

Abstract
Objectives: Defects of the mitochondrial genome (mtDNA) cause a series of rare, mainly neurological disorders. In addition, they have been implicated in more common forms of movement disorders, dementia and the ageing process. In order to try to model neuronal dysfunction associated with mitochondrial disease, we have attempted to establish a series of transmitochondrial mouse embryonic stem cells harbouring pathogenic mtDNA mutations.
Materials and methods: Transmitochondrial embryonic stem cell cybrids were generated by fusion of cytoplasts carrying a variety of mtDNA mutations, into embryonic stem cells that had been pretreated with rhodamine 6G, to prevent transmission of endogenous mtDNA. Cybrids were differentiated into neurons and assessed for efficiency of differentiation and electrophysiological function.
Results: Neuronal differentiation could occur, as indicated by expression of neuronal markers. Differentiation was impaired in embryonic stem cells carrying mtDNA mutations that caused severe biochemical deficiency. Electrophysiological tests showed evidence of synaptic activity in differentiated neurons carrying non‐pathogenic mtDNA mutations or in those that caused a mild defect of respiratory activity. Again, however, neurons carrying mtDNA mutations that resulted in severe biochemical deficiency had marked reduction in post‐synaptic events.
Conclusions: Differentiated neurons carrying severely pathogenic mtDNA defects can provide a useful model for understanding how such mutations can cause neuronal dysfunction.
Introduction
Mitochondria are essential organelles, critically involved in the life and programmed death of the eukaryote cell. One of their major functions, oxidative phosphorylation, occurs at the inner mitochondrial membrane and is mediated by the four enzyme complexes of the respiratory chain, and a fifth enzyme complex, ATP synthetase. Four of these five multi‐subunit complexes contain subunits encoded by the mitochondrial genome (mtDNA). Not surprisingly, therefore, mtDNA mutations can often be pathogenic and the associated disease can affect any organ system of the body. Critically, however, neurological symptoms are an almost invariant feature. These can be mild, but are more usually severe and progressive, leading to disability and death. Typical mtDNA syndromes are rare (1), but mitochondrial dysfunction is implicated in other more common neurodegenerative diseases, such as Parkinson's disease (2) and in the ageing process itself (3, 4). Exact mechanisms of damage to neurons in mtDNA disease are unknown, and study of such processes at a cellular level may lead to improved treatment and greater understanding of the role of mtDNA mutations in neurodegenerative disease and ageing.
Obtaining fresh neuronal material from patients with mtDNA disease is impossible because neurosurgery is not a treatment for the disease; thus, a model system whereby neuronal function can be assessed in cells carrying mutated mtDNA could be very informative. One possible source would be from transgenic mice carrying defective mtDNA in neurons. To date, however, there have been difficulties in producing transgenic mice carrying profound pathogenic mtDNA mutations (5) (R.N.L., personal observation), and it has not been possible to completely recapitulate, in mice, all the symptoms of any form of neuronal disorder associated with mtDNA defects. Thus, we have focussed on embryonic stem (ES) cell expertise to produce neurons that can be studied in culture, derived from ES cells carrying pathogenic mtDNA defects.
Development of transmitochondrial technology, where enucleated somatic cells harbouring pathological mtDNA mutations are fused with a cell line in which the endogenous mtDNA has been chemically ablated (a ρ° cell line), has enabled the study of consequences of mtDNA mutations at the cellular level (6, 7, 8). However, conventional cybrids using patient‐derived platelets or fibroblasts tell us nothing about the cellular pathophysiology of defined tissue types, particularly about the neurological component of mtDNA disease. We have now used this set of skills to produce a series of murine ES cell lines harbouring defective mtDNA. Sequence variants of the mitochondrial genome present in mouse fibroblast cell line L929 were introduced into mouse ES cells (ES‐I and ES‐VI). One ES cell line has two mutations in different Mtnd genes, which severely reduced (< 10%) residual complex I activity. Two others were created with a mutation in Mtco1 and a moderate complex IV defect (~35% residual activity) (9). As control, another ES cell line contains a polymorphic variant in the Mttr gene encoding mt‐tRNAArg (10). ES cell‐derived cybrids retained their pluripotency, and were able to differentiate into neurons when deprived of leukaemia inhibitory factor (LIF) and supplemented with retinoic acid (11). The resulting neurons had small cell bodies and long neurites and expressed typical markers of neuronal and glial cells, while patch clamp analysis demonstrated action and synaptic potentials. We report here that neuronal differentiation was severely affected in the ES cell line carrying the most profound defect. In addition, single cell electrophysiological measurements were consistent with compromised post‐synaptic events in neurons from the profoundly deficient cell line, although further analysis is needed to confirm this observation.
Materials and methods
Cell culture conditions
Cells were cultured in a humidified incubator maintained at 37 °C with 5% CO2. Mouse fibroblasts were grown in Dulbecco's modified Eagle's medium (DMEM, Sigma, St. Louis, MO, USA) and 10% foetal calf serum (FCS, Biosera, Ringmer, UK), supplemented 2 mm l‐glutamine (Invitrogen, Carlsbad, CA, USA). Mouse ES cells for fusions were grown on mitotically inactivated SNL fibroblast (gift of Dr Allan Bradley, Sanger Centre, Cambridge, UK) feeder layers in DMEM and 10% FCS, supplemented with uridine (50 µg/ml), non‐essential amino acids and 0.1 mm 2‐mercaptoethanol. The feeder layer cells contain a LIF expression construct (12) and although express LIF abundantly (13), the medium was further supplemented with mouse recombinant LIF (1 ng/ml, Chemicon, Millipore (Chemicon) Watford, UK). ES cells destined for differentiation were grown on a surface treated with 0.1% (w/v) porcine skin gelatine (Sigma) in Glasgow modified Eagle's medium (Invitrogen) and 10% FCS, supplemented with 1 mm sodium pyruvate, 2 mm l‐glutamine, non‐essential amino acids, 0.1 mm 2‐mercaptoethanol and mouse recombinant LIF (1 ng/ml, Chemicon).
Parental cell lines
Mouse ES cells used in this study were ES‐I (CC9.3.1) and ES‐VI (MPI‐VI (14) a gift of Prof. Anne Voss, Walter and Eliza Hall Institute, Melbourne, Australia). Mouse fibroblasts carrying pathogenic mtDNA mutations were derived from the mouse L929 cell line, which has fixed several mutations over long‐term culture. Nucleotides are numbered according to the C57BL/6J mouse mtDNA sequence (GenBank AY172335) (15). Clones with different sequence variations were used as mitochondrial donor cell lines: E9 with a 6589T>C (V421A) mutation in Mtco1, one of the three mtDNA encoded complex IV genes (9); C5 with two different mutations in mtDNA genes encoding complex I subunits, a frameshift mutation, 13887Cins in Mtnd6 (9, 16) and a point mutation 12273G>A in Mtnd5. The Mtnd5 mutation was heteroplasmic (90% mutant); all others were present in homoplasmic form. Donor cell line for the polymorphic marker was mouse C57BL/6JC57, which carried a variation (8As) in the length of a stretch of As at a highly polymorphic site in the DHU loop of mt‐tRNAArg (15).
