

4. Summary of recent advances
The semaphorins, originally discovered as evolutionarily conserved steering molecules for developing axons, also influence neuronal structure and function in the early postnatal and juvenile nervous systems through several refinement processes. Semaphorins control synaptogenesis, axon pruning, and the density and maturation of dendritic spines. In addition, semaphorins and their downstream signaling components regulate synaptic physiology and neuronal excitability in the mature hippocampus, and these proteins are also implicated in a number of developmental, psychiatric, and neurodegenerative disorders. Significant inroads have been made in defining the mechanisms by which semaphorins regulate dynamic changes in the neuronal cytoskeleton at the molecular and cellular levels during embryonic nervous system development. However, comparatively little is known about how semaphorins influence neuronal structure and synaptic plasticity during adult nervous system homeostasis or following injury and disease. A detailed understanding of how semaphorins function beyond initial phases of neural network assembly is revealing novel insights into key aspects of nervous system physiology and pathology.
5. Introduction
The Semaphorins are a large family of evolutionally conserved glycoproteins, and they are divided into eight different subfamilies based on structural motifs and species of origin (Fig. 1A). Although initially identified as inhibitory axon guidance molecules, semaphorins include both attractants and repellents [1]. How a neuron responds to a specific semaphorin family member, including repulsion or attraction, is not absolute but instead regulated by the context in which this cue is encountered. A detailed understanding of semaphorin signaling, including the molecular switches that determine whether a neuron shows a positive (attractive) or negative (inhibitory) response to a particular semaphorin is of considerable interest. These signaling events are likely to have important implications for neuronal plasticity, and also for axonal regeneration following nervous system injury.
Figure 1. Semaphorins and their receptors and signaling pathways in the nervous system.

(a) Semaphorins exist as secreted, membrane-spanning, or glycosylphosphatidylinositol (GPI)-anchored proteins many of which bind to plexin receptors. Invertebrate semaphorins in classes 1 and 2 (Sema1s and Sema2s) utilize PlexinA (PlexA) and PlexinB (PlexB) receptors, respectively. The co-receptor Off-track (OTK) functions with PlexA, and PlexA and PlexB might form heteromultimeric receptor complexes. In vertebrates, PlexinAs are receptors for Sema3s and Sema6s. In contrast to Sema3s, Sema6s do not require neuropilins (Npns) to direct plexinA binding. Ig-superfamily cell adhesion molecules (IgCAMs) function with Npns and plexinAs to mediate neuronal Sema3 functions. Unlike other Sema3s, Sema3E directly binds plexinD1. Sema3E-plexinD1-mediated axon attraction, but not repulsion, requires Npn-1. Sema4s associate with plexinBs and plexinB-ErbB2 interactions are required for certain Sema4 functions. HSPGs are required for Sema5A-mediated axon attraction and CSPGs switch Sema5A from an attractant to a repellent presumably by facilitating interactions with an unknown neuronal receptor. The GPI-linked semaphorin Sema7A binds neuronal integrin receptors. Recent evidence indicates that some membrane-spanning semaphorins might function both as ligands and receptors, and in addition might influence plexin receptors through cis interactions. (b) Sema3A binding to the Npn–plexinA complex promotes FARP2 dissociation from plexinA. Dissociated FARP2 activates Rac1, which facilitates Rnd1–plexinA associations and drives PIPKIγ661-mediated inhibition of integrin function. Active Rac1 also controls actin dynamics through a PAK-LIMK1-cofilin pathway. Rnd1–plexinA interactions stimulate plexinA RasGAP activity which suppresses R-Ras and inactivates PI3K-Akt signaling. Interestingly, Sema3s also regulate PI3K-Akt through PTEN. Downregulation of PI3K-Akt signaling leads to the inhibition of integrin-mediated adhesion, activation of myosin II (MyoII), and reduced phosphorylation of ERM and GSK-3β. Phosphorylation of CRMP2 by GSK-3β inactivates CRMP2 and relies on a Cdk5- and Fyn-dependent priming phopshorylation. CRMP2 regulates microtubule dynamics. (c) Sema4D–plexinB1 interactions promote phosphorylation of plexinB1 and ErbB2. Repulsive Sema4D–plexinB signaling involves four GTPases; Rnd1, R-Ras, Rho and Rac1. Sema4D–plexinB1 binding promotes Rnd1-dependent activation of the plexinB1 GAP domain and transient suppression of R-Ras activity. R-Ras inactivation promotes PI3K and Akt inactivation followed by GSK-3β activation and CRMP2 inactivation. In addition, plexinB1 associates with the RhoGEFs PDZ-RhoGEF and LARG to regulate Rho and ROCK activity and to influence cytoskeletal dynamics. Rac1–plexinB1 binding might sequester Rac1 away from PAK.
In the central nervous system (CNS), expression of prototypic axon guidance molecules continues beyond the initial phase of process outgrowth, growth cone navigation, and target innervation. Once the initial scaffold of neuronal connectivity has been established, a number of refinement processes continue to sculpt and transform the immature network into the complex and stereotypic pattern of connections observed in the mature brain and spinal cord. Refinement processes include dendritic elaboration, synaptogenesis, experience-dependent remodeling of synaptic connectivity, pruning of exuberant connections, and cell death. In the mature mammalian CNS, neuronal connectivity is not hardwired and many neurons retain a limited degree of structural plasticity throughout adulthood. Dendritic spines undergo morphologic changes and spine shape is regulated, at least in part, by long-lasting changes in synaptic strength [2]. While it has been speculated for some time that guidance molecules expressed in the more mature CNS participate in network refinement processes and neuronal plasticity, recent findings provide solid experimental evidence that members of the semaphorin family and their receptors are important players in several refinement processes that help transform an immature neural network into a fully functional adult nervous system.
Interestingly, altered expression and function of a growing number of semaphorin family members are associated with neurologic disorders and regenerative failure following CNS injury. Thus, a detailed understanding of semaphorin function at the molecular, cellular and systems levels may provide insight into how cytoskeletal remodeling is regulated during axonal outgrowth and growth cone steering in early development, during the subtle and more localized structural changes at synapses in the mature nervous system, and following neural injury or disease. Here, we highlight recent advances in our understanding of how semaphorins shape and influence nervous system structure and function in the juvenile and adult CNS. We discuss the significance of these findings and speculate about their importance for nervous system plasticity, regeneration, and disease.
6. Main text
Semaphorins and their receptors
Semaphorins are secreted and membrane-associated (type-1 transmembrane or glycosylphosphatidylinositol (GPI)-anchored) molecules defined by an amino-terminal semaphorin (sema) domain and, with the exception of viral semaphorins, a plexin-semaphorin-integrin (PSI) domain (Fig. 1). Semaphorins are further distinguished by distinct protein domains, including immunoglobulin-like (Ig), type-1 thrombospondin (TSR), and basic C-terminal domains (Semaphorin Nomenclature Committee, 1999; [3].
