mg/l tetracycline. Stage 5 oocytes were then isolated and injected with 50 nl (5-20 ng) each of the appropriate subunit cRNAs. The rat neuronal nAChR and mouse muscle α1, β1, and δ clones were obtained from Dr. Jim Boulter (UCLA, Los Angeles, CA). The mouse ε clone was provided by Dr. Paul Gardener (University of Massachusetts, Worcester, MA), and human nAChR receptor clones from Dr. Jon Lindstrom (University of Pennsylvania, Philadelphia, PA). To improve the expression of α7 nAChR, α7 RNA was routinely coexpressed with human RIC-3 (Halevi et al., 2003). The RIC-3 clone was obtained from Dr. Millet Treinin (Hebrew University, Jerusalem, Israel). After linearization and purification of cloned cDNAs, RNA transcripts were prepared in vitro using the appropriate mMessage mMachine kit from Ambion (Austin, TX).
Voltage-Clamp Recording in X. laevis Oocytes Expressing nAChRs. Experiments were conducted using OpusXpress 6000A (Molecular Devices, Union City, CA). Each oocyte received initial control applications of acetylcholine (ACh), coapplications of ACh and the experimental drugs, and then a follow-up control application of ACh. The standard control ACh concentrations for α7, α4β2, and α3β4 receptors were 60, 10, and 100 μM, respectively. Both peak amplitude and net charge of the responses were measured for each drug application (Papke and Porter Papke, 2002) and calculated relative to the preceding ACh control responses to normalize the data, compensating for the varying levels of channel expression among the oocytes. Net charge values were used to report inhibitory effects. Competition experiments were conducted by generating concentration-response curves to ACh, applied either alone or in the presence of the AQA analog. Responses were initially normalized to the ACh control response values and then adjusted to reflect the experimental drug responses relative to the ACh maximums. Means and standard errors (S.E.M.) were calculated from the normalized responses of at least four oocytes for each experimental concentration. Concentration-response data were fit to the Hill equation, assuming negative Hill slopes.
Brain Slice Preparation and Patch-Clamp Recording. All procedures involving rats were approved by the University of Florida IACUC and were in accord with the Institute of Laboratory Animal Resources (1996). Male Sprague-Dawley rats (postnatal day 12-25) were anesthetized with Halothane (Halocarbon Laboratories, River Edge, NJ) and swiftly decapitated. Transverse (300 μm) whole-brain slices were prepared using a vibrating microtome (Pelco, Redding, CA) and then placed in a high Mg2+/low Ca2+ ice-cold artificial cerebral spinal fluid containing 124 mM NaCl, 2.5 mM KCl, 1.2 mM NaH2PO4, 2.5 mM MgSO4, 10 mM d-glucose, 1 mM CaCl2, and 25.9 mM NaHCO3, saturated with 95% O2/5% CO2. Slices were incubated at 30°C for 30 min and then left at room temperature until they were transferred to a submersion chamber (Warner Instruments, Hamden, CT) for recording. During experiments, slices were perfused at a rate of 2 ml/min with normal artificial cerebral spinal fluid containing 126 mM NaCl, 3 mM KCl, 1.2 mM NaH2PO4, 1.5 mM MgSO4, 11 mM d-glucose, 2.4 mM CaCl2, 25.9 mM NaHCO3, and 0.004 mM atropine sulfate, saturated with 95% O2/5% CO2 at 30°C. Cells were visualized with infrared differential interference contrast microscopy using a microscope (E600FN; Nikon, Tokyo, Japan). Patch-clamp recording pipettes were pulled from borosilicate glass (Sutter Instruments, Novato, CA) using a Flaming/Brown micropipette puller (P-97; Sutter Instruments). Recording pipettes were filled with an internal solution of 125 mM K-gluconate, 1 mM KCl, 0.1 mM CaCl2, 2 mM MgCl2, 1 mM EGTA, 2 mM MgATP, 0.3 mM Na3GTP, and 10 mM HEPES (pH adjusted to 7.3 with KOH). The resistance of the recording pipette when filled with the internal solution was 3 to 5 MΩ. Cells were held at -70 mV, and a -10-mV/10-ms test pulse was used to determine access resistance, input resistance, and whole-cell capacitance. Cells with access resistances >60 MΩ or those requiring holding currents >200 pA were not included in the final analyses. Signals were digitized using a digitizer (Axon Digidata 1322A; Molecular Devices, Sunnyvale, CA) and sampled at 20 kHz on a personal computer (Dell, Round Rock, TX) using Clampex version 8 or 9. Data analysis was done with Clampfit version 8 or 9, Excel 2000 (Microsoft Corp., Redmond, WA), and Prism version 4.02 (GraphPad Software, San Diego, CA). Data are reported as mean ± S.E.M. Statistical analyses were performed using two-tailed Student's t test and one-way analysis of variance.
Drug Application in Brain Slice Preparations. Local somatic applications of ACh (1 mM pipette concentration) were made using single- or double-barrel glass pipettes attached to a picospritzer (General Valve, Fairfield, NJ) with Teflon tubing (10-20 psi for 5-15 ms). Coapplication experiments were performed using a double-barreled pressure application pipette in which one side had 1 mM ACh and the other had 1 mM ACh + 300 μM tkP3BzPB, applied with a 30-s interstimulus interval. Single-barrel pipettes were pulled from borosilicate glass with outer and inner diameters of 1.5 and 0.86 mm, respectively (Sutter Instruments). Pipette opening size of the single barrel was typically 2 to 3 μm. Double-barreled pipettes were pulled from borosilicate theta-shaped glass tubing with an outer diameter of 1.5 mm; pipette opening size was approximately 3 to 4 μm. The application pipette was usually placed within 10 to 15 μm of the cell soma.
In experiments in which AQA analogs were bath-applied, for each cell four ACh baseline-evoked responses were recorded before bath application of the antagonist. ACh-evoked responses were then recorded for 13 to 22 min in the presence of the AQA analog. Each analog was bath-applied at a final concentration of 1 μM. In some septum experiments, dihydro-β-erythroidine (DHβE) was also bath-applied at a final concentration of 1 μM.
When pipettes were loaded with 1 mM ACh, the average net charge of evoked responses was not significantly different in single- and double-barrel experiments (data not shown). Experiments conducted to describe the error produced by alternating pressure applications using double-barreled pipettes showed an 85 ± 8% (n = 5) correspondence in the peak amplitudes between the agonist applications from the two barrels (data not shown). In previous experiments, we determined that pressure application from a pipette containing 1 mM ACh delivered an effective concentration of approximately 30 μM to the surface of the cell (Lopez-Hernandez et al., 2007).
Results
Inhibition of Rat nAChR Responses Expressed in X. laevis Oocytes. The library of 20 tris-AQA analogs (Fig. 1B) was tested at a probe concentration of 1 μM on oocytes expressing α7 receptors. The results shown in Fig. 2 indicate activities ranging from nearly complete inhibition with tPyQB to no significant effect of tPy2PiB at the 1 μM probe concentration. When the compounds were ordered for their relative effectiveness at inhibiting α7 receptors at the probe concentration, a structure-activity relationship was suggested (Fig. 3), relating greater predicted hydrophobicity of the AQA head group to greater inhibition of α7 nAChR. To further test the hypothesis that multiple hydrophobic head groups contribute importantly to α7 inhibition, we also synthesized a tetrakis analog with very hydrophobic head groups, 1,2,4,5-tetra-{5-[1-(3-benzyl)pyridinium]pent-1-yl}benzene tetrabromide (tkP3BzPB), shown in Fig. 1B.
Fig. 2.

