

Abstract
Triplex-forming oligonucleotides (TFOs) can bind specifically to polypurine sequences in double-stranded DNA. A single interruption of this polypurine tract can greatly destabilize triplex formation. The stability of triplexes can be significantly enhanced by covalently linking the TFO to its DNA target with reactive functional groups conjugated to the TFO. Covalently cross-linked TFOs are effective inhibitors of transcription of the target DNA sequence. We have designed a TFO with a platinum-modified base that can interact with and cross-link to a cytosine interruption in the polypurine tract of a target DNA duplex. The TFO contains an N4-(aminoalkyl)cytosine derivatized with cis- or trans-diamminediaquaplatinum(II). When bound to its target, the tethered platinum of the TFO can reach across the major groove and form an adduct with the guanine N7 of the interrupting C·G base-pair. The optimal tether length is five methylene groups, and cross-linking is most efficient when the tether is modified with trans-diamminediaquaplatinum. Cross-linking requires that the TFO is bound to its designated DNA target. Addition of cyanide to the cross-linked TFO product reversed the cross-link, behavior that is consistent with the presence of a Pt-guanine adduct. The kinetics of the cross-linking reaction were studied and the half-life of the cross-linking reaction was approximately three hours. Our results demonstrate that platinum-conjugated TFOs can be designed to cross-link with DNA targets that contain a single pyrimidine interruption. Modifications of this type may prove useful for expanding the DNA sequences that can be targeted by TFOs and increasing the stability of the resulting triplexes.
Keywords: cisplatin, triplex-forming oligonucleotide, cross-link, pyrimidine interruption
Introduction
Oligopyrimidines can bind, via the major groove, to contiguous polypurine tracts in double-stranded DNA to form triple-stranded complexes [1-7]. These triplex-forming oligonucleotides (TFOs), whose backbone orientation is parallel to that of the polypurine tract, bind by forming T·AT and C+·GC triads. There has been considerable interest in triplex-forming oligonucleotides because of their potential ability to control gene expression either by interfering with DNA transcription [6, 8-12] or by creating gene-inactivating mutations in DNA [13-16].
Because TFOs can be displaced by DNA binding proteins such as RNA polymerase, efforts have been made to enhance their biological effectiveness by enabling them to link covalently to their DNA target. For example TFOs have been conjugated with alkylating agents such as bromoacetamide [17, 18] or chlorambucil [19, 20]. Once the TFO has bound to its DNA target, the alkylating group can react with nearby guanine residues to covalently attach the TFO to the target. TFOs conjugated with derivatives of psoralen are able to cross-link with their DNA targets upon irradiation with long wavelength UV light [21-26]. Platinum derivatives of TFOs have also been found capable of forming adducts with DNA. For example TFOs containing transplatin-modified guanine residues [27-29] or transplatin-modified cytosine residues [27, 28, 30] can form adducts through coordination of the platinum with the N7 of a guanine in the target DNA. In addition, platinum derivatized TFOs in which cisplatin is tethered via a linker to the 5′-end of the TFO have been shown to form very stable adducts with their DNA targets [31, 32]. These platinum cross-linked TFOs have the advantage of forming a more stable adduct than those formed by alkylating agents, and unlike psoralen-modified TFOs, do not require activation with UV light.
The requirement of a polypurine tract for stable triplex formation places limitations on the sequences that can be targeted by TFOs. A single pyrimidine interruption in the polypurine tract can greatly reduce triplex stability [33-38]. A considerable effort has been made to design modified nucleotides that are able to address a pyrimidine interruption and thus stabilize the triplex [38-66]. Although these modifications have achieved varying degrees of success, pyrimidine interruptions still present an obstacle to stable triplex formation.
We have previously reported that N4-(2-aminopropyl) and N4-(2-acetamidopropyl)-modified cytosines could address an interrupting cytosine by making contacts across the major groove with the complementary guanine of the interrupting C·G base-pair [41]. Examination of molecular models suggested that tethering a cisplatin group to N4-alkyl-modified cytosine in the TFO could position the platinum to form an adduct with the N7 of the guanine of an interrupting C·G base-pair as shown schematically in Figure 1. Such interaction would irreversibly anchor the TFO to the interrupted target DNA. In this report we demonstrate that such platinum derivatized TFOs can indeed form highly stable adducts when targeted to a purine tract in double-stranded DNA that contains a single cytosine interruption.