The most prominent semaphorin receptors are the plexins (Fig. 1). Plexins are large, phylogenetically conserved, type-1 transmembrane proteins that are subdivided into four classes (PlexinA-D) [4]. Plexins function as both ligand-binding and as signaling receptors for semaphorins. The Plexin ectodomain harbors a sema-like domain, and most interactions with semaphorins are mediated through association with this domain. Class 3 secreted semaphorins (Sema3s), except for Sema3E [1] require neuropilins as obligatory co-receptors to signal through class A plexins (plexinAs) (Fig. 1A). Neuropilin-1 and neuropilin-2 are type-1 transmembrane proteins with overlapping, yet distinct, binding preferences toward Sema3s. In addition to neuropilins, a number of co-receptors have been described that do not directly support semaphorin binding but are part of a functional semaphorin holoreceptor complex. Certain Ig-superfamily cell adhesion molecules (IgCAMs) serve as modulatory co-receptors for specific attractive or repulsive axon guidance events mediated by Sema3s [5]. Furthermore, heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate proteoglycans (CSPGs) bind to the TSRs of Sema5A and are required for Sema5A-mediated axon attraction or repulsion, respectively [6] (Fig. 1A). The only GPI-linked semaphorin, Sema7A, uses integrin receptors to exert its neurite growth promoting and branching effects [7] (Fig. 1A). In addition to integrins, Sema7A also binds plexinC1 [1]. At present, however, it is unclear whether Sema7A-plexinC1 interactions mediate any neuronal functions.
The repertoire of semaphorin-plexin signaling may be further expanded by the ability of plexins belonging to different subclasses to form heterodimers [8]. Accumulating evidence indicates that transmembrane semaphorins (subclasses 1, 4–6) not only serve as ligands but also as receptors, a process termed bi-directional signaling [9]. In Drosophila, Sema-1a, is both a PlexA ligand for forward signaling and a receptor for reverse signaling during olfactory system axon guidance events [10] [11]. In addition, transmembrane semaphorins and plexins may interact in cis, leading to altered downstream signaling events [12] (Fig. 1A). While the functional significance of plexin-plexin interactions and semaphorin-plexin cis associations needs further characterization, they provide important clues about the diversity of interactions in which semaphorins and their receptors may participate.
Semaphorin signaling
Great progress has been made in defining the intracellular signal transduction pathways that mediate the diverse neuronal functions of semaphorins. Plexins are canonical semaphorin receptors with a large cytoplasmic region that is highly conserved among family members. The Plexin cytoplasmic region contains GTPase-activating motifs that are thought to bind to and inactivate the monomeric G-protein R-Ras upon semaphorin binding to the Plexin ectodomain [13]. The subsequent decrease in active R-Ras, downregulates (phosphoinositide-3 kinase) PI3K activity and triggers the inhibition of integrin β1 signaling, leading to a decrease in growth cone adhesion and allowing collapse responses (Fig. 1B, C). In addition to R-Ras, other GTPases and proteins that regulate their activity, GTPase-exchange factors (GEFs), are known to associate with the cytoplasmic region of plexinAs and plexinBs in neurons (Fig. 1B, C). For example, upon stimulation of growth cones with Sema3A, the RacGEF (FERM, RhoGEF and pleckstrin domain protein 2) FARP2 dissociates from plexinA1 and activates Rac1. Active Rac1 facilitates the association of another GTPase, Rnd1, with plexinA1 thereby stimulating the plexinA1 GAP activity toward R-Ras. Interestingly, a recent study places another FARP family member, FARP1, downstream of Sema6A and plexinA4 in a subset of chick spinal motor neurons. FARP1 is necessary and sufficient to promote dendrite growth in motoneuron. The FARP1 Rho-GEF domain is required for the dendrite growth-promoting effects of Sema6A [14].
In addition, Rac1 has been shown to modulate actin dynamics through the p21-activated kinase (PAK), LIM kinase 1 (LIMK1) and the cofilin pathway, interactions that have been directly implicated in the regulation of neuronal growth [15,16] and synaptic plasticity [17]. The growing list of signaling components regulated by neural Semaphorins reveals new insights into how these cues exert their diverse functions but also poses new challenges. For example, it is far from clear which of the pathway(s) discussed above are most prominent or bare relevance to semaphorin signaling in vivo.
Cytoplasmic and receptor-type protein kinases constitute another important class of semaphorin signaling molecules. In the multi-component receptor complexes that mediate semaphorin function, plexins can alternatively associate with different kinase receptors to elicit divergent signaling pathways and functional responses [4]. For example, upon Sema4D binding, ErbB2 binds to and phosphorylates plexinB1, activating PDZ-RhoGEF and leukemia-associated Rho-GEF (LARG). PDZ-RhoGEF and LARG activate Rho, which then activates Rho kinase (ROCK) causing actin depolymerization and growth cone collapse [18] (Fig. 1C). Cytoplasmic kinases can either bind directly to the cytoplasmic portion of plexins (e.g. Fyn, cyclin-dependent kinase 5 (Cdk5)), or one of its co-receptors, or operate further downstream in the semaphorin signaling cascade (e.g. PI3K, GSK-3β, LIMK1). For example, Sema3A and Sema4D trigger the sequential inhibition of PI3K and Akt, activation of glycogen synthase kinase (GSK)3β and inactivation of collapsin response mediator protein (CRMP)-2 [19–21]. CRMP1-5 define a family of plexinA-interacting phosphoproteins that regulate microtubule dynamics. GSK-3β-mediated CRMP-2 phosphorylation is dependent on a Cdk5- and Fyn-dependent priming phosphorylation. Deactivation of CRMP2 by phosphorylation inhibits microtubule assembly and axon elongation [22] [23].
In addition, several proteins unrelated to kinases and GTPases with important function in neuronal semaphorin signaling have been identified, including Ran small GTPase binding protein (RanBPM), the flavoprotein monooxygenase MICAL, and the A kinase anchoring protein (AKAP) Nervy [4,24,25]. The precise role of these signaling components, where they are located in the semaphorin signaling network, and the cellular context in which they are most important, remains to be established.
Semaphorins regulate synapse development and physiology
In a recent study, members of the class 4 semaphorin family (Sema4s) were identified as important regulators of both glutamatergic and GABAergic synapse development [26]. In primary cultures of hippocampal neurons, RNAi knock-down of Sema4B, but not Sema4D, results in a reduced density of synaptotagmin-1 and GluR2 positive synaptic puncta and is accompanied by a decrease in both frequency and amplitude of AMPA receptor-mediated mEPSPs. Moreover, the number of PSD-95, but not synapsin-1-positive, puncta is selectively reduced (Fig. 2A). This suggests that Sema4B functions postsynaptically, possibly through a direct interaction with PSD-95 [27], to promote synapse maturation [26]. Interestingly, knock-down of Sema4B also causes a decrease in GABAergic synapse number as assessed by GABA-A immunolableing (Fig. 2B) [26]. This suggests that Sema4B may function in the assembly of excitatory and inhibitory postsynaptic specializations.
Figure 2. Class 3 and 4 semaphorins regulate synapse structure and physiology.