Creation of transmitochondrial cybrids
Generation of transmitochondrial ES cybrid cell lines was performed, essentially as described in Sligh et al., with slight modifications (17). To ablate mtDNA, ES cell lines (~ 1 × 106 ES‐I or ES‐VI) were pretreated for 3 days with rhodamine 6G (R6G; 1 µg/ml final concentration) in DMEM containing uridine (50 µg/ml), non‐essential amino acids and 0.1 mm 2‐mercaptoethanol. Two hours prior to fusion, medium was removed and replaced by identical media lacking R6G. Cytoplast donor cell lines (~3 × 106) carrying different mtDNA mutations were incubated in dishes in DMEM containing cytochalasin B (100 µg/ml) for 2 h, and then enucleated by centrifugation at 7000 g for 20 min at 37 °C in a Sorvall SLA‐1500 (Thermo Fisher Scientific, Loughborough, UK) rotor. R6G‐treated cells and donor cytoplasts were harvested following trypsinization and pelleted together at 150 g, then gently suspended in 0.5 ml of pre‐warmed (37 °C) 50% PEG1500 in serum‐free DMEM. Cells were exposed to the PEG for 1 min, serum‐free DMEM (5 ml) was added and cells were pelleted at 100 g, suspended in DMEM containing uridine (50 µg/ml) and plated on a feeder monolayer. Next day the medium was replaced with DMEM without uridine to select against unfused recipient ES cells. After 6–9 days, clones were picked and expanded for further analysis.
Presence of mtDNA mutations
Putative cybrids were screened for presence of the 6589T>C and 12273G>A mutations by polymerase chain reaction–restriction fragment length polymorphism (PCR‐RFLP) analysis. For 6589T>C, DNA was amplified using primers CATGAGCAAAAGCCCACTTCGCCATCATATTCGT AGGCG‐3′ (forward) and 5′‐GTGTTTCATGTGGGTGCGC ATCTGG‐3′ (reverse). This gave a 133‐bp product, which was digested with CfoI to give products of 115 and 18 bp for the wild‐type sequence, and 75, 40 and 18 bp for the mutant sequence. For 12273G>A, primers 5′‐ACTGCAG CCCTACAAGCAATCCTCTATAACCGCCTC‐3′ (forward) and 5′‐TTGTGCTGATTTTCCTGTAGCTGCGATTAAT AGGCC‐3′ (reverse) were used, giving a 182‐bp PCR product. This produced a DdeI restriction site in the mutant sequence, yielding products of 145 and 37 bp on digestion.
For 13887C ins, a 760‐bp region of Mtnd6 was PCR‐amplified using the primers 5′‐CACACAAACATAAC CACTTTAACA‐3′ (forward) and 5′‐GTAGGTCAATGA ATGAGTGGTT‐3′ (reverse) (17). Remaining free nucleotides were removed and the PCR product used as a template for primer extension using a primer with a 5′ D3 fluorescent conjugate (5′(D3)CGTATATCCAAAC ACAACCAACAT‐3′). Analysis of primer extension products was carried out on a Beckman CEQ8000 Genetic Analysis System (Beckman/Coulter, High Wycombe, UK). For the 9821Adel polymorphism, primer extension was used to verify the length of a track of A residues starting at position 9821 in Mttr, as detailed in Bayona‐Bafaluy et al. (15). A 377‐ to 379‐bp fragment containing this region was amplified by PCR with the following primers: (i) forward CTACTTCCACTACCATGAGC (positions 9672–9691); (ii) reverse GTATGGAGCTTATGGAGTTGG (positions 10 028–10 048). The 32P‐labeled primer used for primer extension was Arg‐PE, GGATTAGAATGAACAGAG TAAATGGTAATTAGTTT (positions 9786–9820). Primer‐extended products generated were analysed using 8% polyacrylamide/7 m urea sequencing gels, dried and exposed for autoradiography.
Cell lines harbouring a high level of the sequence variations were selected for further studies.
Microsatellite marker analysis
A candidate informative marker, D6mit102, predicted to discriminate cell lines used in this study, was identified from the Jackson Laboratories informatics website (http://www.informatics.jax.org). D6Mit102 amplified a 177‐bp sequence in MPI‐VI ES cells, a 177‐ and 145‐bp doublet in CC9.3.1 ES cells, and the expected 125‐bp sequence in the L929 fibroblasts. For D6Mit102 analysis, DNA from parental cell lines and putative cybrids was PCR‐amplified using the primers 5′‐CCATGTGGATATCTTCCCTGG‐3′ (forward) and 5′‐GTATACCCAGTTGTAAATCTTGTGTG‐3′ (reverse).
Maintenance of pluripotency
Cybrids were cultured in the presence or absence of LIF for 9 days before extraction of RNA. Reverse transcription of RNA (1 µg) was carried out using the Superscript First‐Strand Synthesis System kit (Invitrogen) using random hexamers as primers. Oct‐4 and β‐actin were PCR‐amplified from the resulting cDNA using the primers 5′‐GGCGTTCTCTTT GGAAAGGTGTTC‐3′ (OCT‐4 forward) and 5′‐CTCGA ACCACATCCTTCTCT‐3′ (OCT‐4 reverse) and 5′‐GGCC CAGAGCAAGAGAGGTATCC‐3′ (β‐actin forward) 5′‐ACGCACGATTTCCCTCTCAGC‐3′ (β‐actin reverse).
Respiratory chain enzymology
Respiratory chain enzymes were measured in hypotonically treated mitochondrial fractions prepared from the undifferentiated cybrids and the parental ES cells as described (18, 19) All assays were performed in duplicate.
Production of cybrid‐derived neurons
The parental ES and the cybrid cell lines were differentiated into neurons using the 4–/4+ protocol of Bain et al. (11). Briefly, cells were allowed to form embryoid bodies in suspension in non‐tissue culture plastic, 10 cm diameter dishes, by deprivation of LIF for the remainder of differentiation and addition of all‐trans retinoic acid (1 × 10−6 m, Sigma) for the last 4 days of the 8 days in suspension. A single cell suspension was then derived from the embryoid bodies by trypsinization. Cells (1 × 106 per coverslip) were plated onto glass coverslips (22 mm diameter) coated with poly‐d‐lysine (0.001%, Sigma) and laminin (0.005%, Sigma), and allowed to differentiate in the presence of fibroblast growth factor (bFGF, 10 ng/ml, Chemicon) for 2 days in a 4 : 1 mixture of Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) and DMEM : F12 (1 : 1) (Invitrogen) supplemented with modified N2 (20). The medium was then replaced with 4 : 1 medium without bFGF, and then half the medium was replaced every 2 days.
