

Summary
Endolymph is the specialised extracellular fluid present inside the inner ear. In mammals, disruptions to endolymph homeostasis can result in either collapse or distension of the endolymphatic compartment in the cochlea, with concomitant hearing loss. The zebrafish little ears (lte) mutant shows a collapse of the otic vesicle in the larva, apparently owing to a loss of endolymphatic fluid in the ear, together with an over-inflation of the swim bladder. Mutant larvae display signs of abnormal vestibular function by circling and swimming upside down. The two available alleles of lte are homozygous lethal: mutant larvae fail to thrive beyond 6 days post-fertilisation. Patterning of the otic vesicle is apparently normal. However, the expression of several genes thought to play a role in endolymph production is downregulated, including the sodium-potassium-chloride cotransporter gene nkcc1 (slc12a2) and several Na+/K+-ATPase channel subunit genes. We show here that lte mutations correspond to lesions in nkcc1. Each allele has a point mutation that disrupts splicing, leading to frame shifts in the coding region that predict the generation of truncated products. Endolymph collapse in the lte/nkcc1 mutant shows distinct parallels to that seen in mouse Nkcc1 mutants, validating zebrafish as a model for the study of endolymph disorders. The collapse in ear volume can be ameliorated in the to27d allele of lte by injection of a morpholino that blocks splicing at an ectopic site introduced by the mutation. This exemplifies the use of morpholinos as potential therapeutic agents for genetic disease.
Keywords: Zebrafish, Ear, Swim bladder, Endolymph, Nkcc1 (Slc12a2)
INTRODUCTION
Endolymph is the highly specialised extracellular fluid that bathes the sensory organs of the inner ear. High in K+ and low in Na+, it is generated and maintained largely by marginal cells of the stria vascularis in the cochlea and dark cells of the vestibular system in the mammalian ear. K+ ions cycle through sensory hair cells into the perilymph, from where they are taken back up through a series of ion channels, transporters and gap junctions to be secreted apically back into the endolymph (Lang et al., 2007; Wangemann, 1995). Mutations have been described in several genes encoding proteins involved in this K+ transit, with a common phenotype encompassing a loss of the endocochlear potential, a reduction in endolymphatic volume, and profound sensorineural deafness owing to hair cell degeneration (Casimiro et al., 2001; Delpire et al., 1999; Dixon et al., 1999; Flagella et al., 1999; Letts et al., 2000). Mutant mice exhibit classic vestibular dysfunction behaviour, such as head bobbing and circling. As a result of the loss of endolymph volume, membranous collapse within the inner ear is a common outcome; in the Nkcc1 mutant mouse, Reissner's membrane becomes closely apposed to the organ of Corti (Delpire et al., 1999; Deol, 1963; Dixon et al., 1999), while in Kcnq1 and Kcne1 mutant mice, the utricular membrane shrinks and the semicircular canal lumina are narrowed (Casimiro et al., 2001; Letts et al., 2000).
These genes provided good candidates for little ears (lte), a zebrafish mutant with a similar spectrum of phenotypic anomalies to those observed in mice. Two lte alleles were isolated from the Tübingen 1996 ENU mutagenesis screen (Whitfield et al., 1996). The lte ear is phenotypically normal until 3 days post-fertilisation (dpf), when it undergoes a collapse owing to an apparent loss of endolymph. The lte mutation was mapped to linkage group (LG) 10 by the Tübingen Mapping Consortium (Geisler et al., 2007). The zebrafish kcnq1 and kcne1 orthologues map to LG7 and LG22, respectively. Zebrafish mutations in Na+/K+-ATPase genes have already been identified and were ruled out on account of chromosomal location and phenotype (Blasiole et al., 2006; Lowery and Sive, 2005). The nkcc1 (slc12a2) gene, however, maps to LG10 and was pursued as the most promising candidate.
NKCC1 (SLC12A2) is a cation-chloride-cotransporter that predominantly mediates the electroneutral transport of Na+ and K+ with 2×Cl- across basolateral membranes of secretory epithelia (Payne et al., 1995; Xu et al., 1994). This contrasts with the related NKCC2 (SLC12A1) cotransporter, expression of which is restricted to the kidney medulla, where it exhibits apical localisation and absorptive properties (Payne and Forbush, 1995). NKCC1 function in the ear has been demonstrated by mouse knockout studies (Delpire et al., 1994; Dixon et al., 1999; Flagella et al., 1999) and reduced NKCC1 immunoreactivity is implicated in the endolymph collapse seen in the mutant German waltzing (gw) guinea pig (Jin et al., 2008). The loop diuretics bumetanide and furosemide, which inhibit NKCC1 function, are known to cause reversible hearing loss in humans (Bourke, 1976; Vargish et al., 1970); they cause an increase in the intrastrial K+ concentration, which abolishes the endolymphatic potential (Marcus et al., 1987). In teleosts, Nkcc1 has been demonstrated in acclimation assays to have a role in osmoregulation and salt-water tolerance in several species (Cutler and Cramb, 2002; Lorin-Nebel et al., 2006) and was initially identified as the entry site for Na+ and Cl- across the basolateral membrane of `chloride' cells in the operculum and gills (Evans et al., 2005). The sea bass has basolateral Nkcc1 immunoreactivity in embryonic skin ionocytes, with a few cells expressing Nkcc1 in the operculum and branchial slits (Lorin-Nebel et al., 2006). Expression of Nkcc1 is upregulated in response to seawater acclimation in several teleost species (Lorin-Nebel et al., 2006; Mackie et al., 2007; Hyndman and Evans, 2008). Two Nkcc1 isoforms, Nkcc1a and Nkcc1b, have been identified in the eel (Cutler and Cramb, 2002), of which Nkcc1a is the more closely related to mammalian homologues and to the zebrafish sequence reported here. Nkcc1a is expressed in various tissues, including the oesophagus, stomach and gill epithelium, whereas Nkcc1b is expressed in the gills and the brain; otic expression has not been described for either isoform (Cutler and Cramb, 2002).
In this study, we show that zebrafish lte mutations disrupt the nkcc1 gene. The two alleles carry point mutations that lead to splicing errors in the cognate mRNA and predict truncated protein products. The predominant phenotype is a gradual reduction in ear size from 3 dpf, despite apparently normal early otic patterning. Some mutants also display the rare and unusual phenotype of an increased swim bladder volume. Localisation of the Nkcc1 protein in wild-type embryos reveals a previously unknown regionalisation of semicircular canal pillar tissue; expression is restricted to the medial face of the ventral pillar. We demonstrate a rescue of the lteto27d phenotype using a morpholino to block an ectopic splice site introduced by the mutation. This forces the use of the nearby wild-type site, restoring Nkcc1 RNA and protein expression and ameliorating the endolymph collapse phenotype.
MATERIALS AND METHODS
Zebrafish husbandry
Standard zebrafish husbandry methods were employed (Westerfield, 1995). Wild-type strains used were AB, TL, WIK and LWT. For the swim bladder caging experiments, embryos were caged using wire mesh and polypropylene rings adhered to Petri dish bases with silicon grease.
Gut function assays
Embryos at 5 dpf were incubated with beads coated in 0.3 mg/ml PED6 (Invitrogen) for 3 hours at room temperature (RT), to assay for gut function and lipid metabolism as described (Farber et al., 2001). Fluorescent latex microspheres (Polysciences) were used as a control assay for ingestion (Farber et al., 2001).
Isolation and sequencing of nkcc1 cDNA and genomic DNA
Total RNA was extracted from zebrafish embryos using TRIzol (Invitrogen) and converted to cDNA using the Superscript III Kit (Invitrogen) with oligo(dT) primers. A full-length nkcc1 clone was isolated by PCR using the following primers (5′ to 3′) based on the GenBank record NM_001002080, incorporating a 5′ EcoRI site and a 3′ XbaI site (underlined): forward (F), GGAATTCCCTTACCATGTCGGCGTCA; reverse (R), GCTCTAGAGCAGTTGTGTCTGTGAGCTC. GenBank accession number for the nkcc1 cDNA sequence: GQ259737. Primers for the amplification of exon 20a were: F, AGGATGATGATGGCAAAGCC; R, GTCATCAAAGAGCCACCACA.
Genomic DNA from the to27d allele was amplified using the following primer pair: F, TAATGCTGTGCCCCTTCTC (intron 4); R, CTACAGCAACAGCATTGGCA (exon 5). Sequencing was performed by the University of Sheffield Genetics Core Facility, or by Lark Technologies, using an ABI 3730 capillary sequencer. Sequence data were analysed using the Biology Workbench (http://workbench.sdsc.edu/). Splice site prediction and analysis were carried out using NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html) (Reese et al., 1997) and ESEfinder (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home) (Cartegni et al., 2003; Smith et al., 2006).
Genotyping of lte alleles
The cDNA insert from lteto27d embryos was detected by PCR using the following primer pair: F, GCCAGGCTGGAATTGCATAT (exon 4); R, CTACAGCAACAGCATTGGCA (exon 5). The cDNA deletion from ltetg414b embryos was detected by amplification of cDNA using the following primer pair: F, AAAAGAGCCCGACAGTTCCT (exon 21); R, CCTCAGACTTTGGCTTTGTG (exon 23). Products were separated on a 2% agarose gel. The RFLP introduced by the point mutation in ltetg414b genomic DNA was identified by amplification with the following primer pair: F, AGCAGTGAAGTGGTTCTGCA (intron 21); R, ATAGCTAACAAAGGCCCTGG (intron 22); this was followed by digestion with NlaIII (NEB). Digest products were separated on a 4.5% agarose gel.