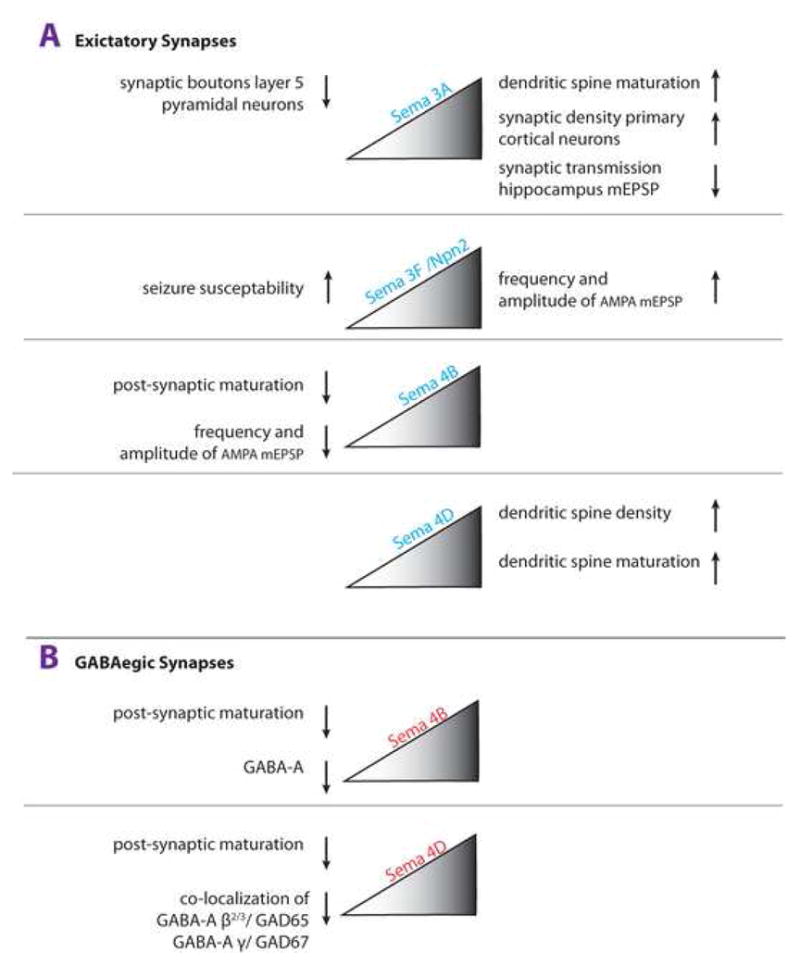
A growing number of semaphorins have been implicated in the regulation of synaptogenesis, dendritic spine density, dendritic spine maturation, and synaptic transmission. Effects of decreased semaphorin signaling are shown on the left and effects in the presence of increased semaphorin signaling are shown on the right. (a) Excitatory Synapses: In glutamatergic neurons, Sema3A increases the density of clusters of synapsin I and postsynaptic density-95 in vitro. Loss of Sema3A results in a dendritic spine phenotype of layer 5 cortical projection neurons in vivo. Physiological studies showed that exogenously applied Sema3F to acute hippocampal slices modulates basal synaptic transmission by increasing the frequency and amplitude of mESPCs in dentate granule cells and CA1 pyramidal neurons. Sema3A and Sema3F appear to have opposite effects on synaptic transmission in the hippocampus, as in the presence of Sema3A CA1 neurons are more depressed. In primary hippocampal neurons, RNAi knock-down of Sema4B, but not Sema4D, attenuates postsynaptic maturation, as assessed by a decrease in the density of synapsin 1/PSD-95 colocalized puncta and a decrease in AMPA-receptor containing synapses. Exogenous application of Sema4D promotes dendritic spine density and spine maturation in vitro. In primary hippocampal neurons, RNAi knock-down of Sema4B (but not Sema4D) results in a decrease of the frequency and amplitude of AMPA-receptor mediated mESPCs.
(b) GABAergic Synapses: In GABAergic neurons, RNAi knock-down of Sema4B or Sema4D attenuates postsynaptic maturation, as assessed by the density of GABA-A receptor puncta. In vivo, loss of Sema4D attenuates the development of GABAergic synapses in the hippocampus, as assessed by co-localization of GABA-A and GAD67 staining. In vivo, Sema3A and Sema3F influence GABAergic function indirectly by regulating tangential migration of cortical interneurons.
Unlike Sema4B, Sema4D RNAi knock-down has no effect on glutamatergic synapses but results in a significant decrease in GABAergic synapse density in hippocampal cultures. Consistent with the idea that Sema4D regulates GABAergic synapse density, Sema4D−/− mice show a ~20% decrease in GAD67 immunoreactivity in the hippocampus in vivo [26]. Supporting a role in synapse development, exogenous application of Sema4D to primary hippocampal neurons causes PlexinB1-dependent activation of the RhoA-ROCK pathway and an increase in spine density [28]. In the same culture system, Sema4D also promotes dendritic spine maturation as revealed by an increase in thin and mushroom shaped spines. Thus, in hippocampal cultures loss of Sema4D appears to only influence GABAergic synapses, but when applied exogenously, Sema4D regulates postsynaptic signaling of excitatory synapses. How does Sema4D bring about these different effects on synapses? Interestingly, Sema4D has the ability to positively or negatively regulate RhoA activity in vitro by either employing PDZ-RhoGEF/LARG or p190RhoGAP, a ubiquitous GAP in the brain [29,30]. Thus, Sema4D may be a bi-functional cue, capable of regulating dendritic spine density in a manner analogous to other semaphorins that exhibit bi-functionality with respect to axon guidance activity. The signaling mechanisms that either lead to activation or inhibition of the RhoA-ROCK pathway downstream of Sema4D in neurons, however, have not yet been defined and little is known about the mechanism of how Sema4D influences dendritic spine development, maturation, or synaptic transmission in the hippocampus in vivo.
Sema3A is a positive regulator of dendritic spine density. In primary cortical neurons, Sema3A increases the density of PSD95 and synaptophysin positive clusters [31]. Fewer synaptic bouton-like structures are present on layer 5 pyramidal neurons of sema3A−/− and fyn deficient mice. Recent work suggests that sema3A modulates dendritic spine density in a Cdk5-dependent manner in vitro and CRMP1-dependent manner in vivo [32].
Semaphorins as coordinators of structural and functional synaptic plasticity?
In the more mature CNS, semaphorins not only regulate synaptic structure but also influence synaptic transmission (Fig. 2). In acute hippocampal slices, Sema3F modulates fast excitatory synaptic transmission by increasing both frequency and amplitude of AMPA receptor-mediated miniature excitatory postsynaptic currents (mEPSCs) in dentate granule cells and CA1 pyramidal neurons [33]. Exogenous application of Sema3A, however, decreases the efficacy of synaptic transmission evoked in the CA1 region of hippocampal slices [34]. This suggests that Sema3A and Sema3F have opposing properties at the CA3-CA1 synapse. Commensurate with a synaptic function for class 3 semaphorins, Npn-1 and Npn-2 are found at synaptic sites. Npn-1 is localized presynaptically and Npn-2 is localized postsynaptically [33]. The molecular events that binding of Sema3s to neuropilins evoke to regulate synaptic transmission are still unknown. Because Sema3A intracellular trafficking and release is regulated in an activity-dependent manner [35], this cue may be part of a negative feedback loop that limits activity-dependent synaptic plasticity. The expression of several Sema3s is regulated in an activity-dependent manner, and as discussed below, loss of Sema3F or its receptor Npn-2 renders mice more susceptible to seizures [33,36].