Data for the inhibition and recovery of α7-mediated responses to the tris-AQA analogs. Each drug was tested at a concentration of 1 μM coapplied with 60 μM ACh. The filled bars are the average responses (n ≥ 4 ± S.E.M.) during the coapplications, normalized to the response to ACh alone applied 5 min earlier to the same cells; the drug was then washed out for 5 min and ACh was applied again. The open bars show the amplitude of the subsequent responses to ACh alone, normalized to the original ACh controls. The data are arranged so that the analogs are in order based on the fractional inhibition produced at the test concentration, with the most effective drugs on the left. The same order is applied to the arrangement of structures in the top of the figure.
Fig. 3.

A structure-activity analysis for the inhibition of α7 by tris-AQA analogs based on the hydrophobicity of AQA head groups. Inhibition was calculated as 1 minus the normalized coapplication response data taken from Fig. 1. LogP values are estimates for head groups only. Compounds with a triple bond in the linker units are black symbols and those with saturated linker units are represented by gray symbols. Two linear regression lines are shown. The R values were 0.912 and 0.863 for the compounds shown in black and gray, respectively.
From the family of tris-AQA compounds, we selected two compounds that varied greatly in their ability to inhibit α7 at the 1 μM probe concentration, tPy2PiB and tPyQB, to study in detail, along with the tetrakis AQA, tkP3BzPB. These AQA analogs were tested on combinations of rat neuronal nAChR α and β subunits (α4β2 and α3β4) and on α7 homomeric receptors expressed in X. laevis oocytes (Fig. 4 and Table 1). Whereas α3β4 nAChRs represent a minimal model for ganglionic nicotinic receptors, α4β2 and α7 nAChRs are the two predominant subtypes of nicotinic receptors in the CNS. All three AQA analogs most potently inhibited α7 nAChRs among the subunit combinations tested. The IC50 value of tPyQB for oocytes expressing α7 subunits was 0.13 ± 0.02 μM, as determined with a simple coapplication protocol, whereas the IC50 values for tPy2PiB and tkP3BzPB were 6.3 ± 0.6 and 1.0 ± 0.1 μM, respectively. Note that there was partial inhibition of the α4β2 receptors at the lowest concentrations of tPy2PIB. This experiment was conducted on cells after the injection of equal amounts of α4 and β2 RNA, which results in a mixed population of receptors with different subunit stoichiometry (Nelson et al., 2003; López-Hernández et al., 2004). The data suggest that the minor population (most likely those with an α4:β2 subunit ratio of 3:2) may have significantly greater sensitivity to tPy2PIB than the major population of receptors in these cells.
Fig. 4.

Inhibition of nAChR responses expressed in X. laevis oocytes. A to C show the averaged normalized mean data (± S.E.M., n ≥ 4) of net charge responses to coapplication of ACh and a range of concentrations of tris- and tetrakis-AQA analogs from oocytes expressing rat α4β2, α3β4, or α7 subunits: tPyQB (A), tPy2PiB (B), and tkP3BzPB (C). The data were normalized to responses to ACh alone obtained 5 min before the coapplication of ACh and antagonist at the indicated concentrations. ○, α4β2; •, α7; ▪ α3β4. IC50 values are provided in Table 1. D, the averaged normalized mean data (± S.E.M., n ≥ 4) of net charge responses to coapplication of ACh and a range of concentrations of MLA from oocytes expressing human α4β2, α3β4, or α7 subunits. IC50 values are provided in Table 1. E, representative data for the effects of low concentrations of MLA coapplied with 60 μM ACh from oocytes expressing human α7. Coapplications of ACh and MLA alternated with control applications of 60 μM ACh alone at 5-min intervals. Although there was relatively little inhibition during the coapplication of ACh and 100 nM MLA, there was a significant decrease in the subsequent ACh control response. F, the effect of preincubation on increasing the potency of MLA inhibition of α7 nAChR. Shown are responses to 300 μM ACh applied alone before a 3-min incubation with 3 nM MLA, then coapplied with 3 nM MLA. Also shown are control responses to 60 μM ACh before and after the MLA treatment, used to follow the rate of recovery. Note that although the peak amplitude of the final 60 μM response shown is similar to that of the first 60 μM control, the net charge of the responses were significantly less than the initial controls (p < 0.05, n = 5).
TABLE 1.
IC50 values for MLA and the AQA analogs in coapplication experiments
|
IC50
|
|||
|---|---|---|---|
| α7 | α4β2 | α3β4 | |
| μM | |||
| tPyQB | 0.13 ± 0.02 | 4.1 ± 1.0 | 1.0 ± 0.1 |
| tPy2PiB | 6.3 ± 0.6 | 86 ± 10 | 10.0 ± 1.4 |
| tkP3BzPB | 1.0 ± 0.1 | 48 ± 11 | 9.2 ± 1.2 |
| MLA | 1.2 ± 0.2 | 34 ± 5 | 2.0 ± 0.2 |
For comparison, we also studied the effects of MLA on human α7, α4β2, and α3β4 nAChRs expressed in X. laevis oocytes (Fig. 4D). When a simple coapplication protocol was used, MLA seemed to be nonselective as an antagonist of α7 and α3β4 receptors and was approximately 20-fold less potent for α4β2, with IC50 values of 1.2 ± 0.2, 2.0 ± 0.2, and 34 ± 5.4, respectively (Table 1). We also evaluated the inhibitory activity of MLA for mouse muscle (α1β1εδ) nAChRs expressed in X. laevis oocytes (data not shown) and obtained an IC50 value (1.5 ± 0.2 μM), comparable with that for the inhibition of α7 receptors in coapplication experiments. This apparent lack of selectivity of MLA for α7 receptors was somewhat surprising. However, when MLA was applied to receptors other than α7, the inhibition produced during the coapplication was fully reversed after a 5-min washout, whereas the inhibition of α7 receptors was not.
Although there was very little inhibition of the α7 responses during coapplications of ACh and concentrations of MLA ≤100 nM, at concentrations ≥100 nM, the oocytes did not recover their full responsiveness to subsequent applications of ACh (representative data are shown in Fig. 4E). For this reason, to test the activity of MLA in coapplication experiments at concentrations greater than 100 nM, separate sets of cells were used for each higher concentration tested. These data suggested that an effective selectivity of low concentrations of MLA for the inhibition of α7 nAChR might be achieved with preapplications and continued application of MLA. As shown in Fig. 4F, when α7 receptors were first pre-exposed to 3 nM MLA for 3 min before the coapplication of MLA and ACh, there was greater than 90% inhibition of the responses to high ACh concentrations. After the 3-min incubation with MLA, with subsequent washout of the MLA, the net charge responses to control applications of 60 μM ACh recovered with a time constant of 24 ± 4 min. When a similar preincubation protocol was used with oocytes expressing α4β2 nAChR (not shown), there was no significant effect of 3 nM MLA on ACh-evoked responses (not shown).
Recovery from Inhibition in X. laevis Oocytes Expressing Rat nAChRs. Under conditions in which coapplications of ACh and antagonist did not significantly reduce subsequent responses to ACh applied alone, our routine protocol (see Materials and Methods) of making alternating applications of ACh and ACh plus antagonist allows single sets of oocytes to be used to generate full concentration-response data sets. However, as was the case with MLA applications to cells expressing α7, slow kinetics of recovery from inhibition required the use of multiple sets of cells and additional experimental protocols. All three of the nAChR subtypes tested recovered fully during the 5-min wash periods after applications of either tPyQB or tPy2PiB (data not shown). However, a difference in recovery was noted for α7-expressing cells treated with tkP3BzPB. As shown in Fig. 5, A and B, α3β4 and α4β2 nAChRs showed no significant residual inhibition 5 min after washout of tkP3BzPB at any of the concentrations tested. In contrast, α7 receptors exhibited decreasing recovery with increasing tkP3BzPB concentrations (Fig. 5C). Therefore, as with MLA, fresh sets of cells were required for each concentration of tkP3BzPB ≥1 μM.
Fig. 5.