Figure 1. Platinated N4-(aminoalkyl)cytosine TFOs.
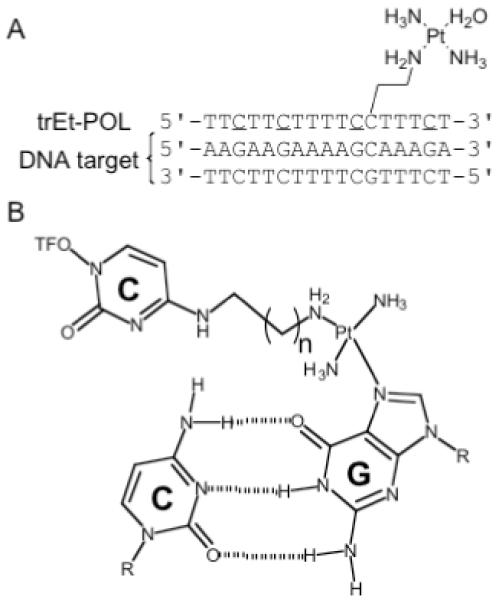
(a) Sequence of Pol DNA target duplex and POL TFO, trEt-POL; C is 5-methylcytosine (b) Proposed cross-linked CPt·CG triad.
Materials and Methods
Protected 2′-deoxy- and 2′-O-methylribonucleoside-3′-O-bis(diisopropylamino)-β-cyanoethylphosphoramidites, 2′-deoxy- and 2′-O-methylnucleoside derivatized controlled pore glass supports were purchased from Glen Research Inc. (Sterling, VA). The base protecting groups were benzoyl (A, C) or isobutyryl (G). 4,5-dicyanoimidazole, 5-ethylthiotetrazole, Cap Mix A (acetic anhydride/THF/pyrimidine), Cap Mix B (16% 1-methylimidazole in THF) and oxidizing solution were purchased from EMD Chemicals (Gibbstown, NJ). 2′-O-methyl-5-methylcytosine phosphoramidite and 2′-O-methyl-5-methyluridine derivatized CPG were purchased from ChemGenes Corp. (Ashland, MA). Phosphoramidite solutions were prepared using synthesis-grade acetonitrile that was dried and stored over calcium hydride. 5 mM solutions of cis- and trans-diamminedichloroplatinum(II) (cis- and trans-DDP) were incubated with 10 mM silver nitrate overnight at 37 °C. The resulting precipitate was centrifuged, and the supernatant containing cis- or trans-diamminediaquaplatinum(II) dinitrate was stored in the dark at room temperature. MALDI-TOF mass spectrometry analysis was performed at the AB Mass Spectrometry/Proteomics Facility at The Johns Hopkins School of Medicine, with support from the National Center for Research Resources Shared Instrumentation, Grant 1S10-RR14702.
Synthesis of oligonucleotides
The sequences of the TFOs are shown in Table 1. The sequence of the target duplex is shown in Figure 1a. The sequence of the scrambled duplex is (5′-AGAAGAAACAAGAAAAG-3′ and 5′-CTTTTCTTGTTTCTTCT-3′). These oligonucleotides were synthesized on an ABI DNA synthesizer (model 392 or 3400), using standard automated phosphoramidite chemistry and procedures. The phosphoramidite solutions were made up to a concentration of 0.15 M, and 5-ethylthiotetrazole was used as the activating agent. Coupling times were 120 s for deoxyribonucleoside phosphoramidites and 360 s for 2′-O-methylribonucleoside phosphoramidites. The oligonucleotides were deprotected and removed from the solid support by incubating the CPG in a solution containing 300 μL of concentrated ammonium hydroxide and 100 μL of 95% ethanol for 3.5 hr at 55 °C. The supernatant from this reaction was evaporated to dryness, and the residue redissolved in 200 μL of 50% aqueous acetonitrile. The oligonucleotides were purified by strong anion exchange HPLC on a Dionex DNAPac PA 100-4 5 250 mm column (Dionex Corp., Sunnyvale, CA), using a 30 min linear gradient of 0 to 0.5 M sodium chloride in a solution containing 100 mM Tris, pH 7.8, and 10% acetonitrile at a flow rate of 1.0 mL/min. The purified oligonucleotides were desalted on a C-18 Clarity Tube (Phenomenex, Inc., Torrance, CA). The concentration of purified oligomers was determined from their UV absorbance at 260 nm, using calculated extinction coefficients based on nearest neighbor approximations [67]. The compositions of the oligonucleotides were confirmed by MALDI-TOF mass spectrometry (see Supplementary Table 1).