Semaphorins regulate stereotypic pruning
An important event in the sculpting of neuronal connectivity is pruning of exuberant axonal projections. Examples of neuronal pruning include stereotypic pruning of the distal portion of the hippocampal mossy fiber infrapyramidal bundle (IPB) and the elimination of axon collateral branches extending from layer 5 pyramidal cell projections to specific subcortical targets. During early development, layer 5 cortical projection neurons from both motor and visual areas have subcortical connections that are nearly identical. Neurons from each cortical area send intermingling axon branches to the spinal cord, superior colliculus (SC), and inferior colliculus (IC) (Fig. 3). Stereotyped removal of upper motor neuron axon collaterals to the SC, IC, and removal of visual cortical axon collaterals to the IC and spinal cord results in the connectivity observed in the mature CNS (Fig 3A, B). Studies in Npn-2 mutant mice, initially focusing on hippocampal projections, identified an abnormal mossy fiber projection phenotype [37,38]. Upon closer examination, it became clear that aberrant mossy fiber projections observed along the outer rim of CA3 pyramidal neurons in Npn-2 and Plexin A3 null mice are the result of impaired pruning of the IPB [39,40]. More recent studies reveal that stereotypic pruning of corticospinal axon collaterals from visual area cortical neurons, but not collicular collaterals of motor area cortical neurons, are eliminated in a manner dependent upon Npn-2, plexinA3 and plexinA4 [41]. Based on previously reported interactions with the Npn-2/PlexinA3 complex and its expression in areas from which axon collaterals are removed, Sema3F is a strong candidate for providing the “punishing” signal that eventually leads to loss of exuberant axonal collaterals (Fig. 3). Along these lines, it is also of interest that in plexinA3 null mice, cell death of sensory neurons is decreased. Sema3A-plexinA3 signaling is not required for axonal pathfinding of sensory peripheral projections, however, it has been shown to trigger apoptosis in sensory neurons [42]. Thus, it is tempting to speculate that like Sema3A, Sema3F-mediated activation of PlexinA3 in axon collaterals from visual cortical projection neurons triggers local axonal death and pruning. These exciting observations may provide a new entry point for future studies aimed at the elucidation of the molecular events that regulate axonal health, stability, and degeneration.
Figure 3. Semaphorins regulate stereotypic pruning.
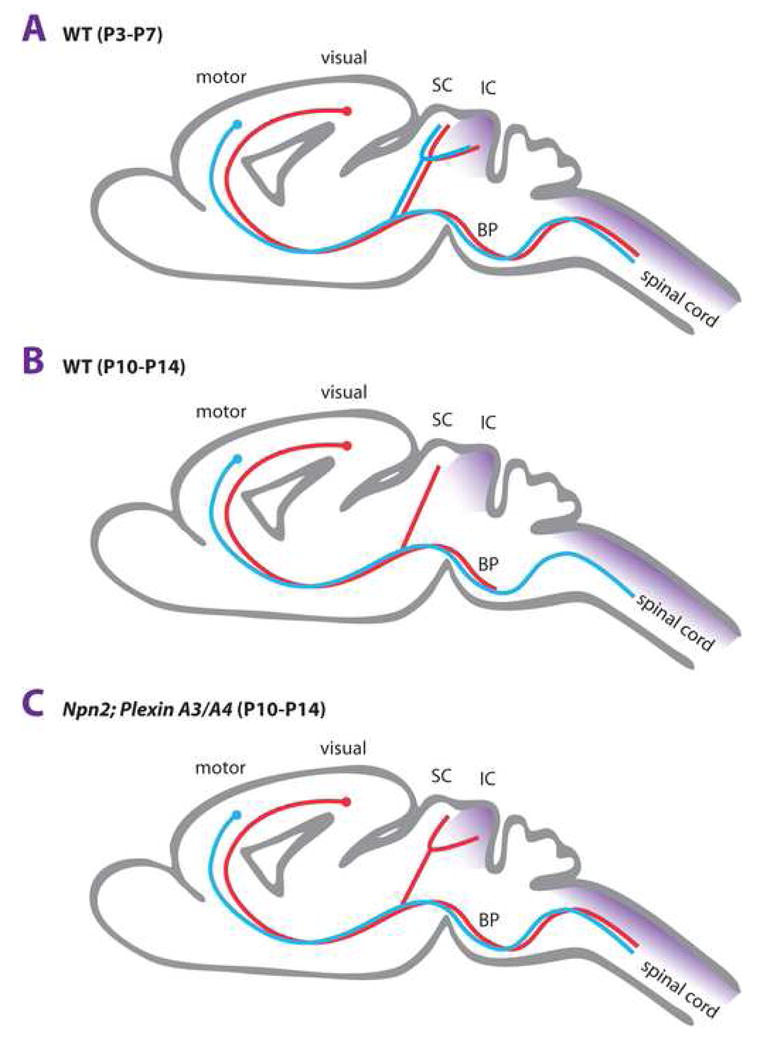
(a) In early mouse postnatal development (P3-P5), layer 5 projection neurons located in the visual cortex or motor cortex extend subcortical projections to similar target areas, including the superior (SC) and inferior colliculus (IC) in the diencephalon, and the dorsal spinal cord. Some of these targets are innervated by virtue of interstitial axon branching. Subsequent elimination (pruning) of specific subsets of axon branches is used to establish the mature innervation pattern. (b) In wild-type animals at P10-14, axon branches of upper motor neurons to the SC and IC and projections of visual cortical neurons to the IC and corticospinal tract (CST) are lost as a result of stereotypic pruning. As a consequence, visual cortical projections terminate in the SC and basilar pons (BP) and layer 5 motor projections give rise to the CST. (c) In Npn-2 null or PlexinA3/PlexinA4 double mutant mice pruning of visual cortical projections to the CST and IC is defective. Pruning of upper motorneuron axon collaterals to the SC and IC is normal and thus, independent of Npn-2 and PlexinA3/PlexinA4. The Npn-2 ligand Sema3F (purple) is expressed in the IC and dorsal spinal cord, and thus, a strong candidate for a ligand that signals pruning of exuberant visual cortical axon projections. The molecular mechanisms that regulate axon pruning of upper motorneuron projections to the SC and IC have not yet been defined.
Neural disease and injury
A growing number of studies implicate semaphorins, their receptors and cytosolic signaling components in psychiatric, neurodegenerative, and neurodevelopmental disorders. Furthermore, semaphorins are thought to contribute to the regenerative failure of severed CNS axons. It should be noted, however, that many of these observations are still correlative in nature and may reflect changes in semaphorin expression as a consequence of disease, rather than as causative role played by semaphorins in disease onset or progression.
A role for Semaphorins in CNS regeneration?
Following injury to the adult mammalian CNS, regenerative growth of severed axons is extremely limited. This lack of axon regeneration is often associated with significant functional deficits, and following spinal cord injury (SCI) may result in paralysis distal to the site of injury. An important factor contributing to the lack of CNS axon regeneration is the growth inhibitory environment near and at the site of injury. Both physical and chemical barriers prevent axons from traversing the lesion site and reconnecting with their original targets. Following SCI, meningeal fibroblasts invade the injury site and populate the center of the neural scar. Injured axons are unable to cross this fibrotic scar and develop swollen endings just proximal to, or within, the zone of reactive astrocytes (Fig. 4A). Sema3s are expressed by scar-associated meningeal cells and based on their growth inhibitory nature may block axon regeneration [43]. At the injury site, Sema3s are co-expressed with several other regeneration inhibitory proteins, including chondroitin sulfate proteoglycans (CSPGs). Intriguingly, Sema3s and CSPGs have been reported to physically associate in the extracellular matrix (ECM), and reduction of proteoglycan levels impedes Sema3A repulsion in vitro [43]. In addition, CSPGs have been shown to convert Sema5A-induced axon attraction of primary thalamic neurons into repulsion [6]. Therefore, it is tempting to speculate that CSPGs exert their inhibitory influences, at least in part, by presenting Sema3s to regenerating axons and/or by modulating semaphorin receptor function. Important future questions are to define how proteoglycans regulate semaphorin function, and to determine the physiological role of such interactions in steady-state (e.g. stabilization of existing networks), and following nervous system injury or disease.
Figure 4.