Recovery from tkP3BzPB inhibition in oocytes expressing rat nAChRs. Recovery experiments were performed after 5-min wash and application at increasing concentrations of tkP3BzPB for cells expressing α3β4 (A), α4β2 (B), or α7 (C) nAChR. There were no significant effects of tkP3BzPB concentration on the recovery of either α3β4- or α4β2-mediated responses. However, there was a tkP3BzPB concentration-dependent accumulation of inhibition for the α7-mediated responses. The IC50 estimated for these recovery data were 1.9 ± 0.7 μM, which was not significantly different from the IC50 estimated from the coapplication experiments (Fig. 2). D, determination of recovery rate for tkP3BzPB-induced inhibition of rat α7 nAChR subunits expressed in X. laevis oocytes. Responses to 60 μM ACh coapplied with 1 μM tkP3BzPB were measured at time = 0 (arrow). Thereafter, responses to ACh alone were recorded at 5-min intervals. Data were normalized to original ACh controls. Data represent the mean responses (± S.E.M., n ≥ 4). Data were fit to an exponential function to estimate the apparent time for recovery.
To evaluate the actual time constant for the recovery of α7 nAChRs from tkP3BzPB inhibition, responses to ACh alone were recorded after 1 μM tkP3BzPB was coapplied with 60 μM ACh. The responses obtained after increasing periods of washout were compared with original ACh controls. The recovery time constant for tkP3BzPB was 26.6 ± 0.8 min (Fig. 5D).
Mechanistic Studies of Inhibition in X. laevis Oocytes Expressing Rat nAChRs. We investigated whether the inhibition of α7 nAChRs by tPyQB, tPy2PiB, or tkP3BzPB was voltage-dependent (Fig. 6A). Cells were held at either -40 or -80 mV and stimulated first with 60 μM ACh alone, followed by 60 μM ACh plus either 300 nM tPyQB, 3 μM tPy2PiB, or 1 μM tkP3BzPB. There was no significant difference in the inhibition of α7 receptors by tkP3BzPB at these two voltages. However, there was a significant effect of voltage on the inhibition by tPyQB and tPy2PiB.
Fig. 6.

Mechanistic studies of AQA analogs-induced inhibition of rat α7 nAChRs expressed in X. laevis oocytes. A, the voltage dependence of α7 nAChR by tPyQB, tPy2PiB, and tkP3BzPB. Cells were held at the indicated holding potentials and then stimulated first by ACh alone and then by ACh plus the tris- and tetrakis-AQA analogs (1 μM for tPyQB, 3 μM for tPy2PiB, and 1 μM for tkP3BzPB). Hyperpolarization did not affect the inhibition produced by tkP3BzPB. However, although 3 μM tPy2PiB produced no significant inhibition at a holding potential of -40 mV, the net charge responses were inhibited 43 ± 3% at a holding potential of -80 mV (***, p < 0.001). Likewise, 1 μM tPyQB, which produced 83 ± 2% inhibition when cells were held at -40 mV, produced significantly more inhibition (97.7 ± 0.1%) at -80 mV (***, p < 0.001). B and C, ACh concentration-response curves from cells expressing α7 nAChRs obtained in the presence of either 300 nM tPyQB or 3 μM tPy2PiB (-60 mV), compared with the data for ACh alone. Each point represents data (± S.E.M.) from at least four cells, normalized to the maximal response obtainable to ACh alone from the same cell. To deliver the compounds effectively in the presence of high concentrations of ACh, which produce very rapid desensitization, the tris-AQA analog was first preapplied to the bath for 30 s and then coapplied with ACh at the indicated concentrations. D, inhibition and recovery of ACh-evoked responses in oocytes expressing rat α7 nAChRs by tkP3BzPB. Two experimental settings were used; solid bars correspond to the experiments in which a 30-s 1 μM tkP3BzPB application preceded ACh and tkP3BzPB coapplication, and dashed bars correspond to coapplication of ACh and tkP3BzPB. ACh was used at two concentrations (60 and 1000 μM). Data are presented as normalized net charge response. E, to determine whether inhibition of α7 by tkP3BzPB was use-dependent, 12-s applications of 3 μM tkP3BzPB were made, with or without coapplication 60 μM ACh. Inhibition of 60 μM ACh-evoked responses was then measured after a 5-min washout. There was no significant difference in the residual inhibition observed between cells treated with tkP3BzPB alone or tkP3BzPB coapplied with ACh.
In addition, coapplication experiments were conducted in X. laevis oocytes expressing rat α7 nAChRs. ACh concentration-response studies of α7 receptors (net charge) were conducted in the presence of either 300 nM tPyQB, the high-potency antagonist, or 3 μM tPy2PiB, the less potent antagonist, and compared with the responses to ACh alone (Fig. 6, B and C). The data obtained with ACh alone were normalized to Imax = 1 and fit with an EC50 = 65 ± 9 μM. In the presence of tPyQB, the Imax was reduced to 0.40 ± 0.01, and the EC50 was 267 ± 8 μM (Fig. 6C). In the presence of 3 μM tPy2PiB, the Imax was reduced to 0.84 ± 0.02, with an EC50 of 105 ± 9 μM (Fig. 6B). In the case of tPyQB and tPy2PiB, both compounds produced a depression of the maximal response of agonist dose-response curves, and this inhibition was not completely overcome by increasing ACh concentrations. These data are consistent with noncompetitive inhibition. However, tPyQB also produced a larger rightward shift of the dose-response curve, and there was a small shift in EC50 value for ACh in the presence of tPy2PiB. These changes in EC50 values in the presence of tPyQB and tPy2PiB suggest a more complex mechanism than just simple voltage-dependent channel block.
Because there was poor recovery of α7 responses after application of tkP3BzPB, it was not practical to generate full ACh concentration-response curves in the presence of this compound. Nonetheless, we wished to determine whether inhibition by 1 μM tkP3BzPB could be surmounted by high concentrations of ACh, which would be consistent with competitive inhibition (Fig. 6D). However, high concentrations of ACh evoke responses that are more rapid than the solution exchange (Papke et al., 2000; Papke and Porter Papke, 2002). Therefore, for tkP3BzPB to be even present at full concentration at the time of the peak of 1 mM ACh-evoked current, 1 μM tkP3BzPB was first preapplied for 30 s and then coapplied with either 60 μM or 1 mM ACh. The data from these experiments were compared with simple coapplication experiments (without 1 μM tkP3BzPB preapplication). As seen in Fig. 6D, there was less inhibition of α7-mediated ACh-evoked responses by 1 μM tkP3BzPB when the ACh concentration was 1 mM than when it was 60 μM, in both experimental settings (30 s preapplication followed by coapplication and in coapplication alone). However, there was no apparent difference in the evoked responses measured after washout regardless of whether 60 μM or 1 mM ACh was coapplied with 1 μM tkP3BzPB (Fig. 6D, right). A concentration of 60 μM ACh is not sufficient to saturate all the binding sites of the receptor, but 1 mM ACh should be sufficient to saturate all the binding sites, so these data support the hypothesis that the inhibition of α7 nAChR by tkP3BzPB is noncompetitive. Radioligand binding data also supports the hypothesis that tkP3BzPB inhibition of α7 nAChR is noncompetitive with ligands binding at the ACh binding site. In particular, using methods published previously (Wilkins et al., 2003), we determined that a 60-min incubation with 100 nM tkP3BzPB displaced no more than 16 ± 8% of the binding of 2.5 nM [3H]MLA to rat brain membranes.