Immunocytochemistry
Differentiated neurons on the coverslips were fixed by treatment with 4% paraformaldehyde for 10 min. Cells were then washed in phosphate‐buffered saline containing 0.1% triton X‐100 (PBST), and blocked for 1 h with 4% BSA in PBST. Primary antibodies in 4% BSA in PBST (β‐tubulin III; glial fibrillary acidic protein, GFAP) or 5% normal goat serum in PBST (GABA; NeuN) were applied for 1–2 h at room temperature or overnight at 4 °C. Cells were stained with antibodies to β‐tubulin III at dilution of 1 : 400 (SDL3D10, Sigma); GFAP at 1 : 400 (MAB360, Chemicon), GABA at 1 : 200 (A2052, Sigma); NeuN at 1 : 200 (MAB377; Chemicon). Cells were washed three times with PBST. Fluorescently labelled secondary antibodies (rabbit anti‐mouse labelled with fluorescein isothiocyanate (Dako, Glostrup, Denmark), or swine anti‐rabbit labelled with TRITC (Serotec, Serotec Ltd, Kidlington, UK), 1 : 100 dilution in 4% BSA in PBST; 1 : 200 goat anti‐rabbit labelled with fluorescein isothiocyanate or 1 : 200 goat anti‐mouse IgG1 labelled with rhodamine in 5% normal goat serum in PBST for GABA and NeuN) were applied for 1 h, then the cells were washed with PBST, PBS, and then water, and mounted on microscope slides with Vectashield mounting medium for fluorescence with 4′,6‐diamidino‐2‐phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). Cells were examined by fluorescence microscopy for β‐tubulin III and GFAP staining using a Zeiss Axioplan 2 imaging system with Axiovision software. GABA and NeuN staining was imaged using an Axioskop FS microscope fitted with a spinning disc confocal head (Visitech International, Sunderland, UK) and photographed using Visitech software. Cells were counted post‐hoc from the confocal images.
Ability of the cells to differentiate into neurons was estimated on day 13 of differentiation by comparing number of cells staining positively with antibodies directed against neuronal marker β‐tubulin III, with number of nuclei stained with DAPI (Fig. 3). The numbers of β‐tubulin III‐positive cells and DAPI‐positive nuclei were counted in 10 randomly selected visual fields, and percentage of β‐tubulin III‐positive cells relative to DAPI‐positive nuclei was calculated. This was performed for two independent differentiation experiments (that is, results represent 2 × 10 fields at ×100 magnification) for all except CY1‐I in which efficiency of differentiation into neurons was quantified only once (1 × 10 fields).
Figure 3.
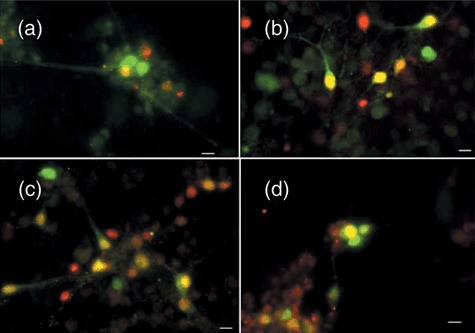
GABAergic neurons are present in subpopulations of differentiated transmitochondrial cells. Following 15–16 days of differentiation, cells were analysed for the expression of an early neuronal marker (NeuN, red) and the presence of GABA (green) as detailed in the Materials and methods. Co‐expression is shown in the image as yellow. The size marker represents 10 µm. (a) Differentiated parental ES‐I; (b) CY1‐I carrying the polymorphic marker; (c) CY2‐I carrying the mild respiratory defect; (d) CY3‐I carrying the severe respiratory defect.
Electrophysiology
Cultures were transferred into a recording chamber, which was mounted on an upright Zeiss Axioskop FS microscope fitted with Luigs & Neumann micromanipulators. There, they were bathed in a continuously flowing stream (1–3 ml/min) of artificial cerebrospinal fluid (in mm: NaCl, 125; NaHCO3, 26; glucose, 10; KCl, 3.5; CaCl2, 1.2; 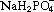 , 1.26; MgSO4, 1). Whole cell patch clamp recordings were made using 5–7 mΩ pipettes (borosilicate glass; Harvard Apparatus, Holliston, MA, USA). The pipette solution contained (in mm) K‐methylsulphate, 125; K‐HEPES, 10; Mg‐ATP, 2.5; NaCl, 6; Na‐GTP, 0.3 (corrected to pH 7.3–7.35 using KOH; 280 mOsm). Data were collected using Axopatch 1D amplifiers (Molecular Devices Ltd, Wokingham, UK), filtered at 3 kHz, digitized at 5 kHz (AD board) and recorded on an Apple Macintosh computer using Axograph software. All recordings were made at room temperature.
, 1.26; MgSO4, 1). Whole cell patch clamp recordings were made using 5–7 mΩ pipettes (borosilicate glass; Harvard Apparatus, Holliston, MA, USA). The pipette solution contained (in mm) K‐methylsulphate, 125; K‐HEPES, 10; Mg‐ATP, 2.5; NaCl, 6; Na‐GTP, 0.3 (corrected to pH 7.3–7.35 using KOH; 280 mOsm). Data were collected using Axopatch 1D amplifiers (Molecular Devices Ltd, Wokingham, UK), filtered at 3 kHz, digitized at 5 kHz (AD board) and recorded on an Apple Macintosh computer using Axograph software. All recordings were made at room temperature.
Glutamate application
Glutamate (1 mm in artificial cerebrospinal fluid) was applied directly from patch pipettes (5–7 megaohms when filled with K‐methylsulphate electrode filling solution (see below)) using a Picospritzer II pressure application system (Parker Instruments, Fairfield, NJ, USA, 10 ms pulses; pressure = 20 psi). Timing of pressure applications was controlled using the Master 8 pulse stimulator (AMPI, Jeruselam, Israel). The pipette tip was in the order of 15–30 µm from the cells, and, unless the pipette was broken, we never detected any movement artefact during glutamate applications, indicating that this means of stimulating cells was relatively atraumatic. We discarded data if the pipette was broken. We calculated that the average bolus for a 10‐ms pressure application through these pipettes was around 1.6 µl.
Results
Transfer of mtDNA variants to ES cells
Cybrid clones were generated and analysed for presence of the mtDNA variants as detailed in the Materials and methods section and nomenclature is provided in Table 1. RFLP analysis confirmed that clones CY2‐I and CY2‐VI contained homoplasmic levels of the 6589T>C mutation in Mtco1, while 12267G>A was present at 90% heteroplasmy in CY3‐I (Fig. 1a,b). Primer extension assays confirmed homoplasmic levels of the 13887Cins and 9821Adel in CY3‐I and CY1‐I, respectively (Fig. 1b,c).
Table 1.
Phenotype and mtDNA genotype of transmitochondrial cybrid cell lines used in this study
| Cybrid cell line | Nuclear background | mtDNA mutation (gene) | Respiratory defect |
|---|---|---|---|
| Cy1‐I | ES‐I | 9821A del Mttr | Non‐pathogenic |
| Cy2‐I | ES‐I | 6589T>C Mtco1 | Mild |
| Cy2‐VI | ES‐VI | 6589T>C Mtco1 | Mild |
| Cy3‐I | ES‐I | 13887C ins Mtnd6 | Severe |
| 12273G>A Mtnd5 |
Figure 1.