Role for semaphorins in regeneration failure and amyotrophic lateral sclerosis (ALS)? (a, b) Schematic representation of the injured rodent spinal cord. Following injury to the central nervous system (CNS), meningeal fibroblasts migrate into the injury site and form the fibrotic core of the neural scar. This fibrotic scar is surrounded by a region of reactive astrocytes and, more distantly, oligodendrocytes. Meningeal fibroblasts and oligodendrocytes in CNS injury sites have been shown to express semaphorins, their receptors and signaling molecules which may act to inhibit regrowth of severed axons. (b) Application of the fungus-derived inhibitor SM-216289 into the lesion site of rats with spinal cord transection injuries enhances anatomical and functional regeneration, presumably by preventing interactions between Sema3A and axonal neuropilin-1 (Npn-1). (c) Schematic representation of the neuromuscular junction (NMJ) of wild-type and G93A-hSOD1 mice. G93A-hSOD1 mice, a mouse model for ALS, display a marked increase of Sema3A expression in terminal Schwann cells (TSCs) at the NMJ. Intriguingly, this increase is limited to TSCs of fast-fatigable type IIb and IIx muscle fibers which are the first muscle subtype to be lost in ALS. Increased expression of Sema3A in TSCs may lead to de-adhesion or repulsion of motor axons away from the NMJ, eventually resulting in axonal denervation and motor neuron degeneration.
Several lines of evidence suggest that regenerating axons can respond to Sema3s. Injured adult neurons express functional Sema3 receptors, sprouting adult sensory axons are responsive to Sema3A repulsion, and knockdown of neuropilin-1 receptors reduces the neurite growth-inhibitory properties of human neural scar tissue in vitro [44,45]. Importantly, the fungus-derived compound SM-216289 inhibits Sema3A signaling and induces considerable anatomical and functional regeneration in spinal cord injured rats [46] (Fig. 4A, B). These observations support the idea that scar-derived Sema3s inhibit regenerative axon growth in vivo and thus may serve as therapeutic targets for improving functional regeneration. However, further work employing genetically manipulated mice and independent means to acutely block Sema3 function will be needed to more firmly establish the contribution of scar-associated Sema3s to regeneration failure in vivo. Membrane-associated semaphorins may also contribute to the regenerative failure of injured CNS axons: Sema4D (CD100), Sema6B, and Sema7A are found in myelin and are strongly upregulated by oligodendrocytes located near the injury site (Fig. 3A, B; [47–49]; R.J.P., unpublished observations). Future studies will undoubtedly analyze the expression and function of membrane-associated semaphorins at sites of CNS injury. A rapidly increasing body of evidence implicates semaphorins and their receptors in modulating physiological and injury induced neuronal plasticity. To what extent targeting of semaphorins results in improved anatomical and behavioral outcomes following CNS injury is the subject of current and future studies. Recent studies have also begun to explore the idea that semaphorins and other axon guidance molecules may not only negatively influence axon regeneration, but could also help to limit inappropriate axon sprouting or redirect regenerating axons to their synaptic targets once they have traversed the injury site in vivo [50].
An important but still underexplored field is the interaction between the nervous system and the immune system following neural injury or disease. At the molecular level, the convergence of the players implicated in nervous system and immune system function is intriguing. Molecular interactions of members of the semaphorin, plexin and neuropilin families, typically implicated in nervous system function, can also be found in cells that control the immune response. Thus, it is likely that there is extensive cross-talk between these two systems and it will be important to determine the extent and functional significance of immune system-nervous system cross-talk with a focus on semaphorin signaling following nervous system injury or disease. Another important function of semaphorins to be considered within the context of neural injury and disease is their ability to induce cell death. Growing evidence suggests that semaphorins function as death molecules for different neural cell populations, including neurons, neural progenitor cells, immature oligodendrocytes and activated microglia. Cell death is prevalent in injury and neurodegeneration. This suggests that an important activity of CNS injury- or disease-induced upregulation of semaphorin expression is in cell death.
Seizures and epilepsy
Epilepsies comprise a remarkably diverse collection of neurological disorders characterized by the periodic and unpredictable occurrence of seizures. One mechanism proposed to facilitate the development of seizures and epilepsy is the pathological reorganization of synaptic connections. These structural aberrations can result from insults during development and also pathological activity in the mature nervous system. Inherited forms of epilepsy account for about 20% of all epilepsies, suggesting that genetic factors contribute to the etiology of this disorder. A similar genetic predisposition for (injury-induced) epilepsy is observed in mice. FVB/NJ mice develop chronic epilepsy and show synaptic reorganization following kainic acid (KA) treatment, however C57Bl/6J mice do not. Examination of the expression of semaphorins and neuropilins in these two mouse strains revealed that genetic background has a profound effect on the expression of semaphorins during injury-induced epileptogenesis. For example, hippocampal Sema3F expression is down-regulated in KA treated FVB/NJ but not in C57Bl/6J mice [51]. Sema3F-Npn2 signaling regulates different aspects of hippocampal development. Both Sema3F−/− and Npn2−/− mice show defects in hippocampal circuitry and interneuron migration, and these mice are prone to seizures [33,36]. PlexinA3 mediates many of these Sema3F neuronal effects, but it is unknown whether plexin expression is regulated during epileptogenesis or whether plexinA3−/− mice develop seizures. In addition to Sema3F, expression of Sema3A and Sema3C is dysregulated in rat models of temporal lobe epilepsy and kainic acid (KA)-induced status epilepticus [52,53]. Additional studies are needed to determine the functional role of Sema3A and Sema3C in epileptogenesis and to assess whether the expression of other semaphorins and their receptors is regulated in epilepsy. Whether changes in semaphorin expression and function contribute to seizures or epilepsy in humans remains to be determined.
Neurodegenerative diseases
Although our understanding of neurodegenerative disease pathogenesis is incomplete, changes in neural connectivity and loss of synaptic contacts and activity are a unifying hallmark of these debilitating brain disorders. It has been suggested that aberrant semaphorin expression, or function, may result in altered neuronal connectivity or synaptic function associated with a number of developmental and degenerative neural disorders. Experimental evidence supporting such a role for semaphorins is currently strongest for Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS).
AD is a chronic, progressive, neurodegenerative disorder that predominantly affects the cerebral cortex and hippocampus, and is accompanied by selective cognitive impairments and behavioral disturbances. A hallmark of AD is the occurrence of intraneuronal structures known as neuritic plaques and neurofibrillary tangles. Compelling evidence for semaphorins playing a role in AD is provided by the isolation and characterization of a multiprotein complex from brain tissue of AD patients that contains phosphorylated microtubule-associated protein (MAP) 1B, Sema3A, CRMP-2, plexinA1 and -A2 [54]. Interestingly, AD brain tissue contains a hyperphosphorylated form of CRMP-2 that exhibits increased phosphorylation on both Ser522 and Thr509 residues [22]. Sequential phosphorylation of CRMP-2 at Ser522 and Thr509 by Cdk-5 and GSK-3β, respectively, reduces its interaction with tubulin and is required for repulsive Sema3A signaling [22,55] [23]. AD pathogenesis is associated with increased levels of aggregated beta-amyloid protein (Aβ), a fragment of APP that at low doses inhibits activity-dependent synaptic transmission and at higher doses is neurotoxic. Interestingly, Aβ reduces neurite length in vitro and regulates phosphorylation of CRMP-2 through a RhoA GTPase-dependent mechanism [56]. Thus, there may be cross-talk between Sema3A and Aβ signaling mechanisms. In AD patients, Aβ aggregation induces hyperphosphorylation of CRMP-2 which then may lead to changes in Sema3A elicited regulation of microtubule dynamics [20].
