

Abstract
Uracil DNA glycosylase (UNG) is the primary enzyme responsible for removing uracil residues from DNA. Although a substantial body of evidence suggests that DNA damage plays a role in cancer cell apoptosis, the underlying mechanisms are poorly understood. In particular, very little is known about the role of base excision repair of misincorporated uracil in cell survival. To test the hypothesis that the repair of DNA damage associated with uracil misincorporation is critical for cancer cell survival, we used small interfering RNA to target the human UNG gene. In a dose- and time-dependent manner, siRNA specifically inhibited UNG expression and modified expression of several genes, at both mRNA and protein levels. In LNCaP cells, p53, p21 and Bax protein levels increased, whereas Bcl2 levels decreased. In DU145 cells, p21 levels were elevated, although mutant p53 and Bax levels remained unchanged. In PC3 cells, UNG inhibition resulted in elevated p21 and Bax levels. In all three cell lines, UNG inhibition reduced cell proliferation, induced apoptosis, and increased cellular sensitivity to genotoxic stress. Furthermore, the in vitro cleavage experiment using uracil-containing double-stranded DNA as template has demonstrated that siRNA-mediated knockdown of UNG expression significantly reduced uracil-excising activity of UNG in human prostate cancer cells, which was associated with DNA damage analyzed by comet assay. Taken together, these findings indicate that RNA interference-directed targeting of UNG is a convenient, novel tool for studying the biological role of UNG and raises the potential of its application for prostate cancer therapy.
Keywords: Uracil DNA glycosylase, DNA repair, apoptosis, prostate cancer
INTRODUCTION
Current cancer therapies are based primarily on radiation and chemotherapy that damage DNA to selectively kill fast-growing tumor cells. This strategy is effective in the treatment of solid tumors at early stages, but suffers from the limitation that a small number of cancer cells commonly withstand therapy and accumulate additional mutations, leading to recurrence of therapy-resistant tumors. Improved understanding of the molecular and cellular biology of cancer may provide a broad range of possible chemo- and radio-sensitizing approaches.
A new and emerging concept designed to sensitize cancer cells to DNA-damaging agents (i.e., chemotherapy and/or radiation) is inhibition of various proteins in the DNA repair pathways (1, 2). Several lines of evidence suggest that DNA damage and repair mechanisms play critical roles in cancer cell survival (3, 4). The genome of cancer cells is more prone to DNA damage due to the high rate of metabolism associated with increased cellular proliferation (5). Recently, DNA-damaging agents have been shown to activate cell death programs in prostate cancer cells (6–10).
Uracilation of DNA represents a constant threat to the survival of many organisms including human cells. Uracil appears in DNA as a result of dUTP misincorporation or by spontaneous deamination of cytosine (11). Hydrolytic deamination of cytosine generates G:U mismatches that cause G:C to A:T transition mutations. To maintain genome integrity, most prokaryotic and eukaryotic cells rapidly eliminate uracil from DNA by the base excision repair (BER) pathway, which is initiated by uracil DNA glycosylase (UNG). The UNG enzyme hydrolyzes the N-glycosidic bond between the uracil residue and the deoxyribose sugar of the DNA backbone generating an apurinic-apyrimidinic (AP) site (12, 13). The AP site is then repaired by the classical BER system (14). The human UNG gene encodes two alternatively spliced isoforms, UNG1 and UNG2. These isoforms differ in their N-terminal sequences, resulting in one form that enters the nucleus (UNG2) whereas the other enters the mitochondria (UNG1) (15–17).
The exploitation of small interfering RNAs (siRNA) to selectively inhibit gene expression has been used previously to knockdown DNA repair activities and to examine their biological importance (18–20). In the present study, we have used RNA interference (RNAi) to knockdown UNG levels in human prostate cancer cell lines and to determine the relative impact of UNG inhibition on DNA damage, cell survival and genotoxic stress. Our results demonstrate that UNG function is essential to the survival of human prostate cancer cells and that knockdown of UNG results in a DNA damage response that induces apoptosis.
RESULTS
Efficient knockdown of UNG gene expression in human prostate cancer cells using RNAi
To examine the effect of direct inhibition of the expression of the UNG gene in prostate cancer cells, a pool of four individual siRNAs against the UNG gene (siUNG) was transfected into UNG-positive prostate cancer cell lines LNCaP, DU145 and PC3. To investigate the specificity of the RNAi system, we transfected prostate cells with the pool of four mismatch control siRNA (siMM). As shown in Fig. 1A, in each of the tested cell lines, the siUNG inhibited protein expression of the UNG gene in a dose-dependent manner, whereas the siMM had no appreciable effect, indicating the specific effect of this set of siRNA in knocking down the expression of the UNG gene in human prostate cancer cells. We also observed the time-dependent nature of this inhibition, with a significant effect being noted after 24 h (Fig. 1B). Next, we performed RT-PCR analysis to examine the expression of UNG mRNA in prostate cancer cells transfected with siUNG or controls. Consistent with the results of the immunoblotting, UNG mRNA was inhibited by siUNG in a dose-, time-, and sequence-dependent manner in all three prostate cancer cells (Figs. 1C and 1D).
Figure 1. UNG siRNA inhibits expression of the UNG gene in human prostate cancer cells.