Table 1. Thermal denaturation of non-platinated and platinated N4-(aminoalkyl)C-TFOs.
| mr-TTCTTCTTTTCXTTTCTa | ||
|---|---|---|
| Oligonucleotide | Xb | Tm (°C)c |
| POL | dC | 57 |
| Et-POL | dCEt | 54 |
| Pr-POL | dCPr | 53 |
| Bu-POL | dCBu | 51 |
| Pe-POL | dCPe | 50 |
| He-POL | dCHe | 52 |
| diEt-POL | dCEt-d | 33 |
| trPe-POL | dCPe-tr | ND |
mr, 2′-O-methylribo and C, 5-methylcytosine.
d, 2′-deoxyribo; CEt, N4-(2-aminoethyl)cytosine; CPr, N4-(3-aminopropyl)cytosine; CBu, N4-(4-aminobutyl)cytosine; CPe, N4-(5-aminopentyl)cytosine; CHe, N4-(6-aminohexyl)cytosine; CEt-d, [Pt(dien)Cl] derivative of N4-(2-aminoethyl)cytosine; CPe-tr, transplatin derivative of N4-(5-aminopentyl)cytosine.
Melting temperatures were determined at pH 6.5 in a buffer containing 100 mM 3-(N-morpholino)propanesulfonic acid, 100 mM sodium acetate, 2.5 mM magnesium acetate; ND, not determined.
Preparation of aminoalkyl-modified TFOs and platinum-modified TFOs
2′-O-methyl TFOs (Et-, Pr-, Bu-, Pe-, He-POL, see Table 1) were prepared as previously described [68]. Briefly, the CPG bound oligonucleotide, which contained O4-triazole-deoxyuridine at position X in the oligomer (see Table 1), was incubated with a 0.25 M solution of diaminoalkane in dry pyridine for 30 min at room temperature. The derivatized CPG was washed with dry acetonitrile and the N4-(aminoalkyl)deoxycytidine-modified oligomers were deprotected and purified, as described above.
The platinum-modified TFOs were prepared from the deprotected and purified N4-(aminoalkyl)deoxycytidine-modified TFOs, as previously described [68]. Briefly, a 10 μM solution of the aminoalkyl-modified TFO in water was incubated with 100 μM cis- or 200 μM trans-diamminediaquaplatinum(II) dinitrate at 37 °C for 2-3 hr. The platinated TFOs were purified by SAX-HPLC as described above and their compositions were confirmed by MALDI-TOF mass spectrometry (Supplementary Table 1).
Thermal denaturation experiments
A solution of 1 μM target duplex and 1 μM TFO was prepared in triplex buffer that contained 100 mM MOPS, pH 6.5, 100 mM sodium acetate, and 2.5 mM magnesium acetate. The solution containing the three strands was heated to 90 °C for 10 min, allowed to cool slowly to room temperature and stored overnight at 4 °C to anneal the triplex. Absorbance at 260 nm was monitored using a Cary 3E spectrophotometer as the solution was heated from 5 to 80 °C at a rate of 0.4 °C/min. The melting temperature was determined by plotting the first derivative of the absorbance vs. temperature and is accurate to ±1 °C.