ALS is a devastating disease characterized by the progressive degeneration of motor neurons in brain and spinal cord. Motor neuron loss results in impaired voluntary movement, paralysis, and death by respiratory failure. As is thought about AD, it is plausible that defects in neuronal connectivity, axonal transport, or synaptic function could predispose individuals to ALS. Semaphorins are instrumental in regulating motor axon pathfinding during development [57] and have also been implicated in axonal transport and synaptic function [35]. Single-nucleotide polymorphisms (SNPs) in semaphorins and other genes encoding axon guidance molecules in ALS patients are of diagnostic value for disease susceptibility, onset and severity, suggesting the existence of semaphorin genetic risk factors [58]. Additional support for semaphorins in ALS stems from a study reporting that SOD1G93A transgenic mice, in comparison to control mice, display a marked increase of Sema3A expression in terminal Schwann cells (TSCs) at the neuromuscular junction (NMJ). Intriguingly, this increase is limited to TSCs of fast-fatigable type IIb and IIx muscle fibers [59]. This subtype of muscle fiber is characterized by its inability to stimulate nerve sprouting after injury and is the first muscle subtype that is lost in ALS. Therefore, it is possible that increased expression of Sema3A in TSCs leads to de-adhesion or repulsion of motor axons at the NMJ, eventually resulting in axonal denervation and motor neuron degeneration (Fig. 4C).
7. Conclusions and future directions
An overriding theme of the diverse functions exerted by semaphorins is their ability to regulate cytoskeletal dynamics and thereby influence cell morphology and function. This is most apparent in the nervous system, where complex neuronal structure is the cellular substrate of an elaborate and highly sophisticated network of connections. The importance of semaphorins in nervous system development and neural network assembly has been appreciated for some time, and considerable progress has been made in defining receptor systems and the associated downstream signaling pathways essential for semaphorins to exert their effects on the neuronal cytoskeleton. The rapidly expanding role for semaphorins in the more mature nervous system poses a number of new questions: How similar are the mechanisms and signaling pathways employed by semaphorins to regulate axon guidance events early in development, network refinement processes during early postnatal life, and neuronal plasticity in adulthood? What are the underlying molecular mechanisms that facilitate semaphorin-mediated axonal pruning and cell death? How do semaphorins influence synaptic maturation and dendritic spine shape? Are semaphorin-induced changes in synaptic transmission simply a reflection of altered spine structure, or do semaphorins have more direct means to regulate synaptic function? Do semaphorins regulate the presynaptic transmitter release machinery or postsynaptic surface expression levels and turnover of NMDA or AMPA receptors? An intriguing possibility is that semaphorins function as coordinators of both structural and functional neuronal plasticity. It appears likely that the cellular context in which specific semaphorins are encountered, e.g neuronal cell type, age, and growth cone or synapse, determines which downstream signaling pathways will predominate. Thus, a major challenge for future studies will be to define which of the semaphorin signaling pathways identified are most relevant for the execution of specific functions in vivo.
Accumulating evidence implicates semaphorins in the neuronal network changes that characterize brain disease. Aberrant semaphorin function and misexpression has been proposed to trigger neuronal structural changes observed during neurodegenerative disorders, but may also mediate more subtle alterations of neuronal structure such as those underlying neurodevelopmental and psychiatric disorders. Moreover, altered semaphorin expression is likely to have a marked impact on immune system function and cell death. While a growing number of semaphorins have been implicated in nervous system injury and disease, it should be noted that many of these observations are still correlative in nature and may simply reflect changes in semaphorin expression that are a consequence of disease rather than a direct causative link. Nevertheless, semaphorins and their associated receptors and signaling proteins are likely to represent valuable biomarkers for monitoring disease progression, and possibly therapeutic targets to regulate neuronal connectivity, cell death, and synaptic plasticity following nervous system injury and disease.
Acknowledgments
We apologize to our colleagues for not being able to cite many additional relevant publications owing to space restrictions. We thank Alex Kolodkin and Jon Terman for critical reading of the manuscript, and Jill Lynch for help with figures. Work in the laboratories of the authors is supported by grants from Netherlands Organization of Scientific Research (ZonMW VIDI 91776357; ZonMW TOP 91208017), the Human Frontier Science Program, and ABC Genomics Center Utrecht (R.J.P.) and NIH/NINDS NS047333 (R.J.G.).
Footnotes
9. Ethic in publishing: General statement
The authors declare no conflict of interest (RJP and RJG)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tran TS, Kolodkin AL, Bharadwaj R. Semaphorin regulation of cellular morphology. Annu Rev Cell Dev Biol. 2007;23:263–292. doi: 10.1146/annurev.cellbio.22.010605.093554. [DOI] [PubMed] [Google Scholar]
- 2.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- 3.Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7:211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franco M, Tamagnone L. Tyrosine phosphorylation in semaphorin signalling: shifting into overdrive. EMBO Rep. 2008;9:865–871. doi: 10.1038/embor.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falk J, Bechara A, Fiore R, Nawabi H, Zhou H, Hoyo-Becerra C, Bozon M, Rougon G, Grumet M, Puschel AW, et al. Dual functional activity of semaphorin 3B is required for positioning the anterior commissure. Neuron. 2005;48:63–75. doi: 10.1016/j.neuron.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 6.Kantor DB, Chivatakarn O, Peer KL, Oster SF, Inatani M, Hansen MJ, Flanagan JG, Yamaguchi Y, Sretavan DW, Giger RJ, et al. Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron. 2004;44:961–975. doi: 10.1016/j.neuron.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL. Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature. 2003;424:398–405. doi: 10.1038/nature01790. [DOI] [PubMed] [Google Scholar]
- 8.Ayoob JC, Terman JR, Kolodkin AL. Drosophila Plexin B is a Sema-2a receptor required for axon guidance. Development. 2006;133:2125–2135. doi: 10.1242/dev.02380. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y, Gunput RA, Pasterkamp RJ. Semaphorin signaling: progress made and promises ahead. Trends Biochem Sci. 2008;33:161–170. doi: 10.1016/j.tibs.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 10**.Komiyama T, Sweeney LB, Schuldiner O, Garcia KC, Luo L. Graded expression of semaphorin-1a cell-autonomously directs dendritic targeting of olfactory projection neurons. Cell. 2007;128:399–410. doi: 10.1016/j.cell.2006.12.028. Evidence for bi-directional signaling for the transmembrane Semaphorin-1a (Sema-1a) in Drosophila olfactory neurons. In vivo, Sema1a loss- and gain-of-function studies show that Sema1a acts cell-autonomously as a receptor to direct the dendritic targeting of projection neurons along the dorsolateral to ventromedial axis of the antennal lobe. [DOI] [PubMed] [Google Scholar]