Protein topology predictions were generated using the HMMTOP server (http://www.enzim.hu/hmmtop/) (Tusnàdy and Simon, 1998).
Morpholino injection
Morpholinos (Gene Tools) were dissolved in water to a stock concentration of 10 mg/ml. Sequences (5′ to 3′) were: ATG morpholino, GAGATTGGAGGTGACGCCGACATGG; splice-blocking (rescue) morpholino, ACAGAACTAAAAACAACCCAACATT. Injections were carried out using a microinjection rig (Narishige) into the yolk of 1- to 8-cell stage embryos (0.5-2 nl per injection).
RNA overexpression
Capped mRNA for the complete nkcc1 open reading frame (ORF) was synthesised from the pCS2-MT vector using Sp6 RNA polymerase (Promega) and stored in aliquots at -80°C. Embryos were injected at the 1-cell stage with 1 nl of 75-200 pg mRNA.
Whole-mount in situ hybridisation
Whole-mount in situ hybridisation was carried out essentially as described (Nüsslein-Volhard and Dahm, 2002). Probes used were atp1a1a.4, atp1a1a.5, atp1b1a, atp1b2b (Blasiole et al., 2003), bmp4 (Hammerschmidt et al., 1996), cahz (Peterson et al., 1997), connexin 33.8, eya1 (Sahly et al., 1999), foxa1 (Odenthal and Nüsslein-Volhard, 1998), foxi1 (Solomon et al., 2003), gbx2 (Su et al., 1993), kcnq1, neurod (Korzh et al., 1998), pax2a (Pfeffer et al., 1998), nkcc1, nkcc2, tbx1 (Piotrowski et al., 2003) and ugdh (Busch-Nentwich et al., 2004).
Immunohistochemistry
Embryos were fixed in Dent's fixative (1:4 DMSO:methanol) for 2 hours at RT, then washed in PTx (PBS containing Triton X-100: 1% Triton if younger than 5 dpf; 2% Triton if 5 dpf or older). Embryos were then dehydrated and rehydrated through a methanol series before washing for several hours in PTx. Samples were blocked in PTx containing 10% bovine serum for 1 hour at RT, prior to overnight incubation with mouse anti-human NKCC1 (T4 antibody) at 1:1000 (DSHB, University of Iowa, USA). Following washing and incubation with HRP-coupled anti-mouse IgG at 1:200 (Sigma), staining was visualised using a DAB Kit (Vector Laboratories). For fluorescent imaging, the secondary antibody was TRITC-coupled anti-mouse IgG (Sigma). TO-PRO-3 (Invitrogen) was used as a nuclear stain during the secondary antibody wash. For anti-Myc staining, embryos were fixed in 4% paraformaldehyde for 3 hours at RT, followed by washes in PTx (0.1% Triton) and blocking in 10% bovine serum. Samples were incubated with mouse anti-Myc antibody at 1:200 overnight, and staining was visualised using an HRP-coupled anti-mouse secondary antibody as above.
Acridine Orange staining
Live 5 dpf embryos were incubated in E3 medium supplemented with 5 μg/ml Acridine Orange at 28°C for 30 minutes. After copious washing, embryos were imaged.
Phalloidin, propidium iodide and FM1-43 staining
Embryos were permeabilised in PTx (2% Triton) for 4 days at 4°C. After several rinses in PBS, embryos were incubated overnight in 20 μg/ml propidium iodide (Sigma) and/or 2.5 μg/ml FITC-phalloidin (Sigma), followed by PBS washes and mounting in Vectashield medium. For the vital dye FM1-43, live 5 dpf embryos were embedded in low-melting-point agarose (Sigma) and the ear injected with ∼2 nl 178 nM FM1-43 (Molecular Probes). Samples were visualised by confocal microscopy.
Use of NKCC1 inhibitors
Bumetanide and furosemide (Sigma) were dissolved in methanol to a stock concentration of 5 mg/ml; for use, this was diluted in E3 to 25-100 μg/ml (∼75-300 μM). Embryos were incubated with the inhibitors at various stages for varying lengths of time. Inhibitors were also injected directly into the otic vesicle (1 nl) at 5 dpf.
Microscopy and imaging
Samples were imaged using a BX51 compound microscope and a CELLB camera and software, or an FV1000 confocal microscope (Olympus). Images were compiled using Adobe Photoshop version 8 and are shown with anterior to the left throughout. Movies were captured using an Olympus C-3030 camera and a Leica MZ16 dissecting microscope.
RESULTS
The zebrafish ear is patterned normally, but later collapses, in little ears (lte) mutants
Two recessive alleles of little ears (lte) were isolated in an ENU mutagenesis screen on the basis of their fully penetrant small ear phenotype at 5 dpf (Whitfield et al., 1996). Early development of the ear, however, appears to be normal in lte mutants. Cavitation of the otic placode occurred on time, at 18 hours post-fertilisation (hpf). At 24 hpf, expression of patterning markers for dorsal (gbx2), ventral (eya1), medial (pax2a) and lateral (tbx1) otic epithelium in mutants was indistinguishable from that in phenotypically wild-type sibling embryos (sibs) (see Fig. S1A-C in the supplementary material; data not shown). Statoacoustic ganglion neuroblasts (see Fig. S1D in the supplementary material), semicircular canal projections (see Fig. S1H and Fig. S2 in the supplementary material), otoliths (Fig. 1; see Fig. S2 in the supplementary material), sensory patches (maculae and cristae) (see Fig. S1E,F in the supplementary material; Fig. 2) and the endolymphatic duct (see Fig. S1G in the supplementary material; Fig. 3A,B) all appeared to form normally, suggesting the correct specification of otic tissue. Alcian Blue staining indicated that the cartilaginous otic capsule surrounding the ear is correctly patterned (data not shown). The lte ear increased in size, as in the wild type, by filling with fluid during the second and third days of development (1-2 dpf).
Fig. 1.

lte mutant zebrafish larvae have a collapsed ear at 5 dpf and an increased swim bladder volume. (A-D) Live images of lteto27d homozygous mutant (B,D) and phenotypically wild-type sibling (sib) (A,C) embryos at 5 dpf. (E) Ear collapse in the tg414b allele at 5 dpf is slightly more severe than in to27d. (F-I) Acridine Orange staining (green) indicates that there is no increase in cell death in the ear (white dashed outline) in lte mutants as compared with siblings at 5 dpf (F,H compared with G,I). Background staining in live neuromasts can be seen in both genotypes (green spots). (J-M) FITC-phalloidin (green)/propidium iodide (red) staining reveals similar architecture and number of cells within the otic epithelium of tg414b and siblings at 5 dpf. (L,M) Enlargement of the ventral pillar (white asterisks in J,K). (N,O) Live images showing the increase in swim bladder volume in lteto27d mutants. (P) The average swim bladder length increases from 390 μm in sibling embryos to 418 μm in the mutants (n=12 siblings, n=12 mutants; P=0.03, Student's t-test). Horizontal lines indicate the distribution of swim bladder lengths; vertical lines indicate the population mean; boxes indicate standard deviation. asc, anterior semicircular canal; psc, posterior semicircular canal; usc, utriculosaccular chamber. Scale bars: 125 μm in A,B; 50 μm in C-E,H-K; 200 μm in F,G; 100 μm in N,O.
Fig. 2.

Hair cells do not degenerate and are still functional in lte mutant ears. (A-J) Staining with FITC-phalloidin labels hair cells at 6 dpf. In both siblings (A,C,E,G,I) and lte mutants (lteto27d in B,D,F; ltetg414b in H,J), actin-rich hair bundles are visible in all five sensory patches. Enlargements of the anterior (G,H) and posterior (I,J) maculae show equivalent numbers of differentiated hair cells in siblings and lte mutants. Asterisks (C-F) mark the reduction in the volume of the otic vesicle lumina in the mutant. (K-N) Live staining with FM1-43 marks hair cell endocytosis at 5 dpf, labelling functional cells in the cristae (K,L) and the anterior macula (M,N) in siblings (K,M) and mutants (L,N). A,B,G-J are projections of confocal z-stacks; C-F,K-N are individual optical sections, in which not all sensory patches are visible. am, anterior macula; ac, anterior crista; lc, lateral crista; pc, posterior crista; n, neuromast. Scale bars: 50 μm in A-F; 25 μm in G-N.
Fig. 3.

Expression of ionic homeostasis genes is disrupted in the ears of lte mutants. (A-H) Whole-mount in situ hybridisation showing that expression of alpha (a) and beta (b) Na+/K+-ATPase subunit genes is downregulated in the inner ear of lte mutants at 74 hpf (atp1b1a in E,F; atp1b2b in G,H), 4 dpf (atp1a1a.4 in A,B) and 5 dpf (atp1a1a.5 in C,D). The endolymphatic duct (asterisk, A,B) expresses atp1a1a.4 and appears normal in lte. (I,J) At 4 dpf, otic expression of the potassium channel gene kcnq1 is also reduced, mainly in the central semicircular canal hub (h). (K,L) Conversely, expression of connexin 33.8 (cx33.8) at 5 dpf is expanded in the anterior macula of lte mutants (black arrowheads), but remains unchanged in the lateral crista (white arrowheads; out of plane of focus in K). Scale bar: 50 μm.