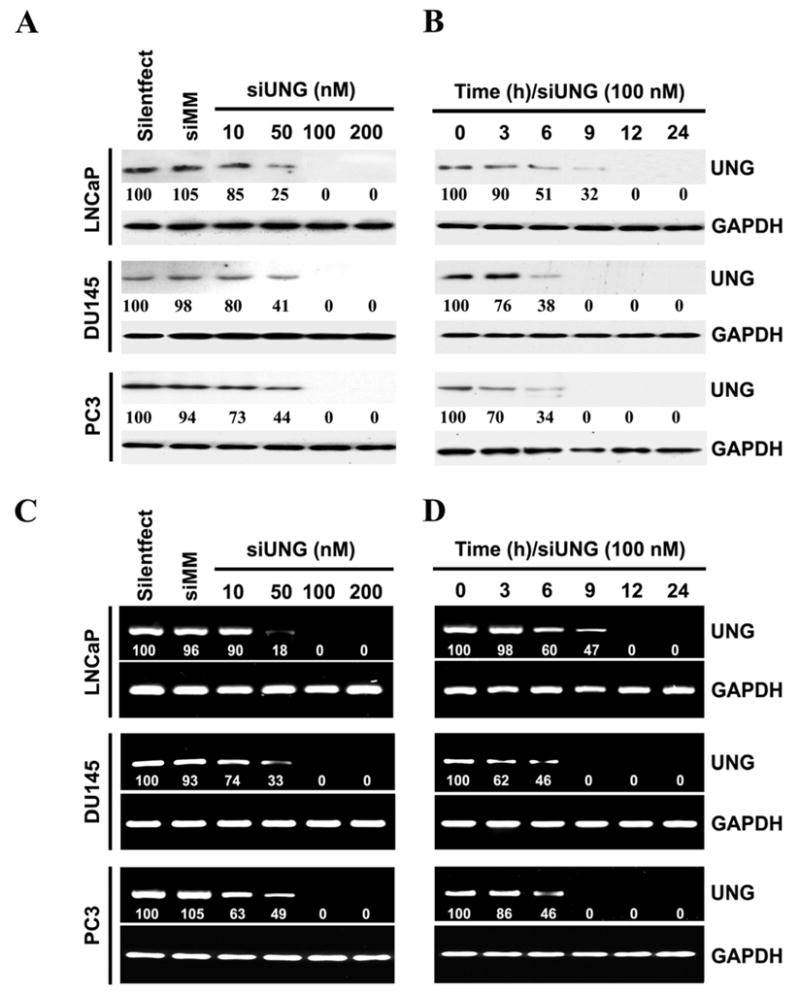
(A) Four individual siRNAs (siUNG) against the UNG gene were combined into one pool, and the human prostate cancer cell lines LNCaP, DU145 and PC3 were transfected with various amounts of the pooled siUNG as indicated. The mismatch control siMM (200 nM) was used as a negative control. Twenty-four hours after transfection, total cell extracts were isolated from controls and siUNG-transfected LNCaP, DU145 and PC3 cells, and the immunoblot analysis was performed using UNG and GAPDH antibodies. GAPDH was utilized as a loading control. The number under each band is expressed as a percentage of Silentfect control, normalized by the corresponding GAPDH level.
(B) Human prostate cancer cell lines LNCaP, DU145 and PC3 were transfected with siUNG (100 nM) and the cells were collected at different time points after transfection as indicated and immunoblot analysis for UNG and GAPDH was performed. The number under each band is expressed as a percentage of Silentfect control, normalized by the corresponding GAPDH level.
(C) Human prostate cancer cell lines LNCaP, DU145 and PC3 were transfected with siUNG (10–200 nM) or siMM (200 nM) as in A. Twenty-four hours after transfection, cells were collected, and the mRNA levels of UNG and GAPDH were examined by RT-PCR. Note that UNG represents the expression of both UNG1 and UNG2. The number under each band is expressed as a percentage of Silentfect control, normalized by the corresponding GAPDH level..
(D) Human prostate cancer cell lines LNCaP, DU145 and PC3were transfected with siUNG (100 nM) and the cells were collected at different time points after transfection as indicated and RT-PCR analysis for UNG and GAPDH was performed. The number under each band is expressed as a percentage of Silentfect control, normalized by the corresponding GAPDH level.
Knockdown of UNG by RNAi suppresses uracil excision activity and induces DNA damage
To analyze the enzymatic activity of the UNG protein in the cell extracts of siRNA transfectants, we used an oligonucleotide cleavage assay. A 34-base pair oligonucleotide with uracil at the 16th nucleotide was incubated with purified uracil DNA glycosylase (control) or extracts from siUNG- and siMM-transfected cells. Fig. 2A shows the intact DNA and cleavage products from each of these reactions. Equal amounts of protein were used in each comparison between siMM-and siUNG-transfected cells. The extracts from siMM-transfected cells show significant enzyme activity levels. In contrast, there was barely detectable enzyme activity levels in siUNG-transfected cells. The weak residual activity observed was probably due to the presence of other cellular UDG activities, such as SMUG1, that are not inhibited by siUNG. The prostate cancer cell lines LNCaP, DU145 and PC3 expressed the SMUG1 protein (Supplementary Figure S1). None of the extracts or purified uracil DNA glycosylase was able to cleave an identical oligonucleotide duplex with normal cytosine at position 16 (data not shown). Based on this assay, we conclude that siRNA directed against UNG specifically blocks UNG cleavage activity in all three prostate cancer cell lines. To assess if UNG contributes to protecting cellular DNA, we measured the induction of DNA fragmentation by exploiting the alkaline comet assay. This assay allows for the detection of both single- and double-stranded DNA breaks, and, therefore, is a highly sensitive method to directly examine the amount of DNA damage incurred in a single cell. Prostate cancer cells transfected with either siMM or siUNG was analyzed for comet expression. After transfection with 200 nM UNG siRNA, the comet tail moment significantly increased in LNCaP, DU145 and PC3 cells. In contrast, the mismatch control siMM had no or minimal effects in all three prostate cancer cell lines, regardless of p53 status (Figs. 2B and 2C). These results suggest that siRNA-mediated inhibition of UNG expression and activity induces DNA damage in prostate cancer cells with various p53 statuses, LNCaP (p53 wild-type), DU145 (p53 mutant) and PC3 (p53 null).