- 11.Sweeney LB, Couto A, Chou YH, Berdnik D, Dickson BJ, Luo L, Komiyama T. Temporal target restriction of olfactory receptor neurons by Semaphorin-1a/PlexinA-mediated axon-axon interactions. Neuron. 2007;53:185–200. doi: 10.1016/j.neuron.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 12.Suto F, Tsuboi M, Kamiya H, Mizuno H, Kiyama Y, Komai S, Shimizu M, Sanbo M, Yagi T, Hiromi Y, et al. Interactions between plexin-A2, plexin-A4, and semaphorin 6A control lamina-restricted projection of hippocampal mossy fibers. Neuron. 2007;53:535–547. doi: 10.1016/j.neuron.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 13.Uesugi K, Oinuma I, Katoh H, Negishi M. Different requirement for Rnd GTPases of R-Ras GAP activity of plexin-C1 and plexin-D1. J Biol Chem. 2009 doi: 10.1074/jbc.M805213200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhuang B, Su YS, Sockanathan S. FARP1 promotes the dendritic growth of spinal motor neuron subtypes through transmembrane Semaphorin6A and PlexinA4 signaling. Neuron. 2009;61:359–372. doi: 10.1016/j.neuron.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oinuma I, Katoh H, Negishi M. Molecular dissection of the semaphorin 4D receptor plexin-B1-stimulated R-Ras GTPase-activating protein activity and neurite remodeling in hippocampal neurons. J Neurosci. 2004;24:11473–11480. doi: 10.1523/JNEUROSCI.3257-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toyofuku T, Yoshida J, Sugimoto T, Zhang H, Kumanogoh A, Hori M, Kikutani H. FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat Neurosci. 2005;8:1712–1719. doi: 10.1038/nn1596. [DOI] [PubMed] [Google Scholar]
- 17.Meng Y, Takahashi H, Meng J, Zhang Y, Lu G, Asrar S, Nakamura T, Jia Z. Regulation of ADF/cofilin phosphorylation and synaptic function by LIM-kinase. Neuropharmacology. 2004;47:746–754. doi: 10.1016/j.neuropharm.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 18.Swiercz JM, Kuner R, Offermanns S. Plexin-B1/RhoGEF-mediated RhoA activation involves the receptor tyrosine kinase ErbB-2. J Cell Biol. 2004;165:869–880. doi: 10.1083/jcb.200312094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chadborn NH, Ahmed AI, Holt MR, Prinjha R, Dunn GA, Jones GE, Eickholt BJ. PTEN couples Sema3A signalling to growth cone collapse. J Cell Sci. 2006;119:951–957. doi: 10.1242/jcs.02801. [DOI] [PubMed] [Google Scholar]
- 20.Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- 21.Ito Y, Oinuma I, Katoh H, Kaibuchi K, Negishi M. Sema4D/plexin-B1 activates GSK-3beta through R-Ras GAP activity, inducing growth cone collapse. EMBO Rep. 2006;7:704–709. doi: 10.1038/sj.embor.7400737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells. 2005;10:165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- 23.Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 24.Terman JR, Mao T, Pasterkamp RJ, Yu HH, Kolodkin AL. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell. 2002;109:887–900. doi: 10.1016/s0092-8674(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 25.Terman JR, Kolodkin AL. Nervy links protein kinase a to plexin-mediated semaphorin repulsion. Science. 2004;303:1204–1207. doi: 10.1126/science.1092121. [DOI] [PubMed] [Google Scholar]
- 26**.Paradis S, Harrar DB, Lin Y, Koon AC, Hauser JL, Griffith EC, Zhu L, Brass LF, Chen C, Greenberg ME. An RNAi-based approach identifies molecules required for glutamatergic and GABAergic synapse development. Neuron. 2007;53:217–232. doi: 10.1016/j.neuron.2006.12.012. A small scale RNAi based screen in primary hippocampal neurons to identify gene products that are important for synapse development and synapse function. This study also demonstrates the feasibility of an RNAi-based screen to identify and characterize molecules that regulate synapse formation and/or maintenance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burkhardt C, Muller M, Badde A, Garner CC, Gundelfinger ED, Puschel AW. Semaphorin 4B interacts with the post-synaptic density protein PSD-95/SAP90 and is recruited to synapses through a C-terminal PDZ-binding motif. FEBS Lett. 2005;579:3821–3828. doi: 10.1016/j.febslet.2005.05.079. [DOI] [PubMed] [Google Scholar]
- 28.Lin X, Ogiya M, Takahara M, Yamaguchi W, Furuyama T, Tanaka H, Tohyama M, Inagaki S. Sema4D-plexin-B1 implicated in regulation of dendritic spine density through RhoA/ROCK pathway. Neurosci Lett. 2007;428:1–6. doi: 10.1016/j.neulet.2007.09.045. [DOI] [PubMed] [Google Scholar]
- 29.Barberis D, Casazza A, Sordella R, Corso S, Artigiani S, Settleman J, Comoglio PM, Tamagnone L. p190 Rho-GTPase activating protein associates with plexins and it is required for semaphorin signalling. J Cell Sci. 2005;118:4689–4700. doi: 10.1242/jcs.02590. [DOI] [PubMed] [Google Scholar]
- 30.Oinuma I, Katoh H, Harada A, Negishi M. Direct interaction of Rnd1 with Plexin-B1 regulates PDZ-RhoGEF-mediated Rho activation by Plexin-B1 and induces cell contraction in COS-7 cells. J Biol Chem. 2003;278:25671–25677. doi: 10.1074/jbc.M303047200. [DOI] [PubMed] [Google Scholar]
- 31.Morita A, Yamashita N, Sasaki Y, Uchida Y, Nakajima O, Nakamura F, Yagi T, Taniguchi M, Usui H, Katoh-Semba R, et al. Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J Neurosci. 2006;26:2971–2980. doi: 10.1523/JNEUROSCI.5453-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Yamashita N, Morita A, Uchida Y, Nakamura F, Usui H, Ohshima T, Taniguchi M, Honnorat J, Thomasset N, Takei K, et al. Regulation of spine development by semaphorin3A through cyclin-dependent kinase 5 phosphorylation of collapsin response mediator protein 1. J Neurosci. 2007;27:12546–12554. doi: 10.1523/JNEUROSCI.3463-07.2007. The authors use a combination of in vitro and in vivo experiments to show that Sema3A regulates dendritic spine development by Cdk5-dependent phosphorylation of CRMP-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33**.Sahay A, Kim CH, Sepkuty JP, Cho E, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J Neurosci. 2005;25:3613–3620. doi: 10.1523/JNEUROSCI.5255-04.2005. First study to show that neuropilins are located synaptically and that the class 3 semaphorin Sema3F regulates synaptic physiology. In acute mouse hippocampal slices, exogenously applied recombinant Sema3F increases excitatory synaptic transmission in granule cells and CA1 pyramidal neurons. Indirect evidence is provided that Sema3F functions postsynaptically at the CA3-CA1 synapse. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouzioukh F, Daoudal G, Falk J, Debanne D, Rougon G, Castellani V. Semaphorin3A regulates synaptic function of differentiated hippocampal neurons. Eur J Neurosci. 2006;23:2247–2254. doi: 10.1111/j.1460-9568.2006.04783.x. [DOI] [PubMed] [Google Scholar]