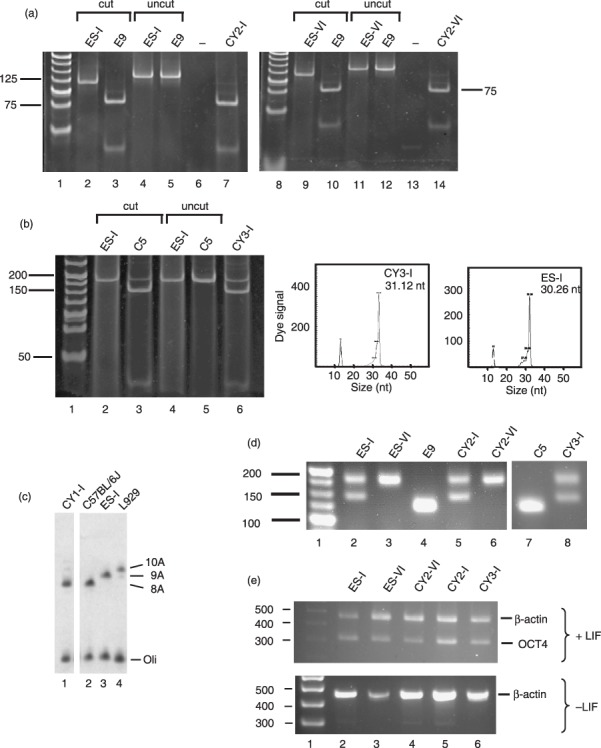
Production of transmitocondrial murine embryonic stem (ES) cell cybrids. (a) Transfer of mtDNA carrying a mild mutation in Mtco1 to pluripotent ES‐I and ES‐VI ES cell lines. A 133‐bp amplicon spanning nt6589 was generated from the indicated ES parental cells (ES‐I, lanes 2 and 4; ES‐VI, lanes 9 and 11), 6589T>C fibroblasts (E9, lanes 3, 5, 10 and 12) and ES cybrids (CY2‐I, lanes 7 and 14) as detailed in the Materials and methods and subjected to digestion with Cfo1. Cleavage generated a 115‐ and 18‐bp fragment for the wild‐type amplicon (lanes 2, 9), with further digestion of the 115‐bp fragment to 75 and 40 bp indicating the 6589T>C mutation (lanes 3, 7, 10 and 14). Uncut amplicon (lanes 4, 5, 11 and 12), negative control (lanes 6 and 13) and molecular weight markers (lanes 1, 8) are also shown. (b) Transfer of mutations in Mtnd5 and Mtnd6 to ES‐I. A 182‐bp amplicon spanning nt12273 was generated from ES parental cells (ES‐I, lanes 2 and 4), 12273G>A fibroblasts (C5, lanes 3 and 5) and ES cell cybrids (CY3‐I, lane 6) as detailed in the Materials and methods and subjected to cleavage with Dde1. Products carrying the mutation were cleaved to generate fragments of 145 and 37 bp (lanes 3 and 6). Uncut amplicon (lanes 4 and 5) and molecular weight ladder (lane 1) are shown. For analysis of mtDNA carrying the 13887C insertion, primer extension was performed as detailed in the Materials and methods. The panels show the length of extension corresponding to the normal mtDNA sequence (ES‐I, ~30 bp) or to the mtDNA carrying the single base insertion (CY3‐I, ~31 bp). The fluorescence signal due to the 5′ D3 fluorescent conjugate is indicated. The conjugated standard (13 nt) is also shown. (c) Transfer of the Mttr polymorphism to ES‐I. Primer extension was used to determine the length of the polyA tract around nt9821 in the mtDNA of various cell lines as detailed in the Materials and methods. Parental ES cells (ES‐I, lane 3), donor fibroblasts (C57BL/6J, lane 2) and cybrids (CY1‐I, lane 1) are shown, as well as the 10A extension found in L929 fibroblasts (lane 4). Radiolabelled oligonucleotide is also shown (oli). (d) Microsatellite markers confirm production of transmitochondrial cybrids. Amplicons spanning the informative marker D6mit102 were prepared from ES parental cells (ES‐I, lane 2; ES‐VI, lane 3), fibroblast donors (E9, lane 4; C5, lane 7) and cybrids (CY2‐I, lane 5; CY2‐VI, lane 6; CY3‐I, lane 8). Molecular weight markers are shown in lane 1. (e) Oct‐4 expression in parental and cybrid ES cells is dependent on leukaemia inhibitory factor (LIF). Cell lines (ES parental lanes 2, 3; cybrids lanes 4–6) were grown in the presence (+) or absence (–) of LIF for 9 days before RNA was isolated and reverse transcribed. Amplicons from the transcripts encoding β‐actin or Oct‐4 were generated as described in the Materials and methods. Molecular weight standards are given (lane 1).
Absence of fibroblast nuclear background in the cybrids was assessed by analysis of the informative polymorphic microsatellite marker D6mit102 (Fig. 1d).
PCR amplification of the ES cell parental DNA gave fragments of 177 bp and 145 bp for ES‐I, or 177 bp alone from the ES‐VI. E9 and C5 fibroblasts gave a product of 125 bp. There was no evidence of a 125‐bp fragment attributable to either the E9 or C5 fibroblast DNA in the cybrids containing the Mtco1 mutation, CY2‐I and CY2‐VI, or those containing the Mtnd mutations, CY3‐I. Thus, nuclear DNA microsatellite markers present in the cybrids corresponded to those present in ES cell parent only, confirming that cell lines contained only the ES cell parent nucleus, and were indeed cybrids, not hybrids. Similar confirmation was also noted with the CY1‐I cybrid (data not shown).
Pluripotency of the ES parental cells and the cybrids was assessed by monitoring expression of OCT‐4, a transcription factor that is expressed in undifferentiated pluripotent cells and not in differentiated cells. OCT‐4 expression was present when both the ES parental cells and the cybrids remained undifferentiated in the presence of LIF. When the cultures were deprived of LIF for 9 days, OCT‐4 expression was absent (Fig. 1e). These results are consistent with the cybrids retaining pluripotency following the fusion, selection and expansion process.
Activities of respiratory chain components in transmitochondrial ES cell lines were similarly affected to the donor cell lines
Biochemical consequences of the mtDNA variations were assessed by measuring activities of the respiratory chain enzymes (complexes I, II, III, and IV) in the ES parental cells and the cybrids (Table 2). Activities of respiratory chain enzymes were determined spectrophotometrically and expressed relative to activity of the mitochondrial matrix marker enzyme citrate synthase (CS ratios), as percentages of the CS ratios in the ES parental cell line. Citrate synthase activities were similar in all cell lines, ranging from 429 to 542 nmol min/mg. Presence of the 9821A(del) polymorphism in Mttr in clone CY1‐I results in reduction of complexes I, II, III and IV, with activities of 56–74% of the parental ES cell line, ES‐I. These reductions are not substantial, and reflect the subtle phenotype reported by Moreno‐Loshuertos et al. (10). Clone CY3‐I with the two mutations in different Mtnd genes (13887Cins in Mtnd6 and 12273 G>A in Mtnd5) has a severe complex I defect (7% of ES‐I activity) as previously reported in fibroblasts with the homoplasmic Mtnd6 mutation (17). The two cybrid clones with the 6589T>C mutation in Mtco1 (CY2‐I MILD and CY2‐VI MILD) both have residual complex IV activity of 37% of their parental ES cell lines, ES‐I and ES‐VI, respectively, consistent with activity reported for the parental L929 mutants (9). Thus, the cybrid cell lines harbour mtDNA variations, which result in a spectrum of biochemical phenotypes (1, 2) ranging from severe (Mtnd5/nd6 mutations), to moderate (Mtco1 mutations), to insignificant (Mttr polymorphism).