Figure 2. Knockdown of UNG by RNAi suppress uracil excision activity and induces DNA damage.

(A) The UNG enzyme activities were analyzed by uracil cleavage assay using the siMM- and siUNG-transfected prostate cell lines LNCaP, DU145 and PC3. Cells were transfected with siUNG (200 nM) or siMM (200 nM) for 24 h. Cell extracts were incubated with a 34-mer double-stranded oligonucleotide containing uracil in the 5′-32P-end labeled strand. Purified UNG and BSA served as positive (+) and negative (−) controls, respectively. Reaction products were run on a denaturing PAGE and detected by autoradiograph. The upper band represents the non-cleaved DNA probe and the lower band represents the cleaved probe.
(B) Human prostate cancer cell lines LNCaP, DU145 and PC3 were transfected with siUNG (200 nM) or siMM (200 nM) for 24 h and then analyzed for fragmented DNA by comet assay under alkaline conditions. Representative pictures are shown.
(C) Quantitation of damaged DNA in siMM- and siUNG-transfected LNCaP, DU145 and PC3 cells as measured by the comet assay image analysis. The extent of the DNA damage induced by UNG knockdown is quantified by determining the tail moment. The tail moment of cells transfected with siMM is expressed as 100%. Results represent the average of three independent experiments with 100 cells (nuclei) analyzed per experiment; bars, ±SD. Significant difference from control (siMM) is represented by asterisks (*, P < 0.01).
Knockdown of UNG by RNAi-modified pro-arrest and pro-apoptotic gene expression in prostate cancer cells
Because p53 is a major modulator of apoptosis and BER has been shown to inhibit p53-mediated apoptosis (21–23), we decided to examine the status of pro-arrest and pro-apoptotic genes in UNG knockdown cell lines with different p53 status.
LNCaP cells
The UNG knockdown resulted in p53 protein elevation in a dose- and time-dependent fashion (Figs. 3A and 3B). However, p53 mRNA levels did not change (Figs. 3C and 3D). The protein and mRNA levels of p21 and Bax were also elevated whereas Bcl2 protein and mRNA levels decreased (Fig. 3).
Figure 3. Knockdown of UNG by RNAi-modified pro-arrest and pro-apoptotic gene expression in prostate cancer cells.

Protein (A and B) and mRNA (C and D) levels of various genes in prostate cancer cells transfected with siUNG or controls. Densitometric analysis of protein or mRNA levels were normalized to corresponding GAPDH level and expressed as percentage of Silentfect control.
(A) The cells were transfected with siUNG (10–200 nM) or siMM (200 nM) for 24 h. Proteins were analyzed by immunoblotting.
(B) Cells were transfected with siUNG (100 nM) for various time periods. Proteins were analyzed by immunoblotting.
(C) Cells were transfected with siUNG (10–200 nM) or siMM (200 nM) for 24 h. Target mRNAs were analyzed by RT-PCR.
(D) Cells were transfected with siUNG (100 nM) for various time periods. Target mRNAs were analyzed by RT-PCR.
DU145 cells
No changes in mutant p53 protein and mRNA were observed (Fig. 3). Interestingly, both p21 protein and mRNA levels were elevated, which is independent of p53. DU145 cells are Bcl2 null and express mutant Bax, which was unchanged after UNG knockdown.
PC3 cells
After UNG knockdown by siRNA, p21 mRNA and protein levels were significantly elevated (Fig. 3). Bax mRNA and protein levels were also elevated. No changes in Bcl2 protein or mRNA levels were found.
Knockdown of UNG resulted in induction of apoptosis, inhibition of cell proliferation, and genotoxic stress sensitization in prostate cancer cells regardless of p53 status
Induction of apoptosis
In a dose-dependent manner, UNG siRNA induced apoptosis in all three prostate cancer cell lines, regardless of p53 status (Fig. 4). After treatment with 200 nM UNG siRNA, the apoptotic index increased by 300% in LNCaP cells (Fig. 4A), 278% in DU145 cells (Fig. 4B), and 451% in PC3 cells (Fig. 4C). The mismatch control siMM had no or minimal effect. We next examined the effect of UNG knockdown on cellular response to various stress conditions including oxidative stress and DNA damage. Pretreatment with 50 nM UNG siRNA, but not the mismatch control siMM, sensitized all of the three cell lines to the genotoxic stress-inducing agents H2O2 (Figs. 4D–F) and doxorubicin (Figs. 4G–I).