Determination of binding of platinum-modified TFOs to DNA
The 5′-hydroxyl group of the pyrimidine strand of the target duplex was labeled using polynucleotide kinase and γ-[32P]-ATP (specific activity of 10 Ci/mmol). The labeled strand was mixed with an equimolar amount of unlabeled complementary strand, and the solution was evaporated to dryness. The residue was redissolved in 100 μL triplex buffer to a final duplex concentration of 1 μM. The duplex solution was heated to 90 °C for 10 min, and then slowly cooled to room temperature. Solutions containing 20-100 pmol of platinated TFO were evaporated to dryness, the residue redissolved in 10 μL of the labeled duplex solution and the solution was stored overnight at 4 °C. A 2 μL aliquot of 50% glycerol was added to each sample, and the samples were loaded on a 20% native polyacrylamide gel that was run at 100 V and 4 °C in triplex buffer. The gel was dried, imaged and radioactivity quantitated using a Fuji FLA-7000 phosphorimager.
Reaction of platinum-modified TFOs with DNA
1 μM [32P]-labeled target duplex (or scrambled duplex) with a specific activity of 10 Ci/mmol was prepared as described above. Solutions containing 40-200 pmol of platinated TFO were evaporated to dryness and redissolved in 10 μL of the labeled duplex solution to a final concentration of 4-20 μM TFO. Each reaction mixture was incubated at 37 °C and quenched with 7 μL of formamide gel loading buffer. The samples were loaded on a 20% denaturing polyacrylamide gel and run at 800 V. The gel was dried, imaged and quantitated using a Fuji FLA-7000 phosphorimager.
Pt-TFO cross-link reversal by cyanide treatment
The platinum-modified TFO, trPe-POL, was cross-linked to the target duplex using the procedure described above. The slower migrating, cross-linked band was excised from the denaturing gel and extracted by shaking in 0.1 M ammonium acetate buffer, pH 6.25, and 20% acetonitrile at 37 °C for 48 hr. The DNA was precipitated in 100% ethanol overnight at -20 °C, and the precipitate was collected by centrifugation for 20 min at 4 °C. The pellet was resuspended in 70% ethanol and centrifuged for 10 min at 4 °C. The pellet was dried under vacuum and dissolved in either 20 μL of water or 20 μL of 200 mM sodium cyanide. Samples were incubated at 37 °C for 48 hr and electrophoresed on a 20% denaturing polyacrylamide gel at 800 V. The gel was dried and imaged using a Fuji FLA-7000 phosphorimager.
Results and Discussion
Binding of N4-(aminoalkyl)C-TFOs and their platinum derivatives to DNA
Figure 1a shows the triplex formed by a double-stranded DNA duplex and a platinum derivatized TFO, trEt-POL. The sequence of the target duplex corresponds to nucleotides 4899-4915 found in the coding region of the HIV-1 polymerase (pol-1) gene [69]. The polypurine strand of the target is interrupted by a single cytosine residue located near the 3′-end of the strand. We have previously shown that a similar target lacking the cytosine interruption forms stable triplexes with unmodified deoxyribo- and 2′-O-methylribo-TFOs, and that the melting temperature of the triplex formed by the 2′-O-methylribo-TFO is reduced from 78 °C to 57 °C at pH 6.5 when the target contains the cytosine interruption [68].
As shown in Figure 1a, the platinum derivatized TFO contains a platinated N4-(aminoalkyl)cytosine residue positioned opposite the interrupting C·G base-pair of the target duplex. This placement was predicted to allow the aminoalkyl tethered platinum group to reach across the major groove of the duplex and form an adduct with the guanine N7 of the interrupting C·G, as shown schematically in Figure 1b. To maximize binding by increasing base stacking and prearranging the oligo in an RNA-like configuration, the TFO contains 5-methylcytosines, C, and consists of 2′-O-methylribonucleotides, with the exception of the platinated nucleotide which contains a deoxyribose sugar. The length of the tether was varied between 2 to 6 methylene groups and the tether was derivatized with both cis- and trans-diamminediaquaplatinum(II).