The data in Fig. 6D suggest that the onset of inhibition by low concentrations of tkP3BzPB is relatively slow compared with the kinetics of the α7 response evoked by 1 mM ACh, at least in regard to the persistent inhibition measured after washout. Inhibition increased throughout the 1 μM tkP3BzPB application regardless of whether ACh was present at high or low concentration, apparently reaching the same equilibrium inhibition before the full washout of this antagonist from the chamber. The data also suggested that tkP3BzPB might produce inhibition in the absence of channel activation. If this is the case, then inhibition will depend on both tkP3BzPB concentration and the amount of time that the antagonist is present. In the simple coapplication experiments, tkP3BzPB was present for 12 s, the same duration as the agonist pulse. As shown above, inhibition by tkP3BzPB is slow to reach equilibrium, so the antagonist effect will increase throughout the duration of application. When using 60 μM ACh, the α7 receptors continue to respond throughout the entire 12-s application (Papke and Porter Papke, 2002), and, in contrast, α7 responses evoked by 1 mM ACh reach a peak and return to baseline rapidly, long before the drug delivery is even complete (Papke and Thinschmidt, 1998). Therefore, during the coapplication of 1 mM ACh and 1 μM tkP3BzPB, inhibition is measured after a very brief exposure to the antagonist, too soon for the inhibition to equilibrate to the degree that it did during the longer 60 μM ACh-evoked responses. However, this effect was diminished with the preapplication protocol.
To test the hypothesis that the inhibition of α7 by tkP3BzPB was use-independent (i.e., that inhibition did not require channel activation), 12-s applications of 3 μM tkP3BzPB were made in either the presence or absence of 60 μM ACh. Inhibition of 60 μM ACh-evoked responses was then measured after a 5-min washout. As shown in Fig. 6E, there was no significant difference in the residual inhibition of α7-mediated responses, whether tkP3BzPB was applied alone for 12 s or coapplied with 60 μM ACh. These data demonstrate that the persistent inhibition of α7 nAChRs induced by tkP3BzPB does not require channel activation. In addition, the time to recovery of 60 μM ACh-evoked responses after the application of 3 μM tkP3BzPB alone was 76 ± 3 min (data not shown).
The use independence (Fig. 6E) and slow kinetics of recovery suggest that tkP3BzPB would show increased potency at inhibiting α7 nAChRs with prolonged application. With our typical coapplication protocol (no preapplication) (Fig. 7A), 100 nM tkP3BzPB produced virtually no inhibition of 60 μM ACh-evoked responses either during the brief coapplication (Fig. 4C, •) or after the washout period (Fig. 5C). To confirm that prolonged application of 100 nM tkP3BzPB could produce substantial inhibition of ACh-evoked responses, we stimulated α7-expressing cells, this time, with 300 μM ACh, a concentration that produces a maximal net charge response, and then switched the bath solution to one containing 100 nM tkP3BzPB for 5 min (preincubation period) before coapplying 100 nM tkP3BzPB and 300 μM ACh. As shown in Fig. 7B, this protocol produced approximately 80% inhibition of the ACh-evoked responses that persisted through an additional 5-min washout period.
Fig. 7.

Inhibition by tkP3BzPB increases with prolonged application to X. laevis oocytes expressing rat α7 nAChRs. A, representative recordings from a cell tested with the coapplication of 100 nM tkP3BzPB and 60 μM ACh. The averaged data for cells treated with this protocol appear in Figs. 2 and 3. B, the raw data traces show representative responses of a cell stimulated strongly with 300 μM ACh and then switched to a bath containing 100 nM tkP3BzPB for 5 min. Averaged data from five cells (± S.E.M.) are shown in the bar graph below the traces, with the leftmost bar representing the normalized net charge responses to 300 μM ACh for each cell obtained before the switch to the tkP3BzPB-containing bath solution. C, the inhibition of the steady-state α7 nAChR activation promoted by bath application of 60 μM choline and 10 μM PNU-120596 by nAChR antagonists. Within 36 h of RNA injection, cells expressing α7 were primed with three 12-s applications of 60 μM ACh in a bath containing 60 μM choline and 10 μM PNU-120596, after which a stable level of steady-state activation was achieved that was approximately equal to the additional peak current that was stimulated by further applications of 60 μM ACh. The dashed lines represent the original baselines (zero nAChR-mediated currents). The steady-state current generated by the bath application of choline and PNU-120596 were several hundred nanoamperes, as indicated by the initial deviations from baseline levels. These receptor-mediated steady-state currents (Papke et al., 2009) were sensitive to nAChR antagonists. As shown, 100 μM mecamylamine produced a transient block of approximately 50% of the current, whereas a high (10 μM) concentration of MLA blocked 100% of the current in a more slowly reversible manner. At 30 μM, tkP3BzPB blocked 100% of the steady-state current nonreversibly on the time scale of the experiment.
As discussed above, with some experimental protocols, the rapid activation and desensitization of α7 receptors can make it difficult to measure and compare the inhibitory effects of agents with differing potency, kinetics, and mechanism. Therefore, we have recently reported a new protocol for studying α7 antagonists under nondesensitizing conditions when the receptors generate steady-state current (Papke et al., 2009) as a result of the combined effects of bath-applied choline and the type 2 positive allosteric modulator PNU-120596 (Papke et al., 2009). Figure 7C shows the ability of tkP3BzPB to block these steady-state currents compared with that of MLA and mecamylamine. In these experiments, steady-state currents were generated with 60 μM choline and 10 μM PNU-120596. Mecamylamine at 100 μM produced only a partial and readily reversible block. The relatively small amount of block produced by mecamylamine might be due to several factors. In particular, in addition to having relatively low potency for the block of α7, mecamylamine is also use-dependent and has relatively rapid reversibility. Although there is a large amount of steady-state current with the experimental paradigm illustrated, the actual single molecule Popen is probably much less than 0.10, based on the 10-fold larger amplitude of the ACh-evoked responses after choline and PNU-120596 were removed (Papke et al., 2009). Although mecamylamine only partially and transiently inhibited the steady-state currents, MLA and tkP3BzPB fully abolished these currents. Consistent with our coapplication experiments, the rapid application of 10 μM MLA was sufficient to produce a complete block of the steady-state activation. However, the inhibition was rapidly reversed with washout, suggesting that MLA may intrinsically be relatively ineffective on receptors modified by PNU-120596; alternatively, when MLA is allowed a long period of time to incubate with the receptors in the absence of PNU-120596, the MLA-receptor complex may change with time, for instance via transitions to longer-lived state(s).
In these experiments, tkP3BzPB, unlike MLA, produced a complete block of the steady-state current that persisted long after the drug was washed out of the chamber.
Effects of tkP3BzPB on Other Cys-loop Ligand-Gated Ion Channel Receptors. To further evaluate the selectivity of tkP3BzPB for α7 nAChR, we also tested its effects on the GABA-evoked responses of both heteromeric and homomeric GABAA receptors and the serotonin-evoked responses of homomeric 5-hydroxytryptamine type 3A (5-HT3A) receptors. When coapplied with 10 μM GABA, 3 μM tkP3BzPB had no inhibitory effect on the GABA-evoked responses of GABAA receptors, formed by the coexpression of α1β2γ2l subunits, or on the homomeric GABAC receptors, formed by the expression of the ρ subunit. Likewise, there were no residual inhibitory effects on GABA-evoked responses after the coapplication of 10 μM GABA and 3 μM tkP3BzPB, because there was no inhibition of responses to subsequent GABA applications (data not shown). Because the anion-conducting GABA receptors show reversed charged distribution in the extracellular vestibule and conduction pathway compared with nAChR and 5-HT3A receptors (Corringer et al., 1999; Jensen et al., 2005; Wang et al., 2008), these results are consistent with the hypothesis that the positively charged headgroups of tkP3BzPB interact at sites within the negatively charged rings of the α7 vestibule.
Like α7 nAChR, 5-HT3A receptors can function as homopentamers, although they lack the rapid concentration-dependent desensitization characteristic of α7 nAChR (Maricq et al., 1991). However, 5-HT3A receptors do show sufficient homology to α7 nAChR that functional α7-5-HT3A chimeric subunits are expressed effectively in oocytes and retain the high Popen characteristic of native 5-HT3A receptors (Bertrand et al., 2008). We found that when coapplied with 10 μM serotonin, 3 μM tkP3BzPB did produce an inhibition of 5-HT3A that was greater than the inhibition of ACh-evoked responses of α3β4 and α4β2 nAChR (p < 0.0001) and not significantly different from the inhibition of α7 ACh-evoked responses (data not shown) during coapplication with 60 μM ACh. However, although α7 receptors showed poor recovery of their ACh-evoked responses after a 5-min washout (Fig. 5C), the serotonin-evoked responses of 5-HT3A receptors recovered fully, similar to the ACh-evoked responses of α3β4 and α4β2 nAChR (Fig. 5, A and B).