Figure 4. Effects of siUNG alone or in combination with H2O2 or doxorubicin on apoptosis in prostate cancer cells.

The cells were transfected with siUNG or siMM at various concentrations for 12 h (A, LNCaP) or 24 h (B, DU145; C, PC3). In combination treatment with H2O2, the cells were transfected with 50 nM siUNG or siMM for 12 h (D, LNCaP) or 24 h (E, DU145; F, PC3) and then exposed to H2O2 for an additional 12 h (D) or 24 h (E and F). In combination treatment with doxorubicin, the cells were transfected with 50 nM siUNG or siMM for 12 h (G, LNCaP) or 24 h (H, DU145; I, PC3) and then exposed to doxorubicin for an additional 12 h (G) or 36 h (H and I). Cells that stained positive for annexin V-FITC (early apoptosis) and positive for FITC and propidium iodide (late apoptosis) were counted. Relative levels of apoptotic indices were expressed as percentage of Silentfect control. (D–I) Filled circles, Silentfect control; filled triangles, siUNG 50 nM; empty circles, siMM 50 nM.
Inhibition of cell proliferation
In a dose-dependent manner, UNG siRNA inhibited proliferation in all three prostate cancer cell lines, regardless of p53 status (Fig. 5). After treatment with 200 nM UNG siRNA, the proliferation index decreased by 65% in LNCaP cells (Fig. 5A), 60% in DU145 cells (Fig. 5B), and 68% in PC3 cells (Fig. 5C). The mismatch control siMM had no or minimal effect. Pretreatment with 50 nM UNG siRNA, but not the mismatch control siMM, sensitized all of the three cell lines to H2O2 (Figs. 5D–F) and doxorubicin (Figs. 5G–I).
Figure 5. Effects of siUNG on the proliferation of prostate cancer cells as analyzed by BrdUrd incorporation.

Using the same treatment protocol as for Fig. 4, LNCaP (A, D, and G), DU145 (B, E, and H), and PC3 (C, F, and I) cells were transfected with siUNG alone (A–C) or in combination with either H2O2 (D–F) or doxorubicin (G–I). Relative levels of BrdUrd incorporation were expressed as percentage of Silentfect control. (D–I) Filled circles, Silentfect control; filled triangles, siUNG 50 nM; empty circles, siMM 50 nM.
DISCUSSION
Mammalian cells have evolved a diverse defense network to safeguard genomic integrity and to prevent permanent genetic damage induced by endogenous and exogenous mutagens. Cells repair damage done to the DNA by a variety of repair mechanisms, each specific to the type of DNA damage (11). One of the repair mechanisms is the BER pathway that repairs lesions of DNA that involve base modification as well as damage by reactive oxygen species. BER involves a DNA glycosylase that cleaves the damaged base by hydrolysis of the glycosidic bond, producing an abasic site. The abasic site generated is then removed by AP endonuclease and the gap is filled by DNA polymerase and then ligated by DNA ligase (12–14). The first enzyme involved in the BER pathway differs depending upon the lesion introduced in the DNA. UNG is the primary enzyme responsible for removing uracil residues from DNA (15–17).
In the present study, we have demonstrated at least five noteworthy results. First, the siUNG specifically knocked down UNG expression as demonstrated at both the mRNA and protein levels in a dose-, time- and sequence-dependent manner. In LNCaP, DU145 and PC3 cells, we achieved a significant decrease of UNG expression after transfection with siUNG. Second, UNG inhibition resulted in decreased uracil excision activity and enhanced DNA damage in a sequence-specific manner. Third, in a dose- and time-dependent manner, siUNG modified the expression of several genes with varying profiles in cells with different p53 status. In the LNCaP cells (p53 wild-type), UNG inhibition resulted in significant elevation of p53, p21, and Bax protein levels and reduction of Bcl2 protein levels. In DU145 cells (p53 mutant), UNG inhibition also resulted in elevation of p21 at both the mRNA and protein levels. In PC3 cells (p53 null), significant elevation of p21 and Bax protein levels was observed. Fourth, UNG inhibition resulted in enhanced apoptosis and decreased cell proliferation in a dose-dependent, sequence-specific manner, regardless of p53 status. Fifth, UNG inhibition resulted in genotoxic sensitization in prostate cell lines, regardless of p53 status. All these data suggest that UNG activity is essential for the survival of these human prostate cancer cell lines.
A considerable body of evidence exists implicating extensive uracil misincorporation and its associated misrepair as contributing mechanisms of cytotoxicity after UNG inhibition (18, 21, 24, 25). It has been demonstrated that overexpression of DNA glycosylase protects certain cell lines from the resultant DNA damage and toxicity (26). RNAi is a strong tool for silencing the function of specific genes (18–20). In the present study, we used siRNA to knockdown UNG in a dose-, time- and sequence-dependent manner. We found that knockdown of UNG induces apoptosis in LNCaP, DU145 and PC3 cells. This was accompanied by reduced cell proliferation and increased strand breakage. It is important to note that these results were achieved in all three prostate cancer cell lines with different p53 status. Taken together, our results clearly show that a novel UNG blockade system using RNAi is a valid, potentially therapeutic goal.