At ∼75 hpf, the otic vesicle began to collapse owing to an apparent loss of fluid (endolymph), and was much smaller by 5 dpf (Fig. 1A-E), when the fluid-filled lumina of the semicircular canals and utriculosaccular cavity showed nearly complete collapse (Fig. 1D,E; Fig. 2). There was no apparent increase in cell death in the ear, as assayed by Acridine Orange staining at this stage (Fig. 1F-I), indicating that cell death does not contribute to the collapse in ear size (ltetg414b, n=59 mutants, n=55 siblings; lteto27d, n=21 mutants, n=21 siblings). There was no obvious decrease in cell number in the semicircular canal pillars of the mutants (Fig. 1J-M). Staining with FITC-phalloidin indicated that the sensory patches contain normal numbers of hair cells and the maculae have a normal morphology (Fig. 2G-J) but are crowded together in the much smaller luminal space (Fig. 2) (Whitfield et al., 1996). Hair cells in the mutant ear were functional, at least until 5 dpf, based on their uptake of FM1-43, a marker of endocytosis (Fig. 2K-N). At 4 dpf, identified mutants appeared to swim normally (see Movies 1, 2 in the supplementary material), but by 5 dpf the embryos displayed abnormal swimming behaviour characteristic of vestibular dysfunction, with fish swimming and resting upside down and occasional circling behaviour (Whitfield et al., 1996) (see Movies 3, 4 in the supplementary material).
The swim bladder is enlarged in lte mutants
The other notable aspect of the lte phenotype was an expansion of the swim bladder, which showed incomplete penetrance and variable expressivity (Fig. 1N-P). This expansion appeared to result from an over-inflation with gas, rather than an increase in size of the swim bladder primordium, as expression of foxa1 in this region at 50 and 74 hpf appeared to be normal (data not shown). Expression of nkcc1 in the developing swim bladder was not detectable by in situ hybridisation (data not shown). Larvae with over-inflated swim bladders often became `trapped' at the water's surface owing to their increased buoyancy. Zebrafish are thought to initiate inflation of the swim bladder by taking a gulp of air (Goolish and Okutake, 1999). To test whether the swim bladder over-inflation in lte mutants is due to a physiological defect that is independent of air gulping, we carried out caging experiments (Riley and Moorman, 2000) to prevent larvae from taking a gulp of air at the surface of the water. Under these conditions, neither siblings nor lte mutant embryos (100%) were able to inflate their swim bladders at 5 dpf (tg414b, n=30, 22 siblings and 6 mutants; to27d, n=43, 33 siblings and 10 mutants). Embryos within the same dish that were allowed free access to the surface inflated their swim bladders with a 100% success rate (tg414b, n=28, 22 siblings and 6 mutants; to27d, n=55, 46 siblings and 9 mutants).
Of the two alleles, the phenotype in tg414b appeared to be slightly stronger in terms of the rapidity and severity of ear collapse (Fig. 1E). Both alleles are early larval lethal; homozygous mutants die between 6 and 14 dpf. The cause of death is unknown, but the vestibular defects and increased buoyancy of mutants are likely to make feeding difficult. Preliminary data from lipid ingestion and metabolism assays (Farber et al., 2001), however, suggest that gut transit and function are normal in lte (data not shown).
Expression of genes involved in endolymph regulation in the lte mutant ear
The loss of fluid in the lte mutant ear indicates a disruption to the production or regulation of endolymphatic fluid. We characterised the expression of homologues of genes known to be involved in endolymphatic ionic homeostasis in mammals, and found that several were disrupted in lte mutants. Expression of genes encoding both alpha (α1a.4, α1a.5) and beta (β1a, β2b) subunits of Na+/K+-ATPase 1, which normally mark semicircular canal tissue, was downregulated in the lte mutant ear from 74 hpf to 5 dpf (Fig. 3A-H), as was the expression of kcnq1 (Fig. 3I,J) and nkcc1 (see below). These gene expression changes correlate with the timing of ear collapse. In contrast to the Na+/K+-ATPase 1 subunit genes, expression of the gap junction gene connexin 33.8, the homologue of human GJB2/6, was upregulated around the edges of the anterior macula at 5 dpf (Fig. 3K,L).
Expression of nkcc1 mRNA and protein in the wild-type and lte mutant ear
Nkcc1 encodes a sodium-potassium-chloride cotransporter involved in endolymph regulation in the mammal. A 3384 bp cDNA clone encompassing the entire ORF (1121 amino acids) of zebrafish nkcc1 was isolated from embryonic cDNA. This cDNA (GenBank accession number GQ259737) differs from the previously published sequence (NM_001002080) by a 48 bp in-frame deletion corresponding to a complete exon, designated `20a'. This shorter version is likely to be a splice variant, as a fragment containing exon 20a can also be amplified by RT-PCR from wild-type embryonic cDNA (data not shown).
We used our cDNA clone to determine the nkcc1 mRNA expression pattern by in situ hybridisation in phenotypically wild-type and lte mutant embryos. In wild-type embryos at the tailbud stage, expression was confined to the notochord, but by early somitogenesis, nkcc1 was expressed in the developing somites, notochord and intermediate mesoderm (Fig. 4A). By 24 hpf, expression was visible in the otic vesicle (Fig. 4B); this persisted until at least 5 dpf, with highest levels in the epithelial pillars and dorsolateral septum that define the developing semicircular canals (Fig. 4C-E). Expression of nkcc1 was reduced or lacking in lte mutants at all stages assayed (Fig. 4F-J).
Fig. 4.

Expression of nkcc1 transcript and protein immunoreactivity are lost in lte mutants. (A-J) Whole-mount in situ hybridisation for nkcc1 reveals a reduction or loss of expression in lte mutants at all stages examined. (A,F) Four-somite stage (4S). Expression is reduced in the notochord (arrows) and somites (asterisks) in putative mutants. (B-E,G-J) Expression is reduced or lacking in the lte mutant ear from 24 hpf to 5 dpf. (K-V) Immunostaining with the T4 (Nkcc1/2) antibody. Immunoreactivity is lost at 6 dpf in the lte mutant ear (K,L). Expression of Nkcc2 in the pronephric ducts is unaffected in mutants (transverse sections M,N). (O-R) Confocal transverse section through the wild-type ear at 6 dpf; O is a brightfield image and R is an overlay of P and Q. Nkcc1 expression is restricted to the cells lining the medial face of the ventral pillar (P,R, red). TO-PRO-3 staining of nuclei (Q,R, blue) confirms that expression is restricted to the cell membrane and is highest basolaterally (P′). (S-V) Confocal transverse section through the pronephric ducts at 6 dpf showing that Nkcc2 expression is restricted to the apical cell membrane. h, semicircular canal hub; p, pronephric duct; v, ventral pillar; usc, utriculosaccular chamber; lsc, lateral semicircular canal lumen. Scale bars: 200 μm in A,F; 50 μm in B-E,G-L,O-R; 25 μm in M,N; 12.5 μm in S-V.
Nkcc1 protein expression was determined using the T4 antibody raised against human NKCC1 (Lytle et al., 1995), which is known to cross-react with both NKCC1 and NKCC2 in several species (Lytle et al., 1995). Antibody staining matched the combined expression patterns of nkcc1 and nkcc2 mRNAs in the zebrafish embryo (Fig. 4E,K and data not shown). As there is no nkcc2 mRNA expression in the ear (data not shown), we presume that all immunoreactivity here is due to the Nkcc1 protein. In the ear, expression was highest in the dorsolateral septum and ventral pillar of the developing semicircular canal system. In the dorsolateral septum, Nkcc1 protein was present throughout the cell membrane, with highest levels basolaterally (Fig. 4O-R). Expression in the ventral pillar was confined to cells facing the utriculosaccular chamber and was excluded from cells facing the lumen of the lateral semicircular canal. Antibody staining in the ear and notochord was lacking in lte mutants (Fig. 4L and data not shown). Staining was also seen in cells of the gill epithelium in both mutants and siblings, and this was not affected by the mutation (data not shown).
The T4 antibody also cross-reacts with the zebrafish Nkcc2 protein, which is clearly visible as apical staining in the proximal region of the pronephric duct at 5 dpf (Fig. 4S-V), a region corresponding to the thick ascending limb of the mammalian nephron (Wingert et al., 2007). This immunoreactivity was still present in the lte mutants (Fig. 4M,N).
The lte mutation disrupts the nkcc1 gene
The loss of nkcc1 transcript and protein in both lte alleles, coupled with the ear collapse phenotype, which is similar to that seen in the mouse Nkcc1 mutant model (Deol, 1963), made nkcc1 an ideal candidate for lte. The lte mutation was mapped to LG10 to an interval lying between z1145 (54.5cM from the top of LG10) and z17219 (77.3 cM) (Geisler et al., 2007). The first four exons of nkcc1 have been assigned to LG10 (Pre-Ensembl release Zv8), but are apparently outside this interval, although the sequence in this region is currently unfinished; we have been unable to resolve this discrepancy. The remaining 22 exons of nkcc1 are currently unassigned, but lie in order on LGUN. There is conserved synteny between genes in this region of LG10 and the corresponding areas of mouse chromosome 18 and human 5q23.3, the genomic loci for Nkcc1 orthologues in these species.