Activity of tris- and tetrakis-AQA Analogs on Native α7 Receptors on Rat Hippocampal Interneurons. Interneurons in CA1 stratum radiatum of the rat hippocampus show robust responses to the pressure application of ACh. These responses are mediated primarily by α7-type nAChRs (Alkondon et al., 1999; Frazier et al., 2003; Thinschmidt et al., 2005b). We obtained stable 1 mM ACh-evoked responses from hippocampal interneurons in fresh brain slices and then applied 1 μM tPyQB, tPy2PiB, or tkP3BzPB to the bath. In oocytes expressing α7 nAChRs, 1 μM tPyQB was shown to completely inhibit 60 μM ACh-evoked responses (Fig. 2); thus, this concentration was selected for comparing the inhibition of 1 mM ACh-evoked responses induced by the AQA analogs in hippocampal interneurons. Consistent with the oocyte data, the ACh-evoked responses of hippocampal interneurons were effectively reduced by tPyQB and tkP3BzPB but not by tPy2PiB (Fig. 8). Only 4 ± 0.2% of the baseline peak response and 0.4 ± 0.7% of the baseline net charge response remained after 1 μM tkP3BzPB bath application. Thus, prolonged bath application of tkP3BzPB produced a larger inhibition of ACh-evoked responses in rat hippocampal interneurons in terms of both peak amplitude and net charge responses compared with the other AQA analogs, consistent with the results obtained with receptors expressed in oocytes.
Fig. 8.

Inhibition of ACh-evoked responses of hippocampal interneurons. Effects of AQA bath application on α7-mediated responses on hippocampal interneurons are presented in terms of both peak amplitude (A) and net charge (B). Stable baseline responses to the pressure application of 1 mM ACh were obtained from hippocampal interneurons. Cells were stimulated at 30 s intervals, and after four stable responses (1.5 min) either 1 μM tPyQB (○), tPy2PiB (▪), or tkP3BzPB (•) was bath-applied. Solid bar represents the time course of the AQA analog application. C, representative traces of 1 mM ACh-evoked responses and the inhibition of those responses by 1 μM tPyQB, tPy2PiB, and tkP3BzPB. Black traces correspond to ACh baseline responses and gray traces correspond to the ACh-evoked responses at the end of AQA application. Whereas tPyQB and tkP3BzPB effectively reduced ACh-evoked responses, tPy2PiB produced no significant reduction of ACh-evoked responses. Horizontal bars represent 250 ms. Vertical bars represent 50 pA. Data represent the averages of six to seven interneurons.
Differential Inhibition of Medial Septal Neurons by tkP3BzPB. In rat MS/DB, functional nAChR subtypes are expressed that are associated with variations in the neuronal, physiological, and neurotransmitter phenotype (Thinschmidt et al., 2005a). For example, MS/DB neurons that have fast firing rates are likely to be GABAergic, and often have both fast and slow components to their ACh-evoked responses, the fast component being sensitive to MLA blockade, whereas slow firing neurons are putatively cholinergic with nicotinic responses predominantly mediated by MLA-sensitive α7* nAChRs (Thinschmidt et al., 2005a). Different receptors containing α7, α4, and/or β2 subunits may account for most of the variety of nicotinic responses in the MS/DB (Henderson et al., 2005; Thinschmidt et al., 2005a; Liu et al., 2009). These neurons have been classified also according to the kinetics of their nicotinic responses. For example, type I cells have relatively fast transient ACh-evoked responses, and type II cells have slower ACh-evoked responses (Thinschmidt et al., 2005a). To investigate whether the α7-selectivity of tkP3BzPB would discriminate between these types of ACh responses, a double-barreled picospritzer pressure application system was used; one barrel contained 1 mM ACh and the other contained 1 mM ACh + 300 μM tkP3BzPB (Fig. 9). Note that we have previously estimated the concentration of drug delivered to cells with this method to be approximately 30-fold less than the pipette concentration (Lopez-Hernandez et al., 2007). After coapplication, type I cells showed 72 ± 6 and 68 ± 9% of the average baseline peak and net charge response, respectively. On the other hand, type II cells exhibited 86 ± 5 and 93 ± 10% of the average baseline peak and net charge responses, respectively. With this protocol, significant inhibition was induced by tkP3BzPB of the type I responses with relatively less effect on the coapplication responses of the type II cells in medial septum. Note, however, that after the sequence of coapplications, there were statistically significant decreases in the responses of type II cells to applications of ACh alone. It is unclear whether this was due to rundown in the ACh-evoked responses or inhibition of an α7 component in the responses of these cells that was slow to equilibrate but persistent.
Fig. 9.

Differential inhibition of septal neurons by tkP3BzPB coapplication. To investigate whether the α7-selectivity of tkP3BzPB would discriminate between the nicotinic components of ACh-evoked responses in septal neurons, a double-barreled picospritzer pressure application system was used with one barrel containing 1 mM ACh and the other containing 1 mM ACh + 300 μM tkP3BzPB. A, representative traces for the tkP3BzPB coapplication experiments in septum. Three initial responses to ACh alone were obtained at 30 s intervals; the average response is shown in the left side of A for both types of cells. Three applications separated by 30 s were then made from the barrel containing 1 mM ACh + 300 μM tkP3BzPB, and the average of those traces are presented in the middle section of A. After ACh/tkP3BzPB applications, ACh alone was repeatedly applied at 30-s intervals, and the averages of those responses are presented in the right side of A. Horizontal bars represent 250 ms and vertical bars represent 10 pA. B, peak and net charge responses for type I and type II cells, normalized to the average of the first three responses to ACh applied alone (that were acquired during the interval of time delimited by the white bar on left). Subsequently, cells were given coapplications of ACh and tkP3BzPB (pipette concentrations of 1 mM and 300 μM, respectively). The period of time when coapplications were made is delimited by the hatched bars. After the coapplications, there was a recovery period during which ACh was again applied alone (applications beneath the rightmost white horizontal bars in each plot). ○, peak amplitudes; □, net charge responses. Paired student's t tests were performed to compare the normalized responses during coapplication and recovery to baseline; asterisks denote p < 0.05. Data represent the average of 11 neurons for type I and 17 neurons for type II.
In the oocyte coapplication experiments, the IC50 value for tkP3BzPB was higher than that for tPyQB; however, tkP3BzPB produced a prolonged inhibition of α7 nAChR responses. Based on the oocyte data, prolonged application of tkP3BzPB would be expected to produce a greater inhibition than that produced by brief applications. To test this hypothesis, we conducted experiments in which 1 μM tkP3BzPB was bath-applied to medial septal neurons. After bath application of tkP3BzPB, the peak amplitudes of the ACh-evoked responses of type I cells were reduced to only 12 ± 0.6% of the initial evoked responses and the net charge of the responses was reduced below our levels of detection. In contrast, the peak currents of responses of type II were only reduced to 59 ± 1% with net charge values still at 79 ± 2% of the baseline response. Theses differences between type I and type II cells were statistically significant (p < 0.001).
As shown in Fig. 10, after bath application of tkP3BzPB, the residual ACh-evoked responses in type II cells were largely sensitive to DHβE blockade. The difference in the inhibition of α7 nAChR responses by tkP3BzPB between coapplication and prolonged bath application experiments in brain slices is consistent with the data obtained in the oocyte expression system and supports the hypothesis that tkP3BzPB is slow to reach equilibrium and so ultimately produces more inhibition than can be measured with a simple coapplication protocol.