Table 2.
Respiratory chain enzyme activities in undifferentiated cybrids. The activities are expressed relative to the mitochondrial matrix enzyme citrate synthase (CS ratio), as percentages of the CS ratio in the parental embryonic stem (ES) cell line
| Ratio enzymatic activity | Transmitochondrial cybrid % ratio of parental ES cell line | |||
|---|---|---|---|---|
| CY1‐I | CY2‐I | CY2‐VI | CY3‐I | |
| Complex I/CS | 74 | 71 | 97 | 7 |
| Complex II/CS | 56 | 64 | 132 | 83 |
| Complex III/CS | 83 | 114 | 143 | 89 |
| Complex IV/CS | 57 | 37 | 37 | 57 |
All differentiated transmitochondrial cell lines expressed neuronal markers
Cybrids with typical neuronal appearance were apparent within 12 h of plating on to the poly‐d‐lysine/laminin‐coated coverslips. Where neuronal cell bodies were clustered together, numerous processes extended from and between the clusters, suggesting that some synaptic connections may have existed. With extended culture, more and longer neurites were apparent, with more connections, between clusters as well as between distinguishable individual cell bodies.
The differentiated cultures of all four main types (parental, polymorphism, complex I, complex IV) contained cells that not only morphologically appeared to be neurons, but also stained positively with β‐tubulin III from the perinuclear region in the cell body to the ends of all neuronal processes (Fig. 2); β‐tubulin III is only expressed in neurons. These cultures also contained astrocytes, as demonstrated by the astrocytic marker, GFAP (Fig. 2f,g). Many, perhaps all, of the neurons (defined by β‐tubulin III expression) also expressed NeuN, a nuclear marker of mature neurons (Fig. 3, red) (T.K.S‐S., personal observation). In addition, a proportion of the neurons also expressed GABA, a neurotransmitter (Fig. 3, green), in the cell bodies and along the neurites. However, only ~15–20% of total number of neurons (assessed by NeuN expression) expressed GABA (ES‐I 15.2 ± 10.9%; Cy1‐I 16.1 ± 2.7%; Cy2‐I 21.5 ± 11.0%; Cy3‐I, 15.9 ± 4.2%, averages from five independent differentiations and 10 counted fields for each cell type). Thus, there were likely to be neurons in these cultures expressing other neurotransmitters. As the percentage of GABA‐positive neurons did not vary significantly between cultures, irrespective of mutation, respiratory chain dysfunction did not appear to affect which type of neurotransmitter was expressed by the differentiated neurons. Clearly, all transmitochondrial cell lines were able to differentiate into neurons in culture, as assessed by morphology and expression of neuronal markers.
Figure 2.
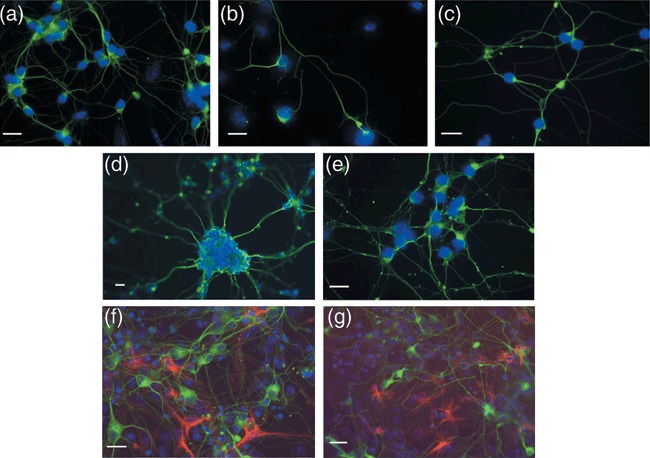
Transmitochondrial embryonic stem (ES) cells can be differentiated into neurons and astrocytes. Parental ES cell lines (a and f, ES‐I; d, ES‐VI) and cybrids (b and g, CY3‐I SEVERE; c, CY2‐I MILD; e, CY2‐VI MILD) were differentiated and fixed on Day 13 for immunohistochemical analysis with the pan‐neuronal anti‐β‐tubulin III antibody (a–g, green) and astrocytic marker anti‐GFAP (F and G, red) as detailed in the Materials and methods. Nuclei are visualized by DAPI staining (blue). The sizing bar represents 20 µm.
Cybrids with the greatest deficiency in respiration were the most affected in differentiation
Expression of β‐tubulin III was used to indicate differentiation into neurons. On day 13 of differentiation (day 5 post‐plating), the number of cells staining positively for β‐tubulin III was counted and compared with the number of nuclei present as visualized by DAPI staining (Fig. 4). The percentage of neurons generated from the cells with the Mttr polymorphism (CY1‐I; 73%) was not significantly different from the number of neurons generated from parental cells (ES‐I, 53%; Mann–Whitney, P = 0.103). For both ES cell cybrids carrying the mild Mtco1 mutation, the percentage of neurons generated (CY2‐I, 69.1%; CY2‐VI, 70.4%) was again comparable to the parental efficiency (ES‐I, 53%, P = 0.095; ES‐VI, 63.9%, P = 0.393, respectively). Any increases in efficiency when compared to parental cells were not significant. However, for the ES cell cybrid CY3‐I, carrying two mtDNA mutations affecting complex I activity, only 21% of cells showed neuronal development, compared with 53% in the parental ES cell line ES‐I (P = 0.0013). Thus, a major effect of the severe respiratory chain defect was to reduce efficiency of neuronal generation.
Figure 4.
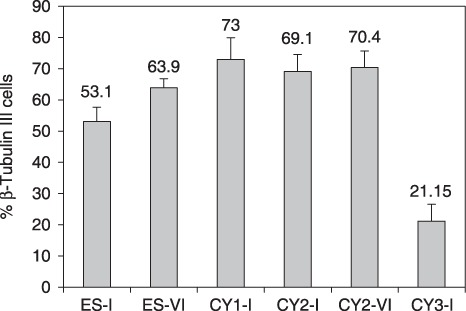
Severe respiratory deficiency affects neuronal differentiation of murine embryonic stem (ES) cells. Cell lines were induced to differentiate as described in the Materials and methods. On day 13, cells were subjected to immunohistochemistry with anti‐β‐tubulin III (late pan‐neuronal marker). Fields (n = 10) were selected and the percentage of cells differentiating into neurons was calculated with reference to the total number of DAPI‐staining nuclei in the field. This was done for two independent differentiation experiments, i.e. the results represent 2 × 10 fields at ×100 magnification, for all except for CY1‐I, in which efficiency of differentiation into neurons was quantified only once (1 × 10 fields). ES‐I and ES‐VI, parental murine ES cells; CY1‐I, polymorphism; CY3‐I, severe respiratory defect; CY2‐I and CY2‐VI, mild respiratory defect.