Sequencing of cDNA from the tg414b allele revealed a 112 bp deletion corresponding to the predicted exon 22 (Fig. 5A); a shorter transcript, which was not present in other strains tested, could be identified by RT-PCR in tg414b mutants and heterozygous siblings (Fig. 5B). This deletion generates a frameshift in the ORF, leading to a premature termination codon at amino acid 972. This predicts a protein that lacks conserved regulatory kinase residues in the C-terminal tail, which is now predicted to be extracellular in the mutant protein rather than intracellular (Fig. 5G). Sequencing of genomic DNA at the 3′ end of exon 22 from ltetg414b mutants revealed a G→A transition mutation resulting in the loss of a splice donor site, thereby accounting for the exon skipping in the nkcc1 transcript (Fig. 5A). This base change introduces a new NlaIII restriction site, forming the basis of a tg414b genotyping protocol for amplified genomic DNA. The resultant RFLP was found in 24/24 tg414b homozygous mutants and 17/24 siblings (presumed heterozygotes), but not in any of the AB, TL or LWT embryos tested (n=12 each), nor in to27d mutant or sibling embryos (n=24 each) (Fig. 5C).
Fig. 5.

Genomic sequencing reveals nkcc1 splice site mutations in both lte alleles. (A) ltetg414b is a G→A transition mutation in the splice donor site of exon 22. The resulting transcript lacks 112 bp and is shifted out of frame. (B) Amplification of nkcc1 cDNA with primers flanking the lost exon (see A; F, forward, R, reverse) reveals a 203 bp band in tg414b homozygotes, compound heterozygotes and full-length subcloned tg414b mutant cDNA, as compared with a 315 bp band amplified from the to27d mutant and sibling embryos, full-length wild-type (WT) and to27d subcloned mutant cDNA. (C) NlaIII digest of PCR products generated with primers flanking the lesion in genomic DNA reveals the introduction of a novel NlaIII site in tg414b mutants, cleaving a 53 bp fragment into 40 bp (asterisk) and 13 bp fragments. (D,E) Sequencing of cDNA from the to27d allele reveals a 13 bp insertion between bases 814 and 815 (D), which can be seen in an RT-PCR reaction using primers flanking this sequence (E): a band of 216 bp is present in the to27d line, as compared with the wild-type 203 bp band, amplified here from the tg414b line. (F) Analysis of to27d genomic DNA reveals a T→G transversion mutation in intron 4, introducing a novel splice site 13 bp upstream of the wild-type site. MO indicates the position of the splice-blocking morpholino used in the rescue experiment (see Fig. 6). (G) Theoretical protein topology models (Tusnàdy and Simon, 1998; Tusnàdy and Simon, 2001). The wild-type protein has 12 transmembrane helices; both the N- and C-termini are predicted to be intracellular. The tg414b protein would be truncated after the eleventh transmembrane helix and lose its C-terminal intracellular localisation. In to27d, a severely truncated isoform is predicted, with only three transmembrane helices remaining. E, extracellular; I, intracellular.
Sequencing of nkcc1 cDNA from the to27d allele revealed a 13 bp insertion of intronic sequence between exons 4 and 5 (Fig. 5D), which forms the basis of an RT-PCR-based genotyping protocol (Fig. 5E). The insertion leads to a shift in the ORF at amino acid 272, followed by 47 amino acids of nonsense sequence prior to a premature termination codon at amino acid 319. This would severely truncate the protein between the predicted third and fourth transmembrane domains (Fig. 5G). Sequencing of the genomic region upstream of exon 5 in lteto27d mutants revealed a T→G transversion mutation, which introduces a new splice acceptor site 13 bp upstream of the wild-type site (Fig. 5F). As all cDNA species sequenced from lteto27d have the insertion, this new site is apparently used in preference to the existing wild-type site, despite the prediction that it is weaker [NNSPLICE program: scores of 0.76 (to27d site) and 0.98 (wild-type site)]. This apparent anomaly might be explained by the prediction that the mutation also causes the loss of an SC35 exon splicing enhancer site (ESEfinder). The mutation is specific to the lteto27d allele: it is not found in genomic DNA from the ltetg414b allele or from several wild-type strains of zebrafish (data not shown). Note that although we were able to amplify mutant cDNA species by PCR, nkcc1 mRNA levels are severely reduced in both alleles (Fig. 4), indicating that much of the mRNA is probably destroyed by nonsense-mediated decay. We cannot estimate the levels of truncated protein produced in either allele as the antibody was raised against the C-terminal region of the human protein; the corresponding region is lost altogether in the predicted protein from the to27d allele and is partially truncated in the tg414b allele.
Morpholino knockdown and treatment with NKCC1 antagonists fail to recapitulate the lte phenotype
To confirm that mutations in nkcc1 are causative for the lte phenotype, we attempted to phenocopy the mutation. We injected a translation blocking (ATG) morpholino into wild-type embryos, which reduced the level of Nkcc1 protein in the notochord at 24 hpf (Fig. 6A-D). As in the mutants, no morphological phenotype was visible at this stage. However, knockdown of the protein was not maintained; immunoreactivity reappeared by 5 dpf, and at this stage the ear and swim bladder size in the morphants was indistinguishable from that in uninjected siblings (data not shown).
Fig. 6.

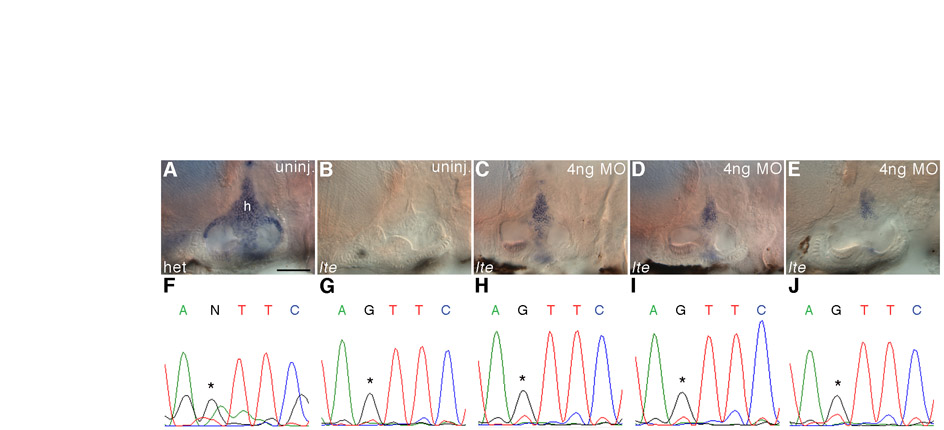
The lte phenotype can be rescued with a splice-blocking morpholino. (A-D) One nanogram of an ATG morpholino knocks down Nkcc1 protein expression in the notochord at 26 hpf (B; arrows in enlargement D) as compared with samples injected with 1 ng of a control morpholino (A; arrows in C). This reduction is not maintained at later stages and no phenocopy of the lte mutation was observed. (E-H) The lte phenotype cannot be rescued with a full-length mRNA encoding nkcc1. Ten picograms of Myc-tagged nkcc1 was injected alongside 10 pg gfp mRNA and embryos stained for Myc expression. At 26 hpf, cells are clearly positive for Myc, indicating expression of the tagged protein (F). Expression is extinguished by 74 hpf (H). No Myc expression is detected in embryos injected with gfp RNA alone (E,G). (I-X) A morpholino directed against the ectopic splice site in to27d can rescue the ear collapse phenotype at 6 dpf. Ear size in live injected mutants (K) can become indistinguishable from that of uninjected siblings (I) and is greatly increased compared with uninjected mutants (J). The sequencing traces (N-P,U-X) below each image confirm the genotype of the pictured embryos; the asterisk indicates the position of the mutation (T, wild-type allele; G, mutant allele). Examples of `mild' and `moderate' rescued embryos (see Table 1) are depicted in L and M, respectively; a rare complete rescue that would have been scored as `wild type' (Table 1) is shown in K. Rescue can also be seen at the protein level: T4 staining shows that an injected homozygous mutant (T) has similar levels of protein to an uninjected wild-type sibling (Q) or an injected heterozygous sibling (S). No antibody staining is present in the ear of an uninjected homozygous mutant (R). Scale bars: 100 μm in A-F; 50 μm in G-M,Q-T.
As an alternative approach, we attempted to inhibit Nkcc1 function by chemical treatment. The loop diuretic bumetanide is a potent antagonist of mammalian NKCC1 (Lytle et al., 1992) and is known to be effective in killifish (Eriksson et al., 1985; Eriksson and Wistrand, 1986) and dogfish (Edwards et al., 1997; Isenring and Forbush, 1997; Isenring et al., 1998a; Isenring et al., 1998b). We incubated wild-type embryos in 50-200 μM bumetanide from 50% epiboly to 5 dpf for varying time windows and also injected the otic vesicle directly with 1 nl of 10 mM bumetanide at 5 dpf (both into the vesicle lumen and into the canal pillars, to target the apical and basolateral sides of the otic epithelium, respectively). However, none of these treatments was able to induce ear collapse or swim bladder expansion (data not shown). This is probably because the binding region is not conserved in zebrafish Nkcc1 - the substitution of a conserved leucine for a tyrosine at position 241 may inhibit binding of the drug (J. Rafferty, personal communication). The related and less potent antagonist of mammalian NKCC1, furosemide, gave a rapid (within 3-4 hours) and dose-dependent phenotype of oedema and circulation failure (data not shown), masking any specific effects on Nkcc1 function in the ear.
Morpholino block of the ectopic splice site in lteto27d mutants rescues the ear phenotype
In an attempt to rescue the lte phenotype, we injected wild-type Myc-tagged nkcc1 mRNA into progeny from crosses between lteto27d/+ heterozygous parents. Although Myc-tagged protein could be seen at 24 hpf, by 48 hpf any trace of the protein had disappeared and no phenotypic rescue of the ear collapse was seen in the 5 dpf mutants (Fig. 6E-H). In contrast to Xenopus embryos (Walters et al., 2008), no effects on axis formation were found in these overexpression experiments.