Fig. 10.

Differential inhibition of ACh-evoked responses in septal neurons by tkP3BzPB bath application. Effects of tkP3BzPB bath application on ΑCh-evoked responses on septum neurons are presented in terms of both peak amplitude (A) and net charge (B). Cells were stimulated at 30-s intervals with 1 mM ACh, and after four stable responses (1.5 min), tkP3BzPB was bath-applied. The solid bar represents the time course of the 1 μM tkP3BzPB application. •, type I cells; ▴, type II cells. Type II cells were inhibited by tkP3BzPB to a lesser extent. C, type II cells displayed a nicotinic component sensitive to DHβE block (n = 3). ▵, average normalized peak amplitude; ▴, average normalized net charge responses. Solid bars represent the time course of the 1 μM tkP3BzPB application, whereas open bars represent the time course of 1 μM DHβE application. D, representative traces of 1 mM ACh-evoked responses for type I and type II cells in septum and the inhibition of those responses by tkP3BzPB. Black traces correspond to the average of the ACh baseline responses, and gray traces correspond to the average of the last four ACh-evoked responses at the end of 1 μM tkP3BzPB application. Horizontal bars represent 250 ms. Vertical bars represent 10 pA for type I cells and 20 pA for type II. Data represent the average of 5 neurons for type I and 11 neurons for type II.
Discussion
In this study, we demonstrate the properties of the novel tris-AQA compounds tPyQB and tPy2PiB and the tetrakis-AQA analog tkP3BzPB as α7 nAChR antagonists. These AQA analogs are, with varying potency, more effective at inhibiting α7 receptors than either α4β2 or α3β4 nAChR subtypes tested in the oocyte expression system. The initial evaluation of the large family of tris-AQA compounds suggested that the effectiveness of these compounds at inhibiting α7 nAChR is correlated to the hydrophobicity of the head group. The relative effectiveness of tPyQB and tPy2PiB was consistent with that hypothesis. The tris-AQA analogs tPyQB and tPy2PiB produced inhibition of α7 ACh-evoked responses that was at least in part noncompetitive, consistent with the voltage-dependence data, which suggests interaction at sites within the membrane's electric field. However, although the inhibition of α7 by tPy2PiB and tPyQB was not fully surmountable by increasing ACh concentration, the compounds did produce apparent shifts in ACh potency, suggesting that the mechanism of inhibition by these compounds could arise from multiple mechanisms. Although voltage-dependence would be consistent with direct channel blocking, because the effects of tPy2PiB and tPyQB were readily reversible and inhibition could only be observed during an ACh application, we could not determine whether their inhibitory effects required channel activation or merely could be measured only during channel activation.
Inhibition by tkP3BzPB was not readily reversible and was voltage-independent, suggesting that the inhibition of α7 nAChR by this compound was qualitatively different from that produced by the tris-AQA analogs. The binding data, as well as the lack of both voltage and use dependence for tkP3BzPB inhibition, suggest that tkP3BzPB's antagonist properties arise from binding to sites that are distinct from the ACh binding sites and not within the membrane's electric field in the ion channel domain. One possibility is that the multiple hydrophobic head groups of tkP3BzPB bind to hydrophobic sites on multiple subunits within the homomeric α7 receptor vestibule, occluding the conduction pathway above the level of the membrane's electric field. Sequence analysis and homology modeling (data not shown) suggest that in the α7 vestibule, there are several hydrophobic domains that are not present in all the subunits of heteromeric nAChR.
In hippocampal CA1 stratum radiatum interneurons, which predominantly
exhibit α7-mediated responses, 1 μM tkP3BzPB or tPyQB effectively
reduced ACh-evoked responses, whereas tPy2PiB effects were negligible at this
concentration. AQA analogs inhibit ACh-evoked peak amplitude and net charge
responses in hippocampal interneurons as follows: tkP3BzPB > tPyQB
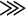 tPy2PiB. On the other hand, in medial septum neurons, there are
different types of nicotinic responses mediated by α7 and/or
non-α7 nAChRs (Henderson et al.,
2005; Thinschmidt et al.,
2005a). The pharmacological isolation of these nicotinic
components is possible with the use of subtype-selective ligands, both
agonists and antagonists. However, available nAChR antagonists show
limitations in their selectivity profiles
(Yum et al., 1996;
Mogg et al., 2002); therefore,
there is a need for better agents to target these receptors, especially in
native systems. For example, we showed previously that the amphipathic blocker
2,2,6,6-tetramethylpiperidin-4-yl heptanoate (TMPH) produced a potent and
long-lasting inhibition of non-α7 receptors, particularly
α4β2 nAChRs, but only transient inhibition of α7 receptors
expressed in X. laevis oocytes
(Papke et al., 2005). TMPH was
shown to be useful in the characterization of complex nicotinic responses,
such as those arising from multiple nAChR subtypes in medial septal neurons by
being able to eliminate the non-α7 nAChR-mediated components of their
ACh-evoked responses (Papke et al.,
2005), essentially exhibiting the complementary effects of
tkP3BzPB.
tPy2PiB. On the other hand, in medial septum neurons, there are
different types of nicotinic responses mediated by α7 and/or
non-α7 nAChRs (Henderson et al.,
2005; Thinschmidt et al.,
2005a). The pharmacological isolation of these nicotinic
components is possible with the use of subtype-selective ligands, both
agonists and antagonists. However, available nAChR antagonists show
limitations in their selectivity profiles
(Yum et al., 1996;
Mogg et al., 2002); therefore,
there is a need for better agents to target these receptors, especially in
native systems. For example, we showed previously that the amphipathic blocker
2,2,6,6-tetramethylpiperidin-4-yl heptanoate (TMPH) produced a potent and
long-lasting inhibition of non-α7 receptors, particularly
α4β2 nAChRs, but only transient inhibition of α7 receptors
expressed in X. laevis oocytes
(Papke et al., 2005). TMPH was
shown to be useful in the characterization of complex nicotinic responses,
such as those arising from multiple nAChR subtypes in medial septal neurons by
being able to eliminate the non-α7 nAChR-mediated components of their
ACh-evoked responses (Papke et al.,
2005), essentially exhibiting the complementary effects of
tkP3BzPB.
The effectiveness of tkP3BzPB to block α7-mediated current and preserve the responsiveness of non-α7 receptors is similar to that of low concentrations of MLA (Thinschmidt et al., 2005a). However, because the inhibition produced by MLA and tkP3BzPB arise from distinctly different mechanisms, comparisons of the effects of these two agents may be of particular use for determining whether all of the effects documented for α7-selective partial agonists such as GTS-21 are strictly dependent on ion channel activation. GTS-21, an α7-selective partial agonist, has relatively low efficacy for human α7 receptors and produces prolonged desensitization (Papke et al., 2009). In addition to its effects on neuronal α7, GTS-21 has also been documented to have α7-mediated effects in non-neuronal cells in which no α7-mediated ion currents have been detected (Giebelen et al., 2007a,b; Wongtrakool et al., 2007; Kageyama-Yahara et al., 2008). In some cases, α7-dependent effects, particularly in non-neuronal cells, have been shown to require downstream events such as modulation of voltage-dependent calcium channels (Ren et al., 2005) or intracellular signal transduction pathways (Marrero et al., 2004; Giebelen et al., 2007a,b). Although tkP3BzPB will block currents evoked by GTS-21 or other α7 agonists, it is not likely to prevent agonist binding and may allow other forms of signal transduction to occur.