Oxidative damage to DNA-cytosine includes isodialuric acid, alloxan and uracil, which are all substrates for the UNG gene (18, 27). Several studies have demonstrated that UNG-deficient mice and cell lines have increased sensitivity to DNA damage and oxidative stress (18, 25, 27, 28). Doxorubicin is a commonly used anticancer drug which causes DNA damage and sensitizes cancer cells to apoptosis (29). In the present study, UNG knockdown by siRNA showed greater sensitivity to H2O2, demonstrating that UNG activity can influence cell fate decisions in response to genotoxic stress. Dizdaroglu and colleagues observed that human UNG is involved in the repair of major modifications caused by oxidative damage to DNA (27). These data support our finding that UNG plays a role in response to genotoxic stress induced by H2O2.
It is well accepted that p53 plays a pivotal role in the maintenance of genomic stability. P53 is believed to function as part of a stress-response pathway, which determines the fate of cells. The options include cell survival, which consists of cell cycle delay accompanied by repair of DNA damage (30–32) or cell suicide through apoptosis (33, 34). A previous study has shown that wild-type p53 is directly involved in BER activity, by using an in vitro experimental assay. Nuclear extracts with wild-type p53 showed an enhanced BER activity in comparison to nuclear extracts expressing the mutant conformation of p53 (35). However, there have been reports of p53 independent activity of BER in mice and cell line models (36, 37). One possibility is that the in vitro assays do not completely reconsititute BER, and suggests that more complex mechanisms influence the p53-BER axis. In the present study, we show that UNG-mediated BER inhibition resulted in enhanced apoptosis, decreased cell proliferation and genotoxic-stress sensitization in a dose-dependent, sequence-specific manner, regardless of p53 status.
The present study also demonstrated changes in expression of multiple genes in UNG knockdown cells. The expression of p21, Bax and Bcl2 were analyzed at both protein and mRNA levels, regardless of p53 status. Although WAF1 (p21) gene is a reporter gene of p53 activity, several reports demonstrate that p53-independent p21 induction is mediated by various stress conditions including DNA damage and oxidative stress (29, 38, 39). Our results from the present study support the concepts that induction of p21 is both p53-dependent and p53-independent and that UNG inhibition affects both p21 protein and mRNA levels. The expression of Bax may also be controlled by p53-dependent (40) or p53-independent mechanisms (30, 41). The ratio of Bcl2/Bax is important in determining which cells undergo apoptosis and which survive after DNA damage (42). In the present study, we demonstrated Bax induction in both LNCaP (p53 wild-type) and PC3 (p53 null) cell lines, after UNG inhibition. In LNCaP cells, UNG inhibition resulted in simultaneous Bcl2 reduction and Bax elevation, explaining partly the significant apoptosis and chemosensitization effects of the RNAi against UNG.
In summary, we have shown that knockdown of UNG expression induces apoptosis in human prostate cancer cells, suggesting the important role of uracil DNA glycosylase activity in malignant cell survival. Furthermore, we have demonstrated that UNG knockdown initiated DNA damage and influenced cell fate through modulation of pro-arrest and pro-apoptotic genes. Additional studies are needed to further elucidate the mechanisms through which UNG suppression leads to critical cellular decisions as well as the proteins and pathways involved in those processes.
MATERIALS AND METHODS
Cell lines and culture
The prostate cancer cell lines LNCaP, DU145, and PC3 were obtained from the American Type Culture Collection (Manassas, VA). LNCaP cells were cultured in RPMI medium supplemented with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, and 1.0 mM sodium pyruvate (Invitrogen, Carlsbad, CA). DU145 and PC3 cells were cultured in advanced Dulbecco’s modified Eagle’s medium. Both media formulations contained 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and 5% penicillin/streptomycin. Cells were transfected with siRNA in the presence of Silentfect (Bio-Rad, Hercules, CA) and 1% FBS for various times before analysis of mRNA and protein levels, apoptosis, and cell proliferation. In combination treatments, siRNA-transfected cells were incubated for an additional 12–36 h after addition of the DNA-damaging agents doxorubicin (Sigma, St Louis, MO) or hydrogen peroxide (H2O2) (Sigma, St Louis, MO) in a 37°C incubator with a 5% CO2 humidified atmosphere. The chosen time at which doxorubicin or hydrogen peroxide was added to the transfected prostate cell lines LNCaP, DU145 and PC3 was based on preliminary drug sensitivity experiments in which different alternative times were tested in combination studies.
Small interfering RNA (siRNA)
Sets of four siRNAs targeting human UNG and its 4-base mismatch control were synthesized and annealed. The siRNA sequences that we used are as follows: UNG siRNA no. 1 (siUNG1), 5′-CAU CAA GCC AAC UCU CAU A-3′; UNG siRNA no. 2 (siUNG2), 5′-CUG UGA GCU UUA UCA GAU A-3′; UNG siRNA no. 3 (siUNG3), 5′CCU UGA UCU UGU UAG CAA U-3′; UNG siRNA no. 4 (siUNG4), 5′-GGG ACA GGA UCC AUA UCA U-3′; UNG mismatch siRNA no. 1 (siMM1), 5′-CAA CAA GCC UUC UCA CAU A-3′; UNG mismatch siRNA no. 2 (siMM2), 5′-CUG AGA GGA UUA UCU GAU A-3′; UNG mismatch siRNA no. 3 (siMM3), 5′-CCU AGA UCU ACU UAC CAA U-3′; UNG mismatch siRNA no. 4 (siMM4), 5′-GGC ACA GGA AGC AUA ACA U-3′. Transfection of siRNA pool (siUNG) and mismatch control pool (siMM) in six-well plates was done using Silentfect (Bio-Rad) according to the manufacturer’s instructions.