Table 1 lists the various non-platinated N4-(aminoalkyl)cytosine-derivatized TFOs and the temperatures (Tm) at which the triplexes formed by these TFOs dissociated into three separate strands. A slight decrease in Tm is seen upon addition of the 2-aminoethyl tether to the cytosine residue. However, there is no significant change in Tm as the length of the tether increases. Derivatization of the 2-aminoethyl tether with a non-reactive platinum group (diEt-POL) resulted in a significant decrease, 21 °C, in the Tm of the third strand. This decrease is presumably due to steric hindrance of the bulky platinum group in the major groove [68].
Platinated derivatives of the N4-(aminoalkyl)C-TFOs were prepared by reacting the N4-(aminoalkyl)C-TFO with cis- or trans-diamminediaquaplatinum(II) dinitrate. The platinated TFOs were purified by strong anion exchange HPLC and characterized by mass spectrometry. The interactions of the platinated TFOs with the DNA duplex target were monitored by electrophoresis on a native polyacrylamide gel. Figure 2a shows the results of a binding experiment in which increasing concentrations of the trans-platinated N4-(aminopentyl)C-TFO, trPe-POL, were added to the target duplex. Under the conditions of the experiment, 4 °C at pH 6.5, 2 μM underivatized TFO, POL, formed approximately 90% triplex, whereas only 30% triplex formation was observed in the presence of 2 μM trPe-POL. Similar experiments were carried out with both the cis- and trans-platinated N4-(aminoalkyl)C-TFOs and the results of these experiments are summarized in Figure 2b. The platinated TFOs with an ethyl, propyl or butyl tether gave comparable levels of triplex formation, ∼30%, at a concentration of 10 μM. On the other hand, approximately 65% triplex formed in the presence of 10 μM trans-platinated N4-(aminopentyl)C-TFO, trPe-POL. This enhanced binding may be due to the increased flexibility of the linker that allows the bulky platinum group to be better accommodated by the major groove of the duplex.
Figure 2. Interaction of trPe-POL with the Pol DNA duplex.
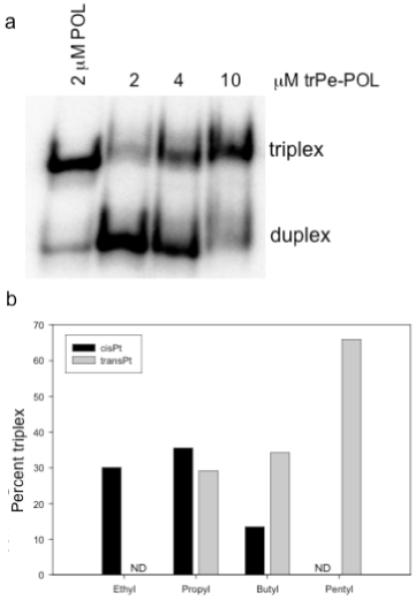
(a) The platinated N4-(aminopentyl)C-TFO was incubated with 1 μM target duplex at 4 °C and the reaction mixture was run on a 20% native polyacrylamide gel. (b) Effect of tether length on binding of platinated N4-(aminoalkyl)C-TFOs to Pol DNA duplex. The platinated TFOs (10 μM) were incubated with the duplex (1 μM) at 4 °C; ND, not determined.
Reaction of platinated N4-(aminoalkyl)C-TFOs with the pyrimidine strand of target DNA
Platinated N4-(aminoalkyl)C-TFOs were tested for their abilities to react with the guanine of the interrupting C·G base-pair of the target duplex. We have previously demonstrated that the N7 of this guanine is accessible to platination by cis-diamminediaquaplatinum(II) within a triple-stranded helix [68]. A 4 to 20 fold molar excess of platinated TFO was incubated with target DNA whose pyrimidine strand was 5′-end-lableled with 32P phosphate. The reaction mixture was then subjected to electrophoresis on a denaturing polyacrylamide gel and the percentage of TFO-cross-linked pyrimidine strand was determined. As shown in Figure 3a, the amount of cross-linked product formed by the trans-platinated N4-(aminopentyl)C-TFO, trPe-POL, increased as the concentration of TFO increased, reaching the level of 7.5% cross-linked at 20 μM TFO. Similar experiments were carried out with the other platinated TFOs. Although the cis-platinated N4-(aminoethyl)C-TFO, cisEt-POL, formed a triplex with the target DNA (see Figure 2b), cross-linking to the pyrimidine strand was not observed as shown in Figure 3b. This could be the result of the aminoethyl tether simply being too short to reach across the major groove and position the platinum near the interrupting guanine. Consistent with this idea is the observation that cross-linking increased as the length of the tether increased from 3 to 5 methylene groups. Further lengthening of the tether, however, diminished cross-linking, which may reflect increased entropic effects that interfere with proper alignment of the platinum with the guanine target. Because the aminopentyl tether provided the greatest percentage of cross-linking, subsequent studies were carried out using the trans-platinated N4-(aminopentyl)C-TFO, trPe-POL.