- 35.de Wit J, Toonen RF, Verhaagen J, Verhage M. Vesicular trafficking of semaphorin 3A is activity-dependent and differs between axons and dendrites. Traffic. 2006;7:1060–1077. doi: 10.1111/j.1600-0854.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 36.Gant JC, Thibault O, Blalock EM, Yang J, Bachstetter A, Kotick J, Schauwecker PE, Hauser KF, Smith GM, Mervis R, et al. Decreased number of interneurons and increased seizures in neuropilin 2 deficient mice: implications for autism and epilepsy. Epilepsia. 2009;50:629–645. doi: 10.1111/j.1528-1167.2008.01725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H, Bagri A, Zupicich JA, Zou Y, Stoeckli E, Pleasure SJ, Lowenstein DH, Skarnes WC, Chedotal A, Tessier-Lavigne M. Neuropilin-2 regulates the development of selective cranial and sensory nerves and hippocampal mossy fiber projections. Neuron. 2000;25:43–56. doi: 10.1016/s0896-6273(00)80870-3. [DOI] [PubMed] [Google Scholar]
- 38.Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS, et al. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron. 2000;25:29–41. doi: 10.1016/s0896-6273(00)80869-7. [DOI] [PubMed] [Google Scholar]
- 39.Bagri A, Cheng HJ, Yaron A, Pleasure SJ, Tessier-Lavigne M. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell. 2003;113:285–299. doi: 10.1016/s0092-8674(03)00267-8. [DOI] [PubMed] [Google Scholar]
- 40.Liu XB, Low LK, Jones EG, Cheng HJ. Stereotyped axon pruning via plexin signaling is associated with synaptic complex elimination in the hippocampus. J Neurosci. 2005;25:9124–9134. doi: 10.1523/JNEUROSCI.2648-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Low LK, Liu XB, Faulkner RL, Coble J, Cheng HJ. Plexin signaling selectively regulates the stereotyped pruning of corticospinal axons from visual cortex. Proc Natl Acad Sci U S A. 2008;105:8136–8141. doi: 10.1073/pnas.0803849105. The authors use mouse genetics to show that Npn-2, PlexinA3, and PlexinA4 signaling is required for the proper stereotypic pruning of visual cortical projections to the spinal cord but not for pruning of motor cortical projections to the visual cortex in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Ben-Zvi A, Manor O, Schachner M, Yaron A, Tessier-Lavigne M, Behar O. The Semaphorin receptor PlexinA3 mediates neuronal apoptosis during dorsal root ganglia development. J Neurosci. 2008;28:12427–12432. doi: 10.1523/JNEUROSCI.3573-08.2008. The authors demonstrate that Sema3A mediated axonal pathfinding and Sema3A mediated cell death can be mechanistically dissociated at the receptor level. Sema3A-PlexinA4 signals guidance and Sema3A-PlexinA3 signals cell death in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasterkamp RJ, Verhaagen J. Semaphorins in axon regeneration: developmental guidance molecules gone wrong? Philos Trans R Soc Lond B Biol Sci. 2006;361:1499–1511. doi: 10.1098/rstb.2006.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Tannemaat MR, Korecka J, Ehlert EM, Mason MR, van Duinen SG, Boer GJ, Malessy MJ, Verhaagen J. Human neuroma contains increased levels of semaphorin 3A, which surrounds nerve fibers and reduces neurite extension in vitro. J Neurosci. 2007;27:14260–14264. doi: 10.1523/JNEUROSCI.4571-07.2007. The authors provide strong evidence that Sema3A contributes to the axon growth inhibitory nature of human neuroma tissue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang XQ, Heron P, Mashburn C, Smith GM. Targeting sensory axon regeneration in adult spinal cord. J Neurosci. 2007;27:6068–6078. doi: 10.1523/JNEUROSCI.1442-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46**.Kaneko S, Iwanami A, Nakamura M, Kishino A, Kikuchi K, Shibata S, Okano HJ, Ikegami T, Moriya A, Konishi O, et al. A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med. 2006;12:1380–1389. doi: 10.1038/nm1505. Elegant work showing that in vivo application of the fungus-derived Sema3A inhibitor SM-216289 facilitates anatomical and functional rat spinal cord regeneration. [DOI] [PubMed] [Google Scholar]
- 47.Moreau-Fauvarque C, Kumanogoh A, Camand E, Jaillard C, Barbin G, Boquet I, Love C, Jones EY, Kikutani H, Lubetzki C, et al. The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J Neurosci. 2003;23:9229–9239. doi: 10.1523/JNEUROSCI.23-27-09229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasterkamp RJ, Kolk SM, Hellemons AJ, Kolodkin AL. Expression patterns of semaphorin7A and plexinC1 during rat neural development suggest roles in axon guidance and neuronal migration. BMC Dev Biol. 2007;7:98. doi: 10.1186/1471-213X-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kury P, Abankwa D, Kruse F, Greiner-Petter R, Muller HW. Gene expression profiling reveals multiple novel intrinsic and extrinsic factors associated with axonal regeneration failure. Eur J Neurosci. 2004;19:32–42. doi: 10.1111/j.1460-9568.2004.03112.x. [DOI] [PubMed] [Google Scholar]
- 50*.Ziemba KS, Chaudhry N, Rabchevsky AG, Jin Y, Smith GM. Targeting axon growth from neuronal transplants along preformed guidance pathways in the adult CNS. J Neurosci. 2008;28:340–348. doi: 10.1523/JNEUROSCI.3819-07.2008. Original study showing that chemoattractive and chemorepulsive guidance cues can steer axons of transplanted neurons, indicating that in the adult mammalian CNS guidance cues can direct growth of regenerating axons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, Houk B, Shah J, Hauser KF, Luo Y, Smith G, Schauwecker E, Barnes GN. Genetic background regulates semaphorin gene expression and epileptogenesis in mouse brain after kainic acid status epilepticus. Neuroscience. 2005;131:853–869. doi: 10.1016/j.neuroscience.2004.09.064. [DOI] [PubMed] [Google Scholar]
- 52.Barnes G, Puranam RS, Luo Y, McNamara JO. Temporal specific patterns of semaphorin gene expression in rat brain after kainic acid-induced status epilepticus. Hippocampus. 2003;13:1–20. doi: 10.1002/hipo.10041. [DOI] [PubMed] [Google Scholar]
- 53.Holtmaat AJ, Gorter JA, De Wit J, Tolner EA, Spijker S, Giger RJ, Lopes da Silva FH, Verhaagen J. Transient downregulation of Sema3A mRNA in a rat model for temporal lobe epilepsy. A novel molecular event potentially contributing to mossy fiber sprouting. Exp Neurol. 2003;182:142–150. doi: 10.1016/s0014-4886(03)00035-9. [DOI] [PubMed] [Google Scholar]
- 54.Good PF, Alapat D, Hsu A, Chu C, Perl D, Wen X, Burstein DE, Kohtz DS. A role for semaphorin 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. J Neurochem. 2004;91:716–736. doi: 10.1111/j.1471-4159.2004.02766.x. [DOI] [PubMed] [Google Scholar]
- 55.Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T, Lim L, Hall C. Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J Neurosci. 2004;24:8994–9004. doi: 10.1523/JNEUROSCI.3184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petratos S, Li QX, George AJ, Hou X, Kerr ML, Unabia SE, Hatzinisiriou I, Maksel D, Aguilar MI, Small DH. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain. 2008;131:90–108. doi: 10.1093/brain/awm260. [DOI] [PubMed] [Google Scholar]
- 57.Huber AB, Kania A, Tran TS, Gu C, De Marco Garcia N, Lieberam I, Johnson D, Jessell TM, Ginty DD, Kolodkin AL. Distinct roles for secreted semaphorin signaling in spinal motor axon guidance. Neuron. 2005;48:949–964. doi: 10.1016/j.neuron.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Lesnick TG, Sorenson EJ, Ahlskog JE, Henley JR, Shehadeh L, Papapetropoulos S, Maraganore DM. Beyond Parkinson disease: amyotrophic lateral sclerosis and the axon guidance pathway. PLoS ONE. 2008;3:e1449. doi: 10.1371/journal.pone.0001449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Winter F, Vo T, Stam FJ, Wisman LA, Bar PR, Niclou SP, van Muiswinkel FL, Verhaagen J. The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol Cell Neurosci. 2006;32:102–117. doi: 10.1016/j.mcn.2006.03.002. [DOI] [PubMed] [Google Scholar]