As an alternative approach, we used a morpholino designed specifically to block the ectopic splice site in the lteto27d mutants, forcing use of the wild-type exon 5 splice donor site (Fig. 5F; Fig. 6I-X; Table 1). Embryos were injected with 1-10 ng morpholino and scored at 6 dpf for the extent of ear collapse, being classified as `wild type', `mild', `moderate' or `complete/severe' (Fig. 6K-M; Table 1). These injections resulted in a dose-dependent rescue of the ear phenotype; the optimal amount for rescue was 4 ng, as toxicity levels increased with higher doses. Genomic sequencing of injected siblings and rescued mutants confirmed that, in some cases, there was complete restoration of ear size at 6 dpf in homozygous mutants (Fig. 6K). Antibody staining showed that Nkcc1 protein levels in the ears of rescued mutant embryos were comparable to those in the wild type (Fig. 6T). No change in ear size or other adverse phenotype was observed in the injected sibling embryos, either wild types or heterozygotes (Fig. 6S), implying that there is no interference with the wild-type splice site. In control injections using the tg414b allele no rescue was seen (Table 1). RT-PCR revealed limited and variable amounts of the wild-type transcript in the rescued mutants, which could also be seen by in situ hybridisation (see Fig. S3 in the supplementary material); the intensity of staining correlated with the degree of rescue in live specimens (data not shown). Although we did not quantify the swim bladder phenotype, injected mutants appeared less buoyant (data not shown). A limited number of injected embryos were monitored until 7 and 8 dpf. By these stages, the ear rescue phenotype was still apparent but had started to decline; of 20 embryos classified as mild/moderate rescue initially, only 15 remained so at 7 dpf (a 25% reduction), whereas the mild/moderate rescue only persisted in 2/9 embryos at 8 dpf (a 78% reduction).
Table 1.
Injection of the to27d splice-blocking morpholino partially rescues the Ite phenotype
| Allele | Dose of MO (ng) | n | Wild type (%) | Mild collapse (%) | Moderate collapse (%) | Severe/complete collapse (%) |
|---|---|---|---|---|---|---|
| to27d | 0 | 917 | 689 (75.1) | 0 (0) | 2 (0.2) | 226 (24.6) |
| 3 | 313 | 237 (75.7) | 5 (1.6) | 59 (18.8) | 12 (3.8) | |
| 4 | 607 | 443 (73.0) | 21 (3.5) | 59 (9.7) | 84 (13.8) | |
| tg414b | 0 | 284 | 196 (69.0) | 0 (0) | 0 (0) | 88 (31.0) |
| 3 | 156 | 106 (67.9) | 0 (0) | 0 (0) | 50 (32.1) | |
| 4 | 184 | 142 (77.2) | 0 (0) | 0 (0) | 42 (22.8) |
Embryos were scored according to their degree of ear collapse at 6 dpf. Similar results were seen at 5 dpf with a graded response from 1 to 7.5 ng morpholino (MO). percentages were rounded to one decimal place.
DISCUSSION
NKCC1 function in the ear is conserved between zebrafish and mammals
We describe here the effects of nkcc1 mutations on the zebrafish inner ear. Both lte alleles are presumed to be severe hypomorphs or nulls on the basis of trace, residual levels of nkcc1 transcript and the predicted truncated protein products. In the mouse, several mutant alleles of Nkcc1, including both spontaneous and targeted knockout mutations, have been described (Delpire et al., 1999; Deol, 1963; Dixon et al., 1999; Evans et al., 2000; Flagella et al., 1999; Pace et al., 2001). These include the shaker-with-syndactylism deletion mutant (Deol, 1963), in which the deafness is attributed to the loss of NKCC1 function (Dixon et al., 1999) and the syndactyly to the simultaneous deletion of the fibrillin 2 gene (Chaudhry et al., 2001). All the murine homozygous Nkcc1 mutants display a consistent vestibulocochlear phenotype, characterised by deafness together with head bobbing and circling behaviour.
The effects of the loss of NKCC1 function in the mouse ear are evident from P0 in homozygotes, with the slackening of Reissner's membrane (Deol, 1963). By P30, Reissner's membrane has collapsed and there is widespread structural damage to the cochlea, with the loss of hair cells and Boettcher cells, together with increased intercellular space in the stria vascularis (Flagella et al., 1999; Pace et al., 2000). Gross development of the ear is normal (Dixon et al., 1999), but the semicircular canals are abnormally thin, with a reduced luminal diameter that eventually solidifies (Deol, 1963). The utricle and saccule are irregular, with a collapsed epithelial lining (Flagella et al., 1999). These latter effects mirror the phenotype of lte. Our data suggest that mechanisms of endolymph generation are conserved between zebrafish and mammals, despite the lack of a cochlea in the fish ear.
In humans, NKCC1 is expressed in the epithelia of several tissues, including the middle and inner ear (Kim et al., 2007; Kakigi et al., 2008; Dzhala et al., 2005; Payne et al., 1995; Dolganov et al., 2001). Mice heterozygous for mutations in Nkcc1 show a progressive, age-related hearing loss (Diaz et al., 2007), and high doses of loop diuretics such as furosemide cause reversible deafness in human patients, presumably through their effects on NKCC1 function (Bourke, 1976; Vargish et al., 1970). These observations, together with the zebrafish model described here, make NKCC1 a good candidate gene for human deafness. However, to our knowledge, no deafness, vestibular or other candidate disorder maps to the human NKCC1 locus (5q23.3). Interestingly, upregulation of NKCC1 expression in the middle ear epithelium has been suggested to underlie the generation of excess middle ear fluid in patients with otitis media with effusion (OME) (Kim et al., 2007).
Nkcc1 protein expression in the zebrafish ear is tightly regulated spatially
In the mouse, Nkcc1 transcripts are present in the embryo from E12.5 (Hubner et al., 2001; Vanden Heuvel et al., 2006) and in the cochlea from E18.5. The T4 antibody shows basolateral expression of NKCC1 protein in marginal cells of the stria vascularis, vestibular dark cells and neurons of the spiral and vestibular ganglia in the adult mouse (Delpire et al., 1999; Dixon et al., 1999), guinea pig (Jin et al., 2008) and gerbil (Crouch et al., 1997; Sakaguchi et al., 1998); protein expression is also seen in the endolymphatic sac of the rat (Akiyama et al., 2007). In the zebrafish ear, Nkcc1 protein expression is restricted to cells in the dorsolateral septum and the ventral semicircular canal pillar facing the utriculosaccular chamber. Cells on the other side of the same pillar, facing the lumen of the lateral (horizontal) canal, do not express Nkcc1. This sharp boundary between expressing and non-expressing cells indicates that cells of the pillar are not homogeneous, but show distinct expression (and probably functional) differences at this early stage in otic development. We conclude that the zebrafish larval semicircular canal epithelium performs an endolymph-generating function equivalent to that of mammalian strial marginal cells and vestibular dark cells. At later stages, it is likely that the Na+/K+-ATPase 1-expressing cells surrounding the maculae also contribute to the generation of endolymph (Shiao et al., 2005).
Feedback control in endolymph homeostasis
We propose that the endolymph collapse phenotype in lte mutants occurs as a result of altered ion homeostasis in the secretory epithelia of the ear. In the mammal, NKCC1 is a component of K+ entry into the cochlear marginal cells of the stria vascularis, and its activity is tightly coupled to that of the basolateral Na+/K+-ATPase, with these proteins providing cellular entry routes for Na+, K+ and Cl-. Apical secretion of K+ is mediated by channels such as KCNE1 and KCNQ1, with an associated transport of water (Lee et al., 2000; Vetter et al., 1996); a loss of K+ recycling with a concomitant loss of water secretion is thought to lead to a loss of fluid in the scala media, causing Reissner's membrane to collapse (Delpire and Mount, 2002; Wangemann, 2002). NKCC1 is probably involved in maintaining endolymph volume in the guinea pig (Jin et al., 2007); the gw mutant has a similar vestibulocochlear impairment to that in mice mutant for Nkcc1, with a collapse of the endolymphatic compartment and degeneration of many cochlear cell types. Mutant animals show a loss of NKCC1 protein expression in the ear, together with a loss of immunoreactivity for the tight junction protein CLDN11 and the potassium channel KCNJ10 (Jin et al., 2008). This demonstrates that the entire ion homeostasis system of the ear can become unbalanced.
In lte homozygotes, we also see decreased expression of other ion channel genes, including those encoding the Na+/K+-ATPase subunits. In the mouse, heterozygous loss-of-function of Na+/K+-ATPase α1 (Atp1a1) delays the onset of hearing loss in mice heterozygous for a mutation in Nkcc1 (Diaz et al., 2007), whereas the Nkcc1+/-; Atp1a2+/- double heterozygote has a restoration to normal hearing thresholds throughout life, suggesting that in the mouse, a downregulation of Na+/K+-ATPase activity might compensate for the effects of reduced NKCC1 function. It is possible that the downregulation of Na+/K+-ATPase subunit genes effects a similar compensatory mechanism in an attempt to maintain ionic homeostasis in the lte mutant ear.
lte mutants have a unique swim bladder phenotype
The increase in swim bladder volume seen in lte mutants represents a unique and rare phenotype. As far as we are aware, this has not been described in any published mutant and provides insight into the mechanisms of swim bladder inflation. The converse situation is well known; failure to inflate the swim bladder is common, and is thought to contribute to the lethality of many mutations (Driever et al., 1996; Haffter et al., 1996; McCune and Carlson, 2004). The only other report of an overfilled swim bladder is in big bladder mutants (McCune and Carlson, 2004). These display a `floating' phenotype at 5 dpf, which resolves by 9 dpf; this is different to lte mutants, in which swim bladder over-inflation is maintained until death.