The current study demonstrates the utility of tkP3BzPB to probe complex patterns of nAChR expression in brain. This approach will be useful for the study of nAChRs in other brain areas such as the ventral tegmental area (VTA). Nicotinic receptor functional expression in the VTA has been demonstrated and is believed to mediate nicotine-evoked dopamine release in the nucleus accumbens (Pidoplichko et al., 1997; Wooltorton et al., 2003) through both nicotine stimulation of VTA action potentials and the stimulation of presynaptic nAChR in the terminal fields of the VTA. It has been proposed that drugs that can inhibit the nicotinic-mediated enhancement of dopamine release could have therapeutic potential for the treatment of nicotine addiction (Dwoskin et al., 2004). Within the VTA, α2, α4, α3, α5, α6, α7, β2, and β4 subunit mRNA expression has been detected (Charpantier et al., 1998; Azam et al., 2002). Given this heterogeneous expression, it has been difficult to unambiguously determine which nAChR combinations are involved in mediating the dopamine release in the nucleus accumbens. Likely candidates are α7, α4*, and α6*, all three subtypes being potentially important within the VTA (Mansvelder et al., 2002) and α4- and α6-containing receptors more important on presynaptic terminals (Salminen et al., 2004, 2007).
In addition, subtype-selective inhibitors, such as the novel AQA analogs described in the present study, will be valuable tools for the identification and isolation of molecular brain nicotinic substrates, because their use can be extended to brain structures and experimental electrophysiological paradigms, such as those associated with the cholinergic components underlying synaptic plasticity. Moreover, the potential clinical development of nicotinic agents for neuropsychiatric indications, such as Tourette's syndrome and nicotine dependence has been limited by the lack of truly selective agents. Although it was a brief application, tkP3BzPB did not seem to be as potent as tPyQB at inhibiting α7 nAChRs expressed in X. laevis oocytes; however, tkP3BzPB still showed a preferential inhibition of α7 responses over α3β4 and α4β2 subtypes in addition to a remarkable long-lasting inhibition of α7 nAChRs. Furthermore, tkP3BzPB was more efficient at inhibiting steady-state currents than either mecamylamine or MLA. This differential inhibition of nAChR subtypes identifies tkP3BzPB in particular as a potentially valuable tool for pharmacological isolation of nicotinic receptor responses. Further development of agents such as TMPH and tkP3BzPB will first aid in identifying the correct molecular targets for neuropsychiatric conditions such as Tourette's syndrome and nicotine addiction and subsequently lead to therapies with reduced side effect liability.
Acknowledgments
We thank Clare Stokes, Lynda Cortés, Shehd Abdullah Abbas Al Rubaiy, and Sara Braley for their valuable input.
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant U19-DA017548], the National Institutes of Health National Institute of General Medical Sciences[Grant R01-GM057481], and the National Institutes of Health National Institute on Aging [Grants P01-AG010485, T32-AG000196].
A portion of this work was previously presented in abstract form: López GY, Thinschmidt JS, Zheng G, Zhang Z, Crooks PA, Dwoskin LP, and Papke RL. Differential inhibition of ACh-evoked responses of neuronal nicotinic receptors in rat brain slices by selective nAChR antagonists. Society for Neuroscience Satellite Symposium on “Nicotinic Acetylcholine Receptors as Therapeutic Targets—Emerging Frontiers in Basic Research and Clinical Science”; San Diego, CA; 2007 Oct 31-Nov 2; Poster 1.31.
The University of Kentucky holds patents on the tris- and tetrakis-azaaromatic quaternary ammonium compounds described herein. A potential royalty stream to L.P.D. and P.A.C. may occur consistent with University of Kentucky policy.
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; CNS, central nervous system; MS/DB, medial septum/diagonal band; MLA, methyllycaconitine; tPy2PiB, 1,3,5,-tri-{5-[1-(2-picolinium)]-pent-1-yn-1-yl}benzene tribromide; tPyQB, 1,3,5-tri-[5-(1-quinolinum)-pent-1-yn-1-yl]-benzene tribromide; tkP3BzPB, 1,2,4,5-tetra-{5-[1-(3-benzyl)pyridinium]pent-1-yl}benzene tetrabromide; AQA, azaaromatic quaternary ammonium; PNU-120596, 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxanol-3-yl)-urea; ACh, acetylcholine; DHβE, dihydro-β-erythroidine; 5-HT, 5-hydroxytryptamine; TMPH, 2,2,6,6-tetramethylpiperidin-4-yl heptanoate; GTS-21, 3-2,4,dimethoxy-benzylidene anabaseine; VTA, ventral tegmental area.
References
- Aiyar VN, Benn MH, Hanna T, Jacyno J, Roth SH, and Wilkens JL (1979) The principal toxin of Delphinium brownii Rydb., and its mode of action. Experientia 35 1367-1368. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Eisenberg HM, and Albuquerque EX (1999) Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci 19 2693-2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Wonnacott S, and Albuquerque EX (1992) Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Mol Pharmacol 41 802-808. [PubMed] [Google Scholar]
- Azam L, Winzer-Serhan UH, Chen Y, and Leslie FM (2002) Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol 444 260-274. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Bertrand S, Cassar S, Gubbins E, Li J, and Gopalakrishnan M (2008) Positive allosteric modulation of the alpha7 nicotinic acetylcholine receptor: ligand interactions with distinct binding sites and evidence for a prominent role of the M2-M3 segment. Mol Pharmacol 74 1407-1416. [DOI] [PubMed] [Google Scholar]
- Buisson B and Bertrand D (2002) Nicotine addiction: the possible role of functional upregulation. Trends Pharmacol Sci 23 130-136. [DOI] [PubMed] [Google Scholar]
- Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, and Bertrand D (1996) Human alpha4beta2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci 16 7880-7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpantier E, Barnéoud P, Moser P, Besnard F, and Sgard F (1998) Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. Neuroreport 9 3097-3101. [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Bertrand S, Galzi JL, Devillers-Thiery A, Changeux JP, and Bertrand D (1999) Molecular basis of the charge selectivity of nicotinic acetylcholine receptor and related ligand-gated ion channels. Novartis Found Symp 225 215-224; discussion 224-230. [DOI] [PubMed] [Google Scholar]
- Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, Valera S, Barkas T, and Ballivet M (1990) A neuronal nicotinic acetylcholine receptor subunit (α7) is developmentally regulated and forms a homo-oligomeric channel blocked by α-BTX. Neuron 5 847-856. [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Sumithran SP, Zhu J, Deaciuc AG, Ayers JT, and Crooks PA (2004) Subtype-selective nicotinic receptor antagonists: potential as tobacco use cessation agents. Bioorg Med Chem Lett 14 1863-1867. [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Wooters TE, Sumithran SP, Siripurapu KB, Joyce BM, Lockman PR, Manda VK, Ayers JT, Zhang Z, Deaciuc AG, et al. (2008) N,N′-Alkane-diyl-bis-3-picoliniums as nicotinic receptor antagonists: inhibition of nicotine-evoked dopamine release and hyperactivity. J Pharmacol Exp Ther 326 563-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, and Heinemann S (1994) Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79 705-715. [DOI] [PubMed] [Google Scholar]
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, and Kellar KJ (1992) A subtype of nicotinic cholinergic receptor in rat brain is composed of α4 and β2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol 41 31-37. [PubMed] [Google Scholar]
- Frazier CJ, Strowbridge BW, and Papke RL (2003) Nicotinic acetylcholine receptors on local circuit neurons in dentate gyrus: a potential role in the regulation of granule cell excitability. J Neurophysiol 89 3018-3028. [DOI] [PubMed] [Google Scholar]
- Giebelen IA, van Westerloo DJ, LaRosa GJ, de Vos AF, and van der Poll T (2007a) Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock 28 700-703. [DOI] [PubMed] [Google Scholar]
- Giebelen IA, van Westerloo DJ, LaRosa GJ, de Vos AF, and van der Poll T (2007b) Stimulation of alpha 7 cholinergic receptors inhibits lipopolysaccharide-induced neutrophil recruitment by a tumor necrosis factor alpha-independent mechanism. Shock 27 443-447. [DOI] [PubMed] [Google Scholar]
- Halevi S, Yassin L, Eshel M, Sala F, Sala S, Criado M, and Treinin M (2003) Conservation within the RIC-3 gene family. Effectors of mammalian nicotinic acetylcholine receptor expression. J Biol Chem 278 34411-34417. [DOI] [PubMed] [Google Scholar]
- Henderson Z, Boros A, Janzso G, Westwood AJ, Monyer H, and Halasy K (2005) Somato-dendritic nicotinic receptor responses recorded in vitro from the medial septal diagonal band complex of the rodent. J Physiol 562 165-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals, 7th ed., Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington DC.