Figure 3. Cross-linking of trPe-POL to the pyrimidine strand of the Pol DNA duplex.
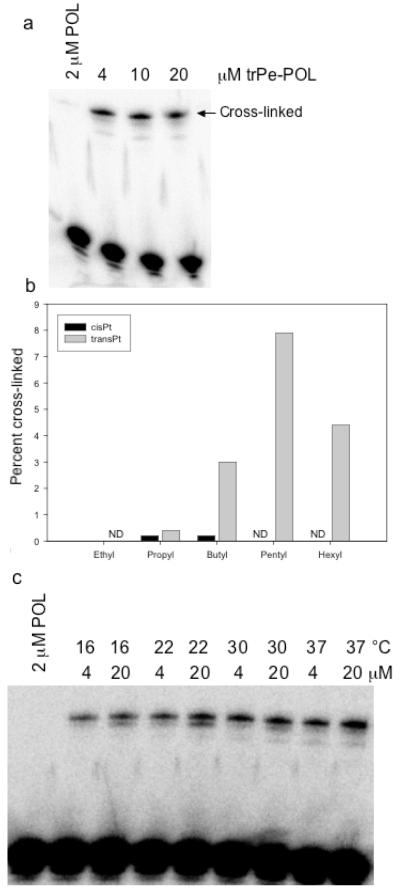
(a) The platinated N4-(aminopentyl)C-TFO was incubated with 1 μM target duplex at 37 °C for 48 hrs and the reaction mixtures were run on a 20% denaturing polyacrylamide gel. (b) Effect of tether length on cross-linking of platinated N4-(aminoalkyl)C-TFOs to Pol DNA duplex. The platinated TFOs (20 μM) were incubated with the duplex (1 μM) at 37 °C for 48 hrs. ND, not determined. (c) Effect of temperature on cross-linking of the platinated trPe-POL to Pol DNA duplex. The platinated N4-(aminopentyl)C-TFO was incubated with 1 μM target duplex at the indicated temperatures for 48 hrs and the reaction mixtures were run on a 20% denaturing polyacrylamide gel.
Figure 3c shows the effect of temperature on the cross-linking by the trans-platinated N4-(aminopentyl)C-TFO. Although increasing temperature would be expected to decrease the stability of the triplex, the amount of cross-linking increased from 16 °C to 37 °C in the presence of 20 μM trPe-POL. Most likely this increase reflects an increase in the rate of platination as the temperature is raised, and implies that the increase in the Pt-guanine reaction more than compensates for the reduction in triplex stability at higher temperatures.