The swim bladder develops as a fluid-filled evagination from a solid endodermal rod (Ober et al., 2003) and inflates with gas from 84 hpf. The inflation mechanism varies between teleosts, as does the composition of the gas filling the chamber (Pelster, 2004). It has been suggested that larvae surface and swallow a bolus of air, which is passed down the oesophagus and into the swim bladder via the pneumatic duct, aided by secreted surfactants (Goolish and Okutake, 1999; Riley and Moorman, 2000). The results of our caging experiments support these findings, suggesting that this is the mechanism used in zebrafish. Although it is clear that the initial inflation of the swim bladder in both lte mutants and siblings is dependent on access to the air/water interface, the mechanism of later over-inflation in lte mutants is still unclear. It is possible that this reflects a later physiological defect in gaseous metabolism [see discussion in Goolish and Okutake (Goolish and Okutake, 1999)]. The cause of lethality in lte mutants remains unknown. The larvae survive to a maximum of 14 dpf and apparently fail to feed successfully. In the mouse, some Nkcc1-/- strains fail to thrive, and die owing to intestinal bleeding (Flagella et al., 1999); however, it has been noted that morbidity varies considerably with genetic background (Pace et al., 2000).
Zebrafish nkcc1 splice variants correspond to those in the mammal
We report here the cloning of a new wild-type splice variant of zebrafish nkcc1. Comparison with the published sequence (NM_001002080) indicated a 48 bp deletion corresponding to a predicted exon denoted 20a (exon 21 in the mouse); this (exon 20a) was found to be missing in the wild-type and in both mutant cDNA species, and probably represents an alternatively spliced variant. This variant is also present in mouse, in which both transcripts are expressed in the brain and the longer transcript also in the cochlea (Dixon et al., 1999; Randall et al., 1997), and in human, in which the ratio of the two transcripts varies between expressing tissues (Vibat et al., 2001). In the mammalian gene, the sequence encoded by exon 21 has been proposed to include a conserved basolateral sorting motif that might direct the correct subcellular localisation of the protein in vitro (Carmosino et al., 2008). We are unable to confirm whether this is the case in zebrafish, as we cannot discern which spliceoform is expressed in the ear.
Splice mutations can be rescued in the zebrafish embryo
The lteto27d allele introduces a novel splice site that results in a 13 bp insertion in the transcript. The major mRNA species found in the mutant embryo is the mutant spliceoform, despite the prediction (NNSPLICE program) that the wild-type site is a `stronger' splice site. Analysis of RNA splicing enhancer sites using ESEfinder suggests that the T→G mutation also removes an SC35 splice enhancer site, which might account for the preferential usage of the mutant site.
The rescue of a splice mutation using morpholinos has been established as a possible therapeutic avenue for several diseases, including Hutchinson-Gilford progeria (Scaffidi and Misteli, 2005), retinal degeneration (Andrieu-Soler et al., 2007), X-linked ocular albinism (Vetrini et al., 2006) and muscular dystrophy (Gebski et al., 2003). Recent work showed proof of principle for this technique in the zebrafish calamity (atp7a) mutant, a model for the copper transport disorder Menkes disease (Madsen et al., 2008). Wild-type protein was restored in mutants injected with a morpholino designed to block an ectopic splice acceptor site. However, an increase in wild-type transcript levels was not found, and it was proposed that only a trace amount of wild-type transcript is required for rescue; alternatively, the morpholino might have relieved the inhibition of translation of the existing small amount of wild-type transcript present in the mutants (Madsen et al., 2008). In rescued lteto27d embryos, we see increased amounts of mRNA, when assayed by in situ hybridisation, and propose that this directly underlies the rescued phenotype.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/136/16/2837/DC1
Supplementary Material
We thank Robert Chandler, Melanie Cotterill and Laina Murphy for technical assistance; Hans-Georg Frohnhöfer (Tübingen Stock Centre) for recovery of the tg414b allele; Robert Geisler for communication of mapping results before publication; John Rafferty for advice on NKCC1 structure and bumetanide binding; Freek van Eeden for suggesting the morpholino rescue experiment; Paul Morcos (Gene Tools) for help with morpholino design; and Matthew Broadhead for comments on the manuscript. Lisa van Hateren and the Sheffield Aquarium staff provided expert care of the fish stocks. The T4 antibody was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NIHCD and maintained by the University of Iowa. This work was funded by the MRC (G0300196). The MRC CDBG zebrafish aquaria and imaging facilities were supported by the MRC (G0400100, G0700091), with additional support from the EU FP6 (ZF-MODELS), Yorkshire Cancer Research and the Wellcome Trust (GR077544AIA). Deposited in PMC for release after 6 months.
References
- Akiyama, K., Miyashita, T., Mori, T. and Mori, N. (2007). Expression of the Na+-K+-2Cl- cotransporter in the rat endolymphatic sac. Biochem. Biophys. Res. Commun. 364, 913-917. [DOI] [PubMed] [Google Scholar]
- Andrieu-Soler, C., Halhal, M., Boatright, J. H., Padove, S. A., Nickerson, J. M., Stodulkova, E., Stewart, R. E., Ciavatta, V. T., Doat, M., Jeanny, J. C. et al. (2007). Single-stranded oligonucleotide-mediated in vivo gene repair in the rd1 retina. Mol. Vis. 13, 692-706. [PMC free article] [PubMed] [Google Scholar]
- Blasiole, B., Degrave, A., Canfield, V., Boehmler, W., Thisse, C., Thisse, B., Mohideen, M. A. and Levenson, R. (2003). Differential expression of Na,K-ATPase alpha and beta subunit genes in the developing zebrafish inner ear. Dev. Dyn. 228, 386-392. [DOI] [PubMed] [Google Scholar]
- Blasiole, B., Canfield, V. A., Vollrath, M. A., Huss, D., Mohideen, M. A., Dickman, J. D., Cheng, K. C., Fekete, D. M. and Levenson, R. (2006). Separate Na,K-ATPase genes are required for otolith formation and semicircular canal development in zebrafish. Dev. Biol. 294, 148-160. [DOI] [PubMed] [Google Scholar]
- Bourke, E. (1976). Frusemide, bumetanide, and ototoxicity. Lancet 1, 917-918. [DOI] [PubMed] [Google Scholar]
- Busch-Nentwich, E., Söllner, C., Roehl, H. and Nicolson, T. (2004). The deafness gene dfna5 is crucial for ugdh expression and HA production in the developing ear in zebrafish. Development 131, 943-951. [DOI] [PubMed] [Google Scholar]
- Carmosino, M., Gimenez, I., Caplan, M. and Forbush, B. (2008). Exon loss accounts for differential sorting of Na-K-Cl cotransporters in polarized epithelial cells. Mol. Biol. Cell 19, 4341-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni, L., Wang, J., Zhu, Z., Zhang, M. Q. and Krainer, A. R. (2003). ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 31, 3568-3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro, M. C., Knollmann, B. C., Ebert, S. N., Vary, J. C. J., Greene, A. E., Franz, M. R., Grinberg, A., Huang, S. P. and Pfeifer, K. (2001). Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc. Natl. Acad. Sci. USA 98, 2526-2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry, S. S., Gazzard, J., Baldock, C., Dixon, J., Rock, M. J., Skinner, G. C., Steel, K. P., Kielty, C. M. and Dixon, M. J. (2001). Mutation of the gene encoding fibrillin-2 results in syndactyly in mice. Hum. Mol. Genet. 10, 835-843. [DOI] [PubMed] [Google Scholar]
- Crouch, J. J., Sakaguchi, N., Lytle, C. and Schulte, B. A. (1997). Immunohistochemical localization of the Na-K-Cl co-transporter (NKCC1) in the gerbil inner ear. J. Histochem. Cytochem. 45, 773-778. [DOI] [PubMed] [Google Scholar]
- Cutler, C. P. and Cramb, G. (2002). Two isoforms of the Na+/K+/2Cl- cotransporter are expressed in the European eel (Anguilla anguilla). Biochim. Biophys. Acta 1566, 92-103. [DOI] [PubMed] [Google Scholar]
- Delpire, E. and Mount, D. B. (2002). Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu. Rev. Physiol. 64, 803-843. [DOI] [PubMed] [Google Scholar]
- Delpire, E., Rauchman, M. I., Beier, D. R., Hebert, S. C. and Gullans, S. R. (1994). Molecular cloning and chromosome localization of a putative basolateral Na(+)-K(+)-2Cl- cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J. Biol. Chem. 269, 25677-25683. [PubMed] [Google Scholar]
- Delpire, E., Lu, J., England, R., Dull, C. and Thorne, T. (1999). Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 22, 192-195. [DOI] [PubMed] [Google Scholar]
- Deol, M. S. (1963). The development of the inner ear in mice homozygous for Shaker-with-Syndactylism. J. Embryol. Exp. Morphol. 11, 493-512. [PubMed] [Google Scholar]
- Diaz, R. C., Vazquez, A. E., Dou, H., Wei, D., Cardell, E. L., Lingrel, J., Shull, G. E., Doyle, K. J. and Yamoah, E. N. (2007). Conservation of hearing by simultaneous mutation of Na,K-ATPase and NKCC1. JARO 8, 422-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, M. J., Gazzard, J., Chaudhry, S. S., Sampson, N., Schulte, B. A. and Steel, K. P. (1999). Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum. Mol. Genet. 8, 1579-1584. [DOI] [PubMed] [Google Scholar]