Uracil DNA glycosylase activity assay
Cell extracts were prepared from prostate cell lines using the PARIS kit (Ambion, Foster City, CA). UNG activity was assayed as described previously (43). Briefly, an oligonucleotide sequence containing a uracil residue at single position (5′-AGC TTG GCT GCA GGT UGA CGG ATC CCC GGG AAT T-3′) was 5′-end labeled with T4 polynucleotide kinase and 32P, then annealed to a 5-fold excess of the complementary strand (5′-AAT TCC CGG GGA TCC GTC TAC CTG CAG CCA AGC T-3′). UNG assay was performed using 10 μg of cell extract in 1X UNG buffer (20 mM Tris-HCl pH8.0, 1 mM EDTA, 1 mM DTT) and 4 pmoles of labeled oligos. The reaction was carried out at 30°C for 45 min. Following incubation, NaOH (0.1 M) was added and the samples were heated at 37°C for 15 min in order to convert the abasic sites created by DNA glycosylases into DNA strand breaks. Formamide containing loading buffer was added, then samples were heated to 65°C for 5 min to stop the reaction. The DNA fragments were then separated on a 15% denaturing polyacrylamide gel. The gel was autoradiographed after electrophoresis to visualize the bands. Ten units of pure UNG (NEB, Ipswich, MA) were used as a positive control and 1 μg of BSA was used as a negative control.
Comet assay
DNA damage was assessed using the alkaline single-cell gel electrophoresis (SCGE) “comet assay” method (8). The comet assay has been shown to be a sensitive and reliable measure of DNA strand breaks associated with incomplete excision repair sites and alkali-labile sites. For comet assay, transfected cells were scraped from plates and pelleted, followed by re-suspension in PBS (10,000 cells/mL). 50 μL of the cell suspension was then mixed with 500 μL of 0.5% low melting point agarose at 37°C. 75 μL of the cell/agarose mixture was transferred onto glass slides. Slides were then immersed in pre-chilled lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10.0, 1% Triton X-100, and 10% Me2SO) for 1 h, followed by equilibration in Tris borate-EDTA (TBE) buffer for 30 min. Slides were electrophoresed in TBE at 1.5 volt/cm for 5 min and stained with Vistra Green (Amersham Biosciences, Pittsburgh, PA). Images were visualized under a fluorescence microscope and captured with a CCD camera. Nuclei with damaged DNA have the appearance of a comet with a bright head and a tail, whereas nuclei with undamaged DNA appear round with no tail. Olive tail moment is one of the parameters that are commonly measured with the comet assay. It represents the product of the amount of DNA in the tail (expressed as a percentage of the total DNA) and the distance between the centers of mass of the head and tail regions as the measure of DNA damage. The comet tail moment was determined using the comet assay image analysis software (Komet 4.0 Kinetic Imaging Ltd.), and the mean ± SD of the olive tail moment was obtained from 100 cells of each treatment group.
BrdUrd cell proliferation assay
BrdUrd incorporation into cells was accomplished using a BrdUrd cell proliferation assay kit from Oncogene (San Diego, CA). Cells were seeded in 96-well plates (5×103 to 1×104 cells per well) and transfected with siRNA for 24 h (12 h for LNCaP). In combination treatments, cells were then exposed to doxorubicin for 36 h (12 h for LNCaP) or H2O2 for 24 h (12 h for LNCaP). BrdUrd was added to the medium 10 h before treatment termination. The levels of BrdUrd incorporated into cells were quantified by anti-BrdUrd antibody, measuring absorbance at dual wavelengths of 450/540 nm with a microplate reader.
Detection of apoptosis
Following a treatment protocol similar to the one described above, cells in early and late stages of apoptosis were detected with an annexin V-FITC apoptosis detection kit from Bio Vision (Mountain View, CA) as described previously (44).
Immunoblot analysis
Cells were lysed in RIPA buffer and proteins were quantified using a BCA assay (Pierce, Rockford, IL). Equal amounts of total protein cell extracts (20–40 μg per lane) were separated on SDS-PAGE gels. Membranes were probed with antibodies against UNG (IMG-403; Imgenex, San Diego, CA) and GAPDH (ab-8245; Abcam, Cambridge, MA). Antibodies against p53 (sc-53394), p21 (sc-51689), Bax (sc-70406) and Bcl2 (sc-65392) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Semiquantitative reverse transcription-PCR (RT-PCR) analysis
RT-PCR was performed with the isolated total RNA (1 μg) using SuperScript One-Step RT-PCR system (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The primer sequences that were used are listed in Supplementary Table 1. Amplification products were resolved by agarose gel electrophoresis and visualized by ethidium bromide staining.
Densitometry
ImageJ software (National Institutes of Health) was used to quantify the mRNA and protein band intensities. Data are represented as relative to the intensity of the indicated loading control.
Statistical analysis
Statistical comparisons were performed using analysis of variance for analysis of significance between different values using GraphPad Prism software (San Diego, CA). Values are expressed as mean ± S.D. from at least three separate experiments, and differences were considered significant at a p value of less than 0.05.