Neurons with the highest level of respiratory deficiency were markedly affected in synaptic activity
Patch clamp recordings were made from 135 cells on 50 culture plates representing all cell lines, at a range of developmental stages between post‐plating days 6 and 20 (Table 3). All cells had extremely high input resistance (101 cells from 34 cultures analysed; Table 3). Given that this generally was within an order of magnitude of the typical seal resistance (5–10 mΩ), the artefact leak current around the tip of the pipette constituted a large fraction of total current at rest. Therefore, this difficult‐to‐measure artefact precluded true estimation of the resting membrane potential. However, firing properties of the cells could be assessed by injecting a small hyperpolarizing current to lower the resting membrane potential to –70 mV. A range of firing responses was subsequently generated in all cell lines at several developmental stages by injecting positive suprathreshold current pulses into the soma (Fig. 5a). The various firing patterns indicate that all cultures included heterogeneous neuronal populations that expressed a range of excitable conductances.
Table 3.
Electrophysiological recordings of differentiated parental embryonic stem (ES) cells and transmitochondrial ES cell cybrids
| ES‐I | ES‐VI | Cy1‐I | Cy2‐I | Cy2‐VI | Cy3‐I | |
|---|---|---|---|---|---|---|
| No. of cells | 25 | 13 | 12 | 8 | 18 | 25 |
| No. of cultures | 9 | 3 | 5 | 3 | 4 | 10 |
| Age range (Post‐plating day) | 6 to 11 | 6 to 20 | 8 to 19 | 7 to 10 | 6 to 20 | 8 to 17 |
| Input resistance (mΩ) | ||||||
| Average | 1.6 | 1.9 | 2.0 | 1.9 | 1.9 | 3.8 |
| Standard deviation | 1.5 | 1.4 | 1.9 | 0.9 | 2.3 | 3.6 |
| Range | (0.2–5.5) | (0.3–4.0) | (0.7–5.4) | (0.8–3.0) | (0.5–10.0) | (0.9–17.2) |
| Synaptic connectivity (% cultures) | 67 | 33 | 60 | 33 | 50 | 20 |
Figure 5.
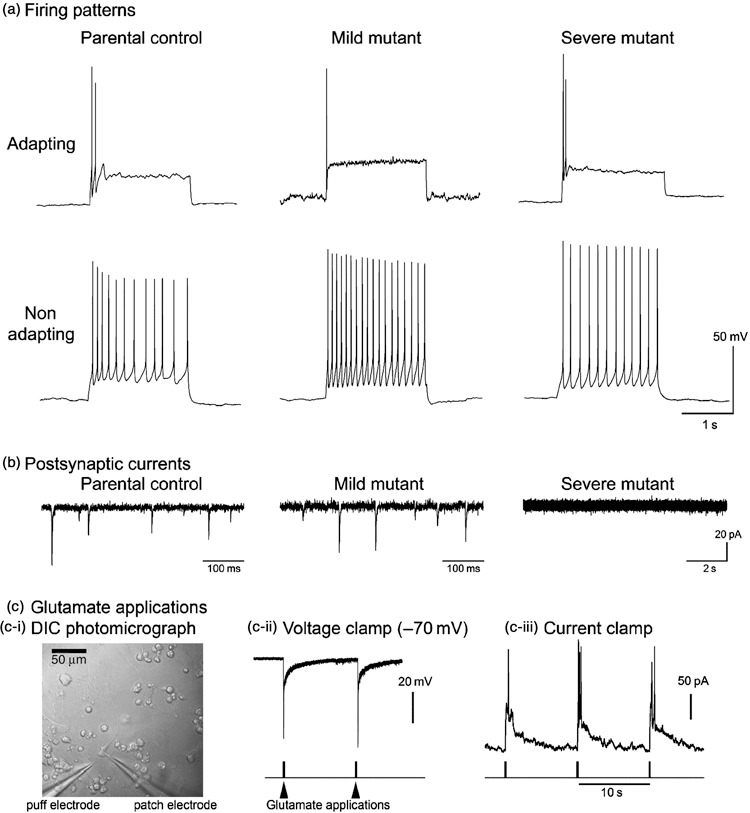
All cell lines produce neurons with a range of firing patterns. (a) Example of adapting and non‐adapting firing patterns are shown in response to square pulses of current injected somatically. Individual cells only ever show one firing pattern, but examples of each type are found in all cell lines. (b) Typical examples of spontaneous synaptic currents recorded in cultured neurons held at –70 mV. In the control and mild mitochondrial mutant cell lines, synaptic currents are routinely recorded in all cells after about the 2nd week after plating (parental recording is at 6 days post‐plating; mild mutant recording – 13 days post‐plating). In contrast, recordings from the severe mutant cell line are routinely devoid of synaptic currents at all ages recorded (example is at post‐plating 17). (c) Glutamate applications. Panel (c‐i) shows a differential interference micrograph of the experimental arrangement with the puff electrode located 10–20 µm from the patched cell. Panels (c‐ii) and (c‐iii) show responses to exogenously applied glutamate (1 mm) in a cell from the severe mutant line that showed no spontaneous synaptic events in more than 5 min of recording.
Connectivity within the networks was assessed by recording spontaneous post‐synaptic currents, holding the cells at –70 mV in voltage clamp mode (Table 3). In control cell lines and the cell lines containing mild mitochondrial deficits, synaptic events could be recorded in cultures only 6–7 days post‐plating, although most cultures of this age were synaptically quiescent. In the various control lines, most cultures showed evidence of synaptic events after this stage (Table 3; Fig. 5b). Even if individual cells did not show evidence of synaptic connectivity, other cells in the same culture could then be recorded, which clearly did receive synaptic inputs. Thus, cultures of control cell lines and those with mild mitochondrial defects routinely formed synaptically interconnected neural networks.
In contrast, synaptic events in the cell lines expressing severe mitochondrial mutation were rarely evidenced. Out of 25 recordings from 10 different culture plates, post‐synaptic currents were recorded in just four cells (two different cultures: one at post‐plating day 8, and one at post‐plating day 15). Recordings from five out of six cells at a relatively late developmental stage (post‐plating days 15–17) were completely quiescent synaptically. It was clear, however, that the synaptic quiescence was not due to a lack of responsiveness in the recorded neuron since large depolarizing currents could be induced in these cells by brief applications of 1 mm glutamate, even driving them to firing (Fig. 5c). Absence of synaptic currents was therefore likely to be due to lack of vesicular release of neurotransmitter. Interpretation of this result, however, was made difficult by many cultures of these severe mutant cells having very low neuronal densities, meaning that recorded cells were often isolated from other neurons. Some of the quiescent neurons did have near neuronal neighbours, and in two cases we patched on to adjacent neurons to test synaptic activity directly by stimulating each in turn: again we could not elicit post‐synaptic responses. Our results are thus consistent with a deficit in synaptic vesicle release, although we cannot completely rule out the apparent paucity of synaptic events reflecting the sparsity of neuronal neighbours.