- Dolganov, G. M., Woodruff, P. G., Novikov, A. A., Zhang, Y., Ferrando, R. E., Szubin, R. and Fahy, J. V. (2001). A novel method of gene transcript profiling in airway biopsy homogenates reveals increased expression of a Na+-K+-Cl- cotransporter (NKCC1) in asthmatic subjects. Genome Res. 11, 1473-1483. [DOI] [PubMed] [Google Scholar]
- Driever, W., Solnica-Krezel, L., Schier, A. F., Neuhauss, S. C. F., Malicki, J., Stemple, D. L., Stainier, D. Y. R., Zwartkruis, F., Abdelilah, S., Rangini, Z. et al. (1996). A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123, 37-46. [DOI] [PubMed] [Google Scholar]
- Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., Delpire, E., Jensen, F. E. and Staley, K. J. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205-1213. [DOI] [PubMed] [Google Scholar]
- Edwards, J., Mackenzie, S., Cutler, C. P. and Cramb, G. (1997). Effects of extracellular sodium concentration on the activity of Na,K-ATPase in dogfish rectal gland epithelial cells. Ann. New York Acad. Sci. 834, 565-568. [DOI] [PubMed] [Google Scholar]
- Eriksson, O. and Wistrand, P. J. (1986). Chloride transport inhibition by various types of loop diuretics in fish opercular epithelium. Acta Physiol. Scand. 126, 93-101. [DOI] [PubMed] [Google Scholar]
- Eriksson, O., Mayer-Gostan, N. and Wistrand, P. J. (1985). The use of isolated fish opercular epithelium as a model tissue for studying intrinsic activities of loop diuretics. Acta Physiol. Scand. 125, 55-66. [DOI] [PubMed] [Google Scholar]
- Evans, D. H., Piermarini, P. M. and Choe, K. P. (2005). The multifunctional fish gill: dominant site of gas exchange, osmoregulation, acid-base regulation, and excretion of nitrogenous waste. Physiol. Rev. 85, 97-177. [DOI] [PubMed] [Google Scholar]
- Evans, R. L., Park, K., Turner, R. J., Watson, G. E., Nguyen, H. V., Dennett, M. R., Hand, A. R., Flagella, M., Shull, G. E. and Melvin, J. E. (2000). Severe impairment of salivation in Na+/K+/2Cl- cotransporter (NKCC1)-deficient mice. J. Biol. Chem. 275, 26720-26726. [DOI] [PubMed] [Google Scholar]
- Farber, S. A., Pack, M., Ho, S. Y., Johnson, I. D., Wagner, D. S., Dosch, R., Mullins, M. C., Hendrickson, H. S., Hendrickson, E. K. and Halpern, M. E. (2001). Genetic analysis of digestive physiology using fluorescent phospholipid reporters. Science 292, 1385-1388. [DOI] [PubMed] [Google Scholar]
- Flagella, M., Clarke, L. L., Miller, M. L., Erway, L. C., Giannella, R. A., Andringa, A., Gawenis, L. R., Kramer, J., Duffy, J. J., Doetschman, T. et al. (1999). Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 274, 26946-26955. [DOI] [PubMed] [Google Scholar]
- Gebski, B. L., Mann, C. J., Fletcher, S. and Wilton, S. D. (2003). Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum. Mol. Genet. 12, 1801-1811. [DOI] [PubMed] [Google Scholar]
- Geisler, R., Rauch, G.-J., Geiger-Rudolph, S., Albrecht, A., van Bebber, F., Berger, A., Busch-Nentwich, E., Dahm, R., Dekens, M. P., Dooley, C. et al. (2007). Large-scale mapping of mutations affecting zebrafish development. BMC Genomics 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goolish, E. M. and Okutake, K. (1999). Lack of gas bladder inflation by the larvae of zebrafish in the absence of an air-water interface J. Fish Biol. 55, 1054-1063. [Google Scholar]
- Haffter, P., Granato, M., Brand, M., Mullins, M. C., Hammerschmidt, M., Kane, D. A., Odenthal, J., van Eeden, F., Jiang, Y.-J., Heisenberg, C. P. et al. (1996). The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 123, 1-36. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt, M., Serbedzija, G. N. and McMahon, A. P. (1996). Genetic analysis of dorsoventral pattern formation in the zebrafish: requirement of a BMP-like ventralizing activity and its dorsal repressor. Genes Dev. 1996, 2452-2461. [DOI] [PubMed] [Google Scholar]
- Hubner, C. A., Lorke, D. E. and Hermans-Borgmeyer, I. (2001). Expression of the Na-K-2Cl-cotransporter NKCC1 during mouse development. Mech. Dev. 102, 267-269. [DOI] [PubMed] [Google Scholar]
- Hyndman, K. A. and Evans, D. H. (2008). Short-term low-salinity tolerance by the longhorn sculpin, Myoxocephalus octodecimspinosus. J. Exp. Zool. Part A Ecol. Genet. Physiol. 311A, 45-56. [DOI] [PubMed] [Google Scholar]
- Isenring, P. and Forbush, B., 3rd. (1997). Ion and bumetanide binding by the Na-K-Cl cotransporter. Importance of transmembrane domains. J. Biol. Chem. 272, 24556-24562. [DOI] [PubMed] [Google Scholar]
- Isenring, P., Jacoby, S. C., Chang, J. and Forbush, B. (1998a). Mutagenic mapping of the Na-K-Cl cotransporter for domains involved in ion transport and bumetanide binding. J. Gen. Physiol. 112, 549-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenring, P., Jacoby, S. C. and Forbush III, B. (1998b). The role of transmembrane domain 2 in cation transport by the Na-K-Cl cotransporter. Proc. Natl. Acad. Sci. USA 95, 7179-7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Mannstrom, P., Jarlebark, L. and Ulfendahl, M. (2007). Malformation of stria vascularis in the developing inner ear of the German waltzing guinea pig. Cell Tissue Res. 328, 257-270. [DOI] [PubMed] [Google Scholar]
- Jin, Z., Ulfendahl, M. and Jarlebark, L. (2008). Spatiotemporal loss of K+ transport proteins in the developing cochlear lateral wall of guinea pigs with hereditary deafness. Eur. J. Neurosci. 27, 145-154. [DOI] [PubMed] [Google Scholar]
- Kakigi, A., Nishimura, M., Takeda, T., Taguchi, D. and Nishioka, R. (2008). Expression of aquaporin1, 3, and 4, NKCC1, and NKCC2 in the human endolymphatic sac. Auris Nasus Larynx 36, 135-139. [DOI] [PubMed] [Google Scholar]
- Kim, S. J., Choi, J. Y., Son, E. J., Namkung, W., Lee, M. G. and Yoon, J. H. (2007). Interleukin-1beta upregulates Na+-K+-2Cl- cotransporter in human middle ear epithelia. J. Cell. Biochem. 101, 576-586. [DOI] [PubMed] [Google Scholar]
- Korzh, V., Sleptsova, I., Liao, J., He, J. and Gong, Z. (1998). Expression of zebrafish bHLH genes ngn1 and nrd defines distinct stages of neural differentiation. Dev. Dyn. 213, 92-104. [DOI] [PubMed] [Google Scholar]
- Lang, F., Vallon, V., Knipper, M. and Wangemann, P. (2007). Functional significance of channels and transporters expressed in the inner ear and kidney. Am. J. Physiol. Cell Physiol. 293, C1187-C1208. [DOI] [PubMed] [Google Scholar]
- Lee, M. P., Ravenel, J. D., Hu, R. J., Lustig, L. R., Tomaselli, G., Berger, R. D., Brandenburg, S. A., Litzi, T. J., Bunton, T. E., Limb, C. et al. (2000). Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J. Clin. Invest. 106, 1447-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letts, V. A., Valenzuela, A., Dunbar, C., Zheng, Q. Y., Johnson, K. R. and Frankel, W. N. (2000). A new spontaneous mouse mutation in the Kcne1 gene. Mamm. Genome 11, 831-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorin-Nebel, C., Boulo, V., Bodinier, C. and Charmantier, G. (2006). The Na+/K+/2Cl- cotransporter in the sea bass Dicentrarchus labrax during ontogeny: involvement in osmoregulation. J. Exp. Biol. 209, 4908-4922. [DOI] [PubMed] [Google Scholar]
- Lowery, L. A. and Sive, H. (2005). Initial formation of zebrafish brain ventricles occurs independently of circulation and requires the nagie oko and snakehead/atp1a1a.1 gene products. Development 132, 2057-2067. [DOI] [PubMed] [Google Scholar]
- Lytle, C., Xu, J. C., Biemesderfer, D., Haas, M. and Forbush, B., 3rd. (1992). The Na-K-Cl cotransport protein of shark rectal gland. I. Development of monoclonal antibodies, immunoaffinity purification, and partial biochemical characterization. J. Biol. Chem. 267, 25428-25437. [PubMed] [Google Scholar]
- Lytle, C., Xu, J. C., Biemesderfer, D. and Forbush, B., 3rd. (1995). Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am. J. Physiol. 269, C1496-C1505. [DOI] [PubMed] [Google Scholar]
- Mackie, P. M., Gharbi, K., Ballantyne, J. S., McCormick, S. D. and Wright, P. A. (2007). Na+/K+/2Cl- cotransporter and CFTR gill expression after seawater transfer in smolts (O+) of different Atlantic salmon (Salmo salmar) families. Aquaculture 272, 625-635. [Google Scholar]
- Madsen, E. C., Morcos, P. A., Mendelsohn, B. A. and Gitlin, J. D. (2008). In vivo correction of a Menkes disease model using antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 105, 3909-3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus, D. C., Marcus, N. Y. and Greger, R. (1987). Sidedness of action of loop diuretics and ouabain on nonsensory cells of utricle: a micro-Ussing chamber for inner ear tissues. Hear. Res. 30, 55-64. [DOI] [PubMed] [Google Scholar]
- McCune, A. R. and Carlson, R. L. (2004). Twenty ways to lose your bladder: common natural mutants in zebrafish and widespread convergence of swim bladder loss among teleost fishes. Evol. Dev. 6, 246-259. [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard, C. and Dahm, R. (2002). Zebrafish: A Practical Approach (ed. B. D. Hames). Oxford: Oxford University Press.