Supplementary Material
Acknowledgments
This research was supported by National Cancer Institute Grant CA75557, CA92393, CA95058, CA116708, CA138409, N.I.N.D.S. NS47699, NS57529, and NS61835 and Caterpillar, Inc., OSF St. Francis, Inc. Peoria, IL (to J.S.R.). Contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
We thank Shellee Abraham for preparing the manuscript and Diana Meister and Sushma Jasti for manuscript review.
Reference List
- 1.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 2.Madhusudan S, Middleton MR. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat Rev. 2005;31:603–17. doi: 10.1016/j.ctrv.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Fishel ML, Seo YR, Smith ML, Kelley MR. Imbalancing the DNA base excision repair pathway in the mitochondria; targeting and overexpressing N-methylpurine DNA glycosylase in mitochondria leads to enhanced cell killing. Cancer Res. 2003;63:608–15. [PubMed] [Google Scholar]
- 4.Rinne M, Caldwell D, Kelley MR. Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol Cancer Ther. 2004;3:955–67. [PubMed] [Google Scholar]
- 5.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 6.Devlin HL, Mack PC, Burich RA, et al. Impairment of the DNA repair and growth arrest pathways by p53R2 silencing enhances DNA damage-induced apoptosis in a p53-dependent manner in prostate cancer cells. Mol Cancer Res. 2008;6:808–18. doi: 10.1158/1541-7786.MCR-07-2027. [DOI] [PubMed] [Google Scholar]
- 7.Hallstrom TM, Laiho M. Genetic changes and DNA damage responses in the prostate. Prostate. 2008;68:902–18. doi: 10.1002/pros.20746. [DOI] [PubMed] [Google Scholar]
- 8.Marquis JC, Hillier SM, Dinaut AN, et al. Disruption of gene expression and induction of apoptosis in prostate cancer cells by a DNA-damaging agent tethered to an androgen receptor ligand. Chem Biol. 2005;12:779–87. doi: 10.1016/j.chembiol.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Pulukuri SM, Rao JS. Activation of p53/p21Waf1/Cip1 pathway by 5-aza-2′-deoxycytidine inhibits cell proliferation, induces pro-apoptotic genes and mitogen-activated protein kinases in human prostate cancer cells. Int J Oncol. 2005;26:863–71. [PubMed] [Google Scholar]
- 10.Trzeciak AR, Nyaga SG, Jaruga P, Lohani A, Dizdaroglu M, Evans MK. Cellular repair of oxidatively induced DNA base lesions is defective in prostate cancer cell lines, PC-3 and DU-145. Carcinogenesis. 2004;25:1359–70. doi: 10.1093/carcin/bgh144. [DOI] [PubMed] [Google Scholar]
- 11.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 12.Kavli B, Sundheim O, Akbari M, et al. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 2002;277:39926–36. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- 13.Krokan HE, Drablos F, Slupphaug G. Uracil in DNA--occurrence, consequences and repair. Oncogene. 2002;21:8935–48. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 14.Nilsen H, Krokan HE. Base excision repair in a network of defence and tolerance. Carcinogenesis. 2001;22:987–98. doi: 10.1093/carcin/22.7.987. [DOI] [PubMed] [Google Scholar]
- 15.Anderson CT, Friedberg EC. The presence of nuclear and mitochondrial uracil-DNA glycosylase in extracts of human KB cells. Nucleic Acids Res. 1980;8:875–88. [PMC free article] [PubMed] [Google Scholar]
- 16.Bharati S, Krokan HE, Kristiansen L, Otterlei M, Slupphaug G. Human mitochondrial uracil-DNA glycosylase preform (UNG1) is processed to two forms one of which is resistant to inhibition by AP sites. Nucleic Acids Res. 1998;26:4953–9. doi: 10.1093/nar/26.21.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otterlei M, Haug T, Nagelhus TA, Slupphaug G, Lindmo T, Krokan HE. Nuclear and mitochondrial splice forms of human uracil-DNA glycosylase contain a complex nuclear localisation signal and a strong classical mitochondrial localisation signal, respectively. Nucleic Acids Res. 1998;26:4611–7. doi: 10.1093/nar/26.20.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.An Q, Robins P, Lindahl T, Barnes DE. C --> T mutagenesis and gamma-radiation sensitivity due to deficiency in the Smug1 and Ung DNA glycosylases. EMBO J. 2005;24:2205–13. doi: 10.1038/sj.emboj.7600689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Priet S, Gros N, Navarro JM, et al. HIV-1-associated uracil DNA glycosylase activity controls dUTP misincorporation in viral DNA and is essential to the HIV-1 life cycle. Mol Cell. 2005;17:479–90. doi: 10.1016/j.molcel.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 20.Turner NC, Lord CJ, Iorns E, et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–77. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruman II, Schwartz E, Kruman Y, et al. Suppression of uracil-DNA glycosylase induces neuronal apoptosis. J Biol Chem. 2004;279:43952–60. doi: 10.1074/jbc.M408025200. [DOI] [PubMed] [Google Scholar]