Discussion
We have succeeded in creating transmitochondrial cybrids with mtDNA variations, on a mouse ES cell nuclear background, which retain the ability to differentiate into neurons. These cell lines maintained pluripotency, as demonstrated by the presence of OCT‐4 expression and the ability of cells to undergo directed differentiation into neurons. The cybrids represent a spectrum of severity of compromised mitochondrial respiratory function. We have generated a cybrid containing a polymorphism that does not substantially affect function, two cybrids in different ES cell backgrounds with the same mutation in Mtco1, which causes a mild complex IV defect on different ES cell backgrounds, and one cybrid with two different mutations in Mtnd genes which cause a severe complex I defect. Cybrids could be differentiated into neurons, as evidenced by staining with neuron‐specific antibodies. Cell lines demonstrated electrophysiological properties of mature neurons, action potentials and synaptic potentials.
The most respiratory compromised cell line, CY3‐I, had the greatest deficiency in differentiation. Neuronal development was severely reduced compared to the parental ES cell line (ES‐I) and with other cybrids harbouring a mutation causing a milder biochemical derangement (CY2‐I and CY2‐VI) or the polymorphism which did not affect function (CY1‐I). In contrast, CY3‐I produced less neurons. For CY2‐I and CY2‐VI, numbers of neurons were similar to those produced by CY1‐I and the parental ES cells, and appearance of the cultures was similar to each other and indistinguishable from those derived from CY1‐I and the parental cell lines. It had less than 10% residual complex I activity and could only generate approximately half the neurons of its ES cell parent. Since the other cybrids exhibit no such compromise of ability to generate neurons, it appears that the number of neurons generated is related to the severity of the biochemical defect and there is a relatively high threshold for impairment (between 60% and 90%) for there to be an effect.
Dependence of neuronal differentiation on complex I function has been previously suggested by Papa et al. (21). They showed a marked increase in complex I activity during differentiation of mouse hippocampal cell cultures into neurons and glia. The authors reported a significantly higher complex I/complex III and complex I/complex IV activity ratios in both isolated brain mitochondria and in vitro differentiated hippocampal neurons than in other mammalian tissues. Complex I activity appears to exhibit strong control strength for respiratory chain activity, and increase in complex I activity observed in these neural tissues allows increase in energy metabolism to cope with the energy demand required for differentiation. Thus, high complex I activity appears to be required to maintain normal neuronal activity by providing a high ATP production rate. The severe complex I defect in CY3‐I would presumably result in insufficient respiratory chain activity and ATP production, leading to the observed decreased differentiation of ES cells into functional neurons.
The findings of Papa et al. (21) would support our observation that decrease in neurons is due to decreased proliferation during differentiation rather than increased cell death, although contributions by both mechanisms cannot be discounted. Complex I is a site of superoxide production; therefore, free radical damage may also be responsible for the paucity of neurons in CY3‐I. In another cybrid model of mtDNA disease affecting complex I, Wong et al. (22) observed decrease in production of both differentiated neurons and glial cells in LHON‐NT2 cell cybrids harbouring two of the most common LHON mutations: 11778G>A and 3460G>A. They saw no difference in morphology, expression of neuronal genes or mitochondrial membrane potential between the parental NT2 cells and the cybrids, but did find increased superoxide production in differentiated neurons derived from the cybrids. Examination of free radical production in our differentiated ES cell cybrids may provide further clues to the mechanisms of decreased neuronal differentiation. If increased reactive oxygen species production is found to be a contributor, this model provides the possibility of studying effects of antioxidants and other treatments on neurons in vitro, and may lead to translation into new and more effective treatments for patients. Unlike this current study, however, Wong et al. did not assess basic electrophysiological properties of their apparently differentiated neurons (22).
We believe the strength of our approach is the relative ease with which these electrophysiological studies can be extended in this model of neuronal dysfunction caused by mtDNA mutations. Here we have provided just a preliminary description of the neuronal phenotypes, the most notable feature being apparent deficit in synaptic function in cells with severely compromised mitochondrial activity. The fact that these cells respond to exogenously applied glutamate suggests that any deficit lies presynaptically, consistent with other studies suggesting a role for mitochondria in synaptic release (23, 24, 25, 26, 27, 28, 29). Mitochondrial dysfunction might affect synaptic function in a variety of ways (reviewed in Ly and Verstreken (30)) both through altered Ca2+ homeostasis within the presynaptic terminals (26, 27, 28, 29), or through deficits in ATP (24), since vesicle cycling and refilling are both ATP‐dependent (31). Our preliminary explorations clearly do not distinguish between these different possibilities, or the contribution of other factors, such as a developmental delay or reduced neuronal density. This cybrid technology will provide the means to examine synaptic and other neuronal function in controlled conditions of mitochondrial impairment, and test whether different respiratory chain defects are equivalent in their effect (32).
A potentially powerful alternative to our in vitro cell culture studies is to model mtDNA defects in transgenic mice, allowing neurophysiological analysis to be performed on brain sections, slices or indeed primary neuronal cultures. A recent publication from Fan et al. (5) has shown that mouse oocytes harbouring mutated mtDNA that underlies a severe respiratory phenotype selectively lose this mtDNA rapidly during oogenesis, suggesting that the production of a mouse model carrying a high mutant load of a highly pathogenic mtDNA mutation may not be possible. However, this was not the case for a mouse generated with the same ‘mild’ missense mutation (6589T>C in Mtco1) as two of our ES cell cybrids (33). Mice homoplasmic for this mutation had decreased cytochrome c oxidase (COX) staining in heart, decreased COX activity in brain, heart, liver and skeletal muscle, growth retardation and lactic acidosis. ES cell cybrids have 50% residual complex IV activity, a similar level to that observed in our cybrids carrying the same mutation. Potentially these mice could provide a suitable model for studying the effects of mild COX mutation on the brain. However, residual COX activity in tissues tested in the mice ranged between approximately 50% and 80%, with brain close to 80%. This may not result in high enough level of mitochondrial impairment to produce a neurological phenotype. The authors noted that patients with MTCO1 and MTCO2 mutations had epilepsy or muscle weakness, neither of which was observed in the mice. Our ES cybrids had 40% residual COX activity, but it was not possible to obtain enough differentiated neurons to perform respiratory chain enzymology in them, so we do not know the level of COX impairment in the differentiated neurons. The differentiated neurons, however, behaved no differently from those derived from the parental ES cells or the cybrids harbouring the polymorphism. It is therefore likely that this mutation is not severe enough to produce a deficit that affects neuronal function, either in our in vitro system, or in vivo in mice.