- Jensen ML, Schousboe A, and Ahring PK (2005) Charge selectivity of the Cys-loop family of ligand-gated ion channels. J Neurochem 92 217-225. [DOI] [PubMed] [Google Scholar]
- Kageyama-Yahara N, Suehiro Y, Yamamoto T, and Kadowaki M (2008) IgE-induced degranulation of mucosal mast cells is negatively regulated via nicotinic acetylcholine receptors. Biochem Biophys Res Commun 377 321-325. [DOI] [PubMed] [Google Scholar]
- Lindstrom J, Anand R, Gerzanich V, Peng X, Wang F, and Wells G (1996) Structure and function of neuronal nicotinic acetylcholine receptors. Prog Brain Res 109 125-137. [DOI] [PubMed] [Google Scholar]
- Liu Q, Huang Y, Xue F, Simard A, DeChon J, Li G, Zhang J, Lucero L, Wang M, Sierks M, et al. (2009) A novel nicotinic acetylcholine receptor subtype in basal forebrain cholinergic neurons with high sensitivity to amyloid peptides. J Neurosci 29 918-929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Hernandez G, Placzek AN, Thinschmidt JS, Lestage P, Trocme-Thibierge C, Morain P, and Papke RL (2007) Partial agonist and neuromodulatory activity of S 24795 for alpha7 nAChR responses of hippocampal interneurons. Neuropharmacology 53 134-144. [DOI] [PubMed] [Google Scholar]
- López-Hernández GY, Sánchez-Padilla J, Ortiz-Acevedo A, Lizardi-Ortiz J, Salas-Vincenty J, Rojas LV, and Lasalde-Dominicci JA (2004) Nicotine-induced up-regulation and desensitization of alpha4beta2 neuronal nicotinic receptors depend on subunit ratio. J Biol Chem 279 38007-38015. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, and McGehee DS (2002) Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron 33 905-919. [DOI] [PubMed] [Google Scholar]
- Maricq AV, Peterson AS, Brake AJ, Myers RM, and Julius D (1991) Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science 254 432-437. [DOI] [PubMed] [Google Scholar]
- Marrero MB, Papke RL, Bhatti BS, Shaw S, and Bencherif M (2004) The neuroprotective effect of 2-(3-pyridyl)-1-azabicyclo[3.2.2]nonane (TC-1698), a novel alpha7 ligand, is prevented through angiotensin II activation of a tyrosine phosphatase. J Pharmacol Exp Ther 309 16-27. [DOI] [PubMed] [Google Scholar]
- Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, and Wonnacott S (2002) Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther 302 197-204. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, and Lindstrom J (2003) Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol 63 332-341. [DOI] [PubMed] [Google Scholar]
- Papke RL (1993) The kinetic properties of neuronal nicotinic receptors: genetic basis of functional diversity. Prog Neurobiol 41 509-531. [DOI] [PubMed] [Google Scholar]
- Papke RL, Buhr JD, Francis MM, Choi KI, Thinschmidt JS, and Horenstein NA (2005) The effects of subunit composition on the inhibition of nicotinic receptors by the amphipathic blocker 2,2,6,6-tetramethylpiperidin-4-yl heptanoate. Mol Pharmacol 67 1977-1990. [DOI] [PubMed] [Google Scholar]
- Papke RL, Kem WR, Soti F, López-Hernández GY, and Horenstein NA (2009) Activation and desensitization of nicotinic α7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J Pharmacol Exp Ther 329 791-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Meyer E, Nutter T, and Uteshev VV (2000) Alpha7-selective agonists and modes of alpha7 receptor activation. Eur J Pharmacol 393 179-195. [DOI] [PubMed] [Google Scholar]
- Papke RL and Porter Papke JK (2002) Comparative pharmacology of rat and human alpha7 nAChR conducted with net charge analysis. Br J Pharmacol 137 49-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL and Thinschmidt JS (1998) The correction of alpha7 nicotinic acetylcholine receptor concentration-response relationships in Xenopus oocytes. Neurosci Lett 256 163-166. [DOI] [PubMed] [Google Scholar]
- Peng X, Katz M, Gerzanich V, Anand R, and Lindstrom J (1994) Human α7 acetylcholine receptor: cloning of the α7 subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional α7 homomers expressed in Xenopus oocytes. Mol Pharmacol 45 546-554. [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, and Dani JA (1997) Nicotine activates and desensitizes midbrain dopamine neurons. Nature 390 401-404. [DOI] [PubMed] [Google Scholar]
- Ren K, Puig V, Papke RL, Itoh Y, Hughes JA, and Meyer EM (2005) Multiple calcium channels and kinases mediate alpha7 nicotinic receptor neuroprotection in PC12 cells. J Neurochem 94 926-933. [DOI] [PubMed] [Google Scholar]
- Role LW and Berg DK (1996) Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16 1077-1085. [DOI] [PubMed] [Google Scholar]
- Salminen O, Drapeau JA, McIntosh JM, Collins AC, Marks MJ, and Grady SR (2007) Pharmacology of alpha-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol Pharmacol 71 1563-1571. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, and Grady SR (2004) Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 65 1526-1535. [DOI] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, and Patrick JW (1993) Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 13 596-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinschmidt JS, Frazier CJ, King MA, Meyer EM, and Papke RL (2005a) Medial septal/diagonal band cells express multiple functional nicotinic receptor subtypes that are correlated with firing frequency. Neurosci Lett 389 163-168. [DOI] [PubMed] [Google Scholar]
- Thinschmidt JS, Frazier CJ, King MA, Meyer EM, and Papke RL (2005b) Septal innervation regulates the function of alpha7 nicotinic receptors in CA1 hippocampal interneurons. Exp Neurol 195 342-352. [DOI] [PubMed] [Google Scholar]
- Wang HL, Cheng X, Taylor P, McCammon JA, and Sine SM (2008) Control of cation permeation through the nicotinic receptor channel. PLoS Comput Biol 4 e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins LH Jr, Grinevich VP, Ayers JT, Crooks PA, and Dwoskin LP (2003) N-n-alkylnicotinium analogs, a novel class of nicotinic receptor antagonists: interaction with α4β2* and α7* neuronal nicotinic receptors. J Pharmacol Exp Ther 304 400-410. [DOI] [PubMed] [Google Scholar]
- Wongtrakool C, Roser-Page S, Rivera HN, and Roman J (2007) Nicotine alters lung branching morphogenesis through the alpha7 nicotinic acetylcholine receptor. Am J Physiol Lung Cell Mol Physiol 293 L611-L618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnacott S (1997) Presynaptic nicotinic ACh receptors. Trends Neurosci 20 92-98. [DOI] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, and Dani JA (2003) Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci 23 3176-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum L, Wolf KM, and Chiappinelli VA (1996) Nicotinic acetylcholine receptors in separate brain regions exhibit different affinities for methyllycaconitine. Neuroscience 72 545-555. [DOI] [PubMed] [Google Scholar]
- Zheng G, Zhang Z, Pivavarchyk M, Deaciuc AG, Dwoskin LP, and Crooks PA (2007) Bis-azaaromatic quaternary ammonium salts as antagonists at nicotinic receptors mediating nicotine-evoked dopamine release: An investigation of binding conformation. Bioorg Med Chem Lett 17 6734-6738. [DOI] [PMC free article] [PubMed] [Google Scholar]