- 22.Offer H, Zurer I, Banfalvi G, et al. p53 modulates base excision repair activity in a cell cycle-specific manner after genotoxic stress. Cancer Res. 2001;61:88–96. [PubMed] [Google Scholar]
- 23.Smith ML, Seo YR. p53 regulation of DNA excision repair pathways. Mutagenesis. 2002;17:149–56. doi: 10.1093/mutage/17.2.149. [DOI] [PubMed] [Google Scholar]
- 24.Studebaker AW, Ariza ME, Williams MV. Depletion of uracil-DNA glycosylase activity is associated with decreased cell proliferation. Biochem Biophys Res Commun. 2005;334:509–15. doi: 10.1016/j.bbrc.2005.06.118. [DOI] [PubMed] [Google Scholar]
- 25.Venkatesh J, Kumar P, Krishna PS, Manjunath R, Varshney U. Importance of uracil DNA glycosylase in Pseudomonas aeruginosa and Mycobacterium smegmatis, G+C-rich bacteria, in mutation prevention, tolerance to acidified nitrite, and endurance in mouse macrophages. J Biol Chem. 2003;278:24350–8. doi: 10.1074/jbc.M302121200. [DOI] [PubMed] [Google Scholar]
- 26.An Q, Robins P, Lindahl T, Barnes DE. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007;67:940–5. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- 27.Dizdaroglu M, Karakaya A, Jaruga P, Slupphaug G, Krokan HE. Novel activities of human uracil DNA N-glycosylase for cytosine-derived products of oxidative DNA damage. Nucleic Acids Res. 1996;24:418–22. doi: 10.1093/nar/24.3.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akbari M, Otterlei M, Pena-Diaz J, Krokan HE. Different organization of base excision repair of uracil in DNA in nuclei and mitochondria and selective upregulation of mitochondrial uracil-DNA glycosylase after oxidative stress. Neuroscience. 2007;145:1201–12. doi: 10.1016/j.neuroscience.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Sankala HM, Hait NC, Paugh SW, et al. Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res. 2007;67:10466–74. doi: 10.1158/0008-5472.CAN-07-2090. [DOI] [PubMed] [Google Scholar]
- 30.Bates S, Vousden KH. p53 in signaling checkpoint arrest or apoptosis. Curr Opin Genet Dev. 1996;6:12–8. doi: 10.1016/s0959-437x(96)90004-0. [DOI] [PubMed] [Google Scholar]
- 31.Janus F, Albrechtsen N, Dornreiter I, Wiesmuller L, Grosse F, Deppert W. The dual role model for p53 in maintaining genomic integrity. Cell Mol Life Sci. 1999;55:12–27. doi: 10.1007/s000180050266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaufmann WK, Levedakou EN, Grady HL, Paules RS, Stein GH. Attenuation of G2 checkpoint function precedes human cell immortalization. Cancer Res. 1995;55:7–11. [PubMed] [Google Scholar]
- 33.Gottlieb TM, Oren M. p53 and apoptosis. Semin Cancer Biol. 1998;8:359–68. doi: 10.1006/scbi.1998.0098. [DOI] [PubMed] [Google Scholar]
- 34.Meulmeester E, Jochemsen AG. p53: a guide to apoptosis. Curr Cancer Drug Targets. 2008;8:87–97. doi: 10.2174/156800908783769337. [DOI] [PubMed] [Google Scholar]
- 35.Offer H, Wolkowicz R, Matas D, Blumenstein S, Livneh Z, Rotter V. Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett. 1999;450:197–204. doi: 10.1016/s0014-5793(99)00505-0. [DOI] [PubMed] [Google Scholar]
- 36.de Souza-Pinto NC, Harris CC, Bohr VA. p53 functions in the incorporation step in DNA base excision repair in mouse liver mitochondria. Oncogene. 2004;23:6559–68. doi: 10.1038/sj.onc.1207874. [DOI] [PubMed] [Google Scholar]
- 37.Sobol RW, Kartalou M, Almeida KH, et al. Base excision repair intermediates induce p53-independent cytotoxic and genotoxic responses. J Biol Chem. 2003;278:39951–9. doi: 10.1074/jbc.M306592200. [DOI] [PubMed] [Google Scholar]
- 38.Russo T, Zambrano N, Esposito F, et al. A p53-independent pathway for activation of WAF1/CIP1 expression following oxidative stress. J Biol Chem. 1995;270:29386–91. doi: 10.1074/jbc.270.49.29386. [DOI] [PubMed] [Google Scholar]
- 39.Somasundaram K, Zhang H, Zeng YX, et al. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature. 1997;389:187–90. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- 40.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 41.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 42.Chaudhary KS, Abel PD, Stamp GW, Lalani E. Differential expression of cell death regulators in response to thapsigargin and adriamycin in Bcl-2 transfected DU145 prostatic cancer cells. J Pathol. 2001;193:522–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH821>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 43.Radany EH, Dornfeld KJ, Sanderson RJ, et al. Increased spontaneous mutation frequency in human cells expressing the phage PBS2-encoded inhibitor of uracil-DNA glycosylase. Mutat Res. 2000;461:41–58. doi: 10.1016/s0921-8777(00)00040-9. [DOI] [PubMed] [Google Scholar]
- 44.Lipscomb EA, Simpson KJ, Lyle SR, Ring JE, Dugan AS, Mercurio AM. The alpha6beta4 integrin maintains the survival of human breast carcinoma cells in vivo. Cancer Res. 2005;65:10970–6. doi: 10.1158/0008-5472.CAN-05-2327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.