Accepting the difficulties in producing heteroplasmic mice carrying severe pathogenic mtDNA mutations, it is clear that differentiated neurons generated from ES cell‐derived cybrids provide an attractive model for human mtDNA disease and for studying the effects of mtDNA mutations on neuronal development and function. These initial studies are promising and allow for longer‐term possibilities of determining the electrophysiological effects of mtDNA mutations on neuronal subtypes, experiments that are currently being pursued.
Acknowledgements
This work was funded from a grant from the European Commission 6th Framework to R.N.L., D.M.T. and J.‐A.E. (EUMITOCOMBAT LSJM‐CT‐2004‐503116). D.M.K. was supported by an NHMRC CJ Martin Postdoctoral Fellowship; R.N.L. and D.M.T. gratefully acknowledge continuing support from the Wellcome Trust (grant no. 074454/Z/04/Z).
Re‐use of this article is permitted in accordance with the Creative Commons Deed, Attribution 2.5, which does not permit commercial exploitation.
References
- 1. Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH et al (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. [DOI] [PubMed] [Google Scholar]
- 3. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE et al (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. [DOI] [PubMed] [Google Scholar]
- 4. Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA et al (2008) DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 40, 392–394. [DOI] [PubMed] [Google Scholar]
- 5. Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE et al (2008) A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. King MP, Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503. [DOI] [PubMed] [Google Scholar]
- 7. Trounce I, Wallace DC (1996) Production of transmitochondrial mouse cell lines by cybrid rescue of rhodamine‐6G pre‐treated 1‐cells. Somat. Cell Mol. Genet. 22, 81–85. [DOI] [PubMed] [Google Scholar]
- 8. Chomyn A, Martinuzzi A, Yoneda M, Daga A, Hurko O, Johns D et al (1992) MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. USA 89, 4221–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Acin‐Pérez R, Bayona‐Bafaluy MP, Bueno M, Machicado C, Fernandez‐Silva P, Pérez‐Martos A et al (2003) An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum. Mol. Genet. 12, 329–339. [DOI] [PubMed] [Google Scholar]
- 10. Moreno‐Loshuertos R, Acin‐Pérez R, Fernández‐Silva P, Movilla N, Pérez‐Martos A, Rodriguez de Cordoba S et al (2006) Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 38, 1261–1268. [DOI] [PubMed] [Google Scholar]
- 11. Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI (1995) Embryonic stem cells express neuronal properties in vitro. Dev. Biol. 168, 342–357. [DOI] [PubMed] [Google Scholar]
- 12. Williams RL, Hilton DJ, Pease S, Willson TA, Stewart CL, Gearing DP et al (1988) Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 336, 684–687. [DOI] [PubMed] [Google Scholar]
- 13. McMahon AP, Bradley A (1990) The Wnt‐1 (int‐1) proto‐oncogene is required for development of a large region of the mouse brain. Cell 62, 1073–1085. [DOI] [PubMed] [Google Scholar]
- 14. Voss AK, Thomas T, Gruss P (1997) Germ line chimeras from female ES cells. Exp. Cell Res. 230, 45–49. [DOI] [PubMed] [Google Scholar]
- 15. Bayona‐Bafaluy MP, Acin‐Pérez R, Mullikin JC, Park JS, Moreno‐Loshuertos R, Hu P et al (2003) Revisiting the mouse mitochondrial DNA sequence. Nucleic Acids Res. 31, 5349–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bai Y, Attardi G (1998) The mtDNA‐encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 17, 4848–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sligh JE, Levy SE, Waymire KG, Allard P, Dillehay DL, Nusinowitz S et al (2000) Maternal germ‐line transmission of mutant mtDNAs from embryonic stem cell‐derived chimeric mice. Proc. Natl. Acad. Sci. USA 97, 14461–14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW et al (1996) Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann. Neurol. 39, 343–351. [DOI] [PubMed] [Google Scholar]
- 19. Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR (1999) Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology 52, 1255–1264. [DOI] [PubMed] [Google Scholar]
- 20. Ying QL, Stavridis M, Griffiths D, Li M, Smith A (2003) Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat. Biotechnol. 21, 183–186. [DOI] [PubMed] [Google Scholar]
- 21. Papa S, Petruzzella V, Scacco S, Vergari R, Panelli D, Tamborra R et al (2004) Respiratory complex I in brain development and genetic disease. Neurochem. Res. 29, 547–560. [DOI] [PubMed] [Google Scholar]
- 22. Wong A, Cavelier L, Collins‐Schramm HE, Seldin MF, McGrogan M, Savontaus ML et al (2002) Differentiation‐specific effects of LHON mutations introduced into neuronal NT2 cells. Hum. Mol. Genet. 11, 431–438. [DOI] [PubMed] [Google Scholar]
- 23. Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C et al (2008) Docking of axonal mitochondria by syntaphilin controls their mobility and affects short‐term facilitation. Cell 132, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ (2005) Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. [DOI] [PubMed] [Google Scholar]
- 25. Nguyen PV, Marin L, Atwood HL (1997) Synaptic physiology and mitochondrial function in crayfish tonic and phasic motor neurons. J. Neurophysiol. 78, 281–294. [DOI] [PubMed] [Google Scholar]
- 26. David G, Barrett EF (2003) Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J. Physiol. 548, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. David G, Talbot J, Barrett EF (2003b) Quantitative estimate of mitochondrial [Ca2+] in stimulated motor nerve terminals. Cell Calcium 33, 197–206. [DOI] [PubMed] [Google Scholar]
- 28. García‐Chacón LE, Nguyen KT, David G, Barrett EF (2006) Extrusion of Ca2+ from mouse motor terminal mitochondria via a Na+‐Ca2+ exchanger increases post‐tetanic evoked release. J. Physiol. 574, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Talbot JD, David G, Barrett EF (2003) Inhibition of mitochondrial Ca2+ uptake affects phasic release from motor terminals differently depending on external [Ca2+]. J. Neurophysiol. 90, 491–502. [DOI] [PubMed] [Google Scholar]
- 30. Ly CV, Verstreken P (2006) Mitochondria at the synapse. Neuroscientist 12, 291–299. [DOI] [PubMed] [Google Scholar]
- 31. Murthy VN, De Camilli P (2003) Cell biology of the presynaptic terminal. Annu. Rev. Neurosci. 26, 701–728. [DOI] [PubMed] [Google Scholar]
- 32. Calabresi P, Gubellini P, Picconi B, Centonze D, Pisani A, Bonsi P et al (2001) Inhibition of mitochondrial complex II induces a long‐term potentiation of NMDA‐mediated synaptic excitation in the striatum requiring endogenous dopamine. J. Neurosci. 21, 5110–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kasahara A, Ishikawa K, Yamaoka M, Ito M, Watanabe N, Akimoto M et al (2006) Generation of trans‐mitochondrial mice carrying homoplasmic mtDNAs with a missense mutation in a structural gene using ES cells. Hum. Mol. Genet. 15, 871–881. [DOI] [PubMed] [Google Scholar]