- Ober, E. A., Field, H. A. and Stainier, D. Y. (2003). From endoderm formation to liver and pancreas development in zebrafish. Mech. Dev. 120, 5-18. [DOI] [PubMed] [Google Scholar]
- Odenthal, J. and Nüsslein-Volhard, C. (1998). fork head domain genes in zebrafish. Dev. Genes Evol. 208, 245-258. [DOI] [PubMed] [Google Scholar]
- Pace, A. J., Lee, E., Athirakul, K., Coffman, T. M., O'Brien, D. A. and Koller, B. H. (2000). Failure of spermatogenesis in mouse lines deficient in the Na(+)-K(+)-2Cl(-) cotransporter. J. Clin. Invest. 105, 441-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, A. J., Madden, V. J., Henson, O. W., Jr, Koller, B. H. and Henson, M. M. (2001). Ultrastructure of the inner ear of NKCC1-deficient mice. Hear. Res. 156, 17-30. [DOI] [PubMed] [Google Scholar]
- Payne, J. A. and Forbush, B., 3rd. (1995). Molecular characterization of the epithelial Na-K-Cl cotransporter isoforms. Curr. Opin. Cell Biol. 7, 493-503. [DOI] [PubMed] [Google Scholar]
- Payne, J. A., Xu, J. C., Haas, M., Lytle, C. Y., Ward, D. and Forbush, B., 3rd. (1995). Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J. Biol. Chem. 270, 17977-17985. [DOI] [PubMed] [Google Scholar]
- Pelster, B. (2004). The development of the swim bladder: structure and performance. In 26th Annual Larval Fish Conference (ed. J. J. Govoni), pp. 37-46. Bergen, Norway: American Fisheries Society.
- Peterson, R. E., Tu, C. and Linser, P. J. (1997). Isolation and characterization of a carbonic anhydrase homologue from the zebrafish (Danio rerio). J. Mol. Evol. 44, 432-439. [DOI] [PubMed] [Google Scholar]
- Pfeffer, P. L., Gerster, T., Lun, K., Brand, M. and Busslinger, M. (1998). Characterization of three novel members of the zebrafish Pax2/5/8 family: dependency of Pax5 and Pax8 expression on the Pax2. 1 (noi) function. Development 125, 3063-3074. [DOI] [PubMed] [Google Scholar]
- Piotrowski, T., Ahn, D. G., Schilling, T. F., Nair, S., Ruvinsky, I., Geisler, R., Rauch, G. J., Haffter, P., Zon, L. I., Zhou, Y. et al. (2003). The zebrafish van gogh mutation disrupts tbx1, which is involved in the DiGeorge deletion syndrome in humans. Development 130, 5043-5052. [DOI] [PubMed] [Google Scholar]
- Randall, J., Thorne, T. and Delpire, E. (1997). Partial cloning and characterization of Slc12a2: the gene encoding the secretory Na+-K+-2Cl- cotransporter. Am. J. Physiol. 273, C1267-C1277. [DOI] [PubMed] [Google Scholar]
- Reese, M. G., Eeckman, F. H., Kulp, D. and Haussler, D. (1997). Improved splice site detection in Genie. J. Comput. Biol. 4, 311-323. [DOI] [PubMed] [Google Scholar]
- Riley, B. B. and Moorman, S. J. (2000). Development of utricular otoliths, but not saccular otoliths, is necessary for vestibular function and survival in zebrafish. J. Neurobiol. 43, 329-337. [DOI] [PubMed] [Google Scholar]
- Sahly, I., Andermann, P. and Petit, C. (1999). The zebrafish eya1 gene and its expression pattern during embryogenesis. Dev. Genes Evol. 209, 399-410. [DOI] [PubMed] [Google Scholar]
- Sakaguchi, N., Crouch, J. J., Lytle, C. and Schulte, B. A. (1998). Na-K-Cl cotransporter expression in the developing and senescent gerbil cochlea. Hear. Res. 118, 114-122. [DOI] [PubMed] [Google Scholar]
- Scaffidi, P. and Misteli, T. (2005). Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 11, 440-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiao, J. C., Lin, L. Y., Horng, J. L., Hwang, P. P. and Kaneko, T. (2005). How can teleostean inner ear hair cells maintain the proper association with the accreting otolith? J. Comp. Neurol. 488, 331-341. [DOI] [PubMed] [Google Scholar]
- Smith, P. J., Zhang, C., Wang, J., Chew, S. L., Zhang, M. Q. and Krainer, A. R. (2006). An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum. Mol. Genet. 15, 2490-2508. [DOI] [PubMed] [Google Scholar]
- Solomon, K. S., Kudoh, T., Dawid, I. B. and Fritz, A. (2003). Zebrafish foxi1 mediates otic placode formation and jaw development. Development 130, 929-940. [DOI] [PubMed] [Google Scholar]
- Su, L., Vogelstein, B., Kinzler, K. W. (1993). Association of the APC tumor suppressor protein with catenins. Science 262, 1734. [DOI] [PubMed] [Google Scholar]
- Tusnàdy, G. E. and Simon, I. (1998). Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 283, 489-506. [DOI] [PubMed] [Google Scholar]
- Tusnàdy, G. E. and Simon, I. (2001). Topology of membrane proteins. J. Chem. Inf. Comput. Sci. 41, 364-368. [DOI] [PubMed] [Google Scholar]
- Vanden Heuvel, G. B., Payne, J. A., Igarashi, P. and Forbush, B., 3rd. (2006). Expression of the basolateral Na-K-Cl cotransporter during mouse nephrogenesis and embryonic development. Gene Expr. Patterns 6, 1000-1006. [DOI] [PubMed] [Google Scholar]
- Vargish, T., Benjamin, R. and Shenkman, L. (1970). Deafness from furosemide. Ann. Intern. Med. 72, 761. [DOI] [PubMed] [Google Scholar]
- Vetrini, F., Tammaro, R., Bondanza, S., Surace, E. M., Auricchio, A., De Luca, M., Ballabio, A. and Marigo, V. (2006). Aberrant splicing in the ocular albinism type 1 gene (OA1/GPR143) is corrected in vitro by morpholino antisense oligonucleotides. Hum. Mutat. 27, 420-426. [DOI] [PubMed] [Google Scholar]
- Vetter, D. E., Mann, J. R., Wangemann, P., Liu, J., McLaughlin, K. J., Lesage, F., Marcus, D. C., Lazdunski, M., Heinemann, S. F. and Barhanin, J. (1996). Inner ear defects induced by null mutation of the isk gene. Neuron 17, 1251-1264. [DOI] [PubMed] [Google Scholar]
- Vibat, C. R., Holland, M. J., Kang, J. J., Putney, L. K. and O'Donnell, M. E. (2001). Quantitation of Na+-K+-2Cl- cotransport splice variants in human tissues using kinetic polymerase chain reaction. Anal. Biochem. 298, 218-230. [DOI] [PubMed] [Google Scholar]
- Walters, Z. S., Haworth, K. E. and Latinkic, B. (2008). NKCC1 (SLC12a2) induces secondary axis in Xenopus laevis embryos independently of its co-transporter function. J. Physiol. 587, 521-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann, P. (1995). Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hear. Res. 90, 149-157. [DOI] [PubMed] [Google Scholar]
- Wangemann, P. (2002). K+ cycling and the endocochlear potential. Hear. Res. 165, 1-9. [DOI] [PubMed] [Google Scholar]
- Westerfield, M. (1995). The Zebrafish Book: a Guide for the Laboratory Use of Zebrafish (Danio rerio). Oregon: University of Oregon Press.
- Whitfield, T. T., Granato, M., van Eeden, F. J., Schach, U., Brand, M., Furutani-Seiki, M., Haffter, P., Hammerschmidt, M., Heisenberg, C. P., Jiang, Y. J. et al. (1996). Mutations affecting development of the zebrafish inner ear and lateral line. Development 123, 241-254. [DOI] [PubMed] [Google Scholar]
- Wingert, R. A., Selleck, R., Yu, J., Song, H. D., Chen, Z., Song, A., Zhou, Y., Thisse, B., Thisse, C., McMahon, A. P. et al. (2007). The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet. 3, 1922-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. C., Lytle, C., Zhu, T. T., Payne, J. A., Benz, E., Jr and Forbush, B., 3rd. (1994). Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc. Natl. Acad. Sci. USA 91, 2201-2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}