Specificity of platinated TFO binding and cross-linking
We examined the interaction of the trans-platinated N4-(aminopentyl)C-TFO, trPe-POL, with a scrambled version of the POL DNA target duplex. This scrambled duplex has the same base composition as the POL DNA target and a single C interruption in the polypurine strand (see Materials and Methods). The pyrimidine strand of the duplex was 5′-end-labeled with 32P phosphate and binding between the duplex and TFO was monitored by electrophoresis on a native polyacrylamide gel. As expected, the unmodified TFO, POL, formed a stable triplex with the POL DNA target duplex, but not with the scrambled duplex, as shown in Figure 4a, lanes 3 and 4, respectively. Similar results were observed with the trans-platinated N4-(aminopentyl)C-TFO. As shown in lane 5, this TFO formed a triplex with the POL DNA duplex. However, as seen in lanes 6-8, no triplex formation was observed with the scrambled duplex, even with 10 μM TFO. This result shows that triplex binding specificity is not affected by platinum modification of the TFO. It also suggests that the platinated TFO does not react indiscriminately with the target duplex. To confirm this, the binding reaction mixtures were also subjected to electrophoresis on a denaturing polyacrylamide gel as shown in Figure 4b. As previously demonstrated and shown in lane 2, the trans-platinated N4-(aminopentyl)C-TFO cross-links to the pyrimidine strand of the POL DNA duplex. In contrast, essentially no cross-linked product was formed when the scrambled duplex was incubated with increasing concentrations of the platinated TFO (lanes 3-5). Furthermore, no cross-linking was observed when the trans-platinated N4-(aminopentyl)C-TFO was incubated with the POL DNA target in the presence of 200 μM unmodified TFO (lanes 6-8). In this case, the unmodified TFO out-competes the platinated TFO for binding to the target thus preventing the cross-linking reaction. Taken together, the experiments described above show that the platinated TFO binds specifically to its designated target and that cross-linking to the target depends upon TFO binding.
Figure 4. Specificity of trPe-POL binding and cross-linking.
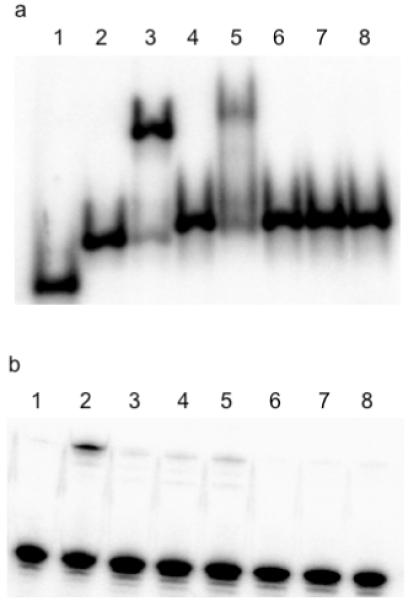
(a) Interaction of trPe-POL with the Pol DNA duplex or the scrambled DNA duplex. The platinated N4-(aminopentyl)C-TFO was incubated with Pol DNA duplex or scrambled duplex at 4 °C and the reaction mixtures were run on a 20% native polyacrylamide gel. Lane 1: single stranded 17-mer, lane 2: target duplex, lane 3: 1 μM target duplex plus 2 μM POL, lane 4: 1 μM scrambled duplex plus 2 μM POL, lane 5: 1 μM target duplex plus 10 μM trPe-POL, lanes 6-8: 1 μM target duplex plus 2, 4, or 10 μM trPe-POL (respectively). (b) Cross-linking of trPe-POL with the Pol DNA duplex or the scrambled DNA duplex. The platinated N4-(aminopentyl)C-TFO was incubated with Pol DNA duplex or scrambled duplex at 4 °C and the reaction mixtures were run on a 20% denaturing polyacrylamide gel. Lane 1: 1 μM target duplex plus 2 μM POL, lane 2: 1 μM target duplex plus 10 μM trPe-POL, lanes 3-5: 1 μM scrambled duplex plus 4, 10 or 20 μM trPe-POL (respectively), lanes 6-8: 1 μM target duplex plus 4, 10 or 20 μM trPe-POL (respectively) and 200 μM POL.
We also determined if the transplatin-modified N4-(aminopentyl)C-TFO was able to react with the purine strand of the target duplex. Approximately 8% cross-linked product was observed when 20 μM trPe-POL was incubated with 1 μM Pol DNA in which the purine strand was labeled with 32P phosphate (Supplementary Figure 1). This cross-linking most likely occurs as a result of platination of the guanine neighboring the interrupting cytosine in the purine strand of the target DNA. Also, there are two cross-linked bands present in the denaturing polyacrylamide gel of this reaction, which suggests the platinum group may also be able to react with the neighboring adenine residue in the purine strand of the target DNA. While not the original intent of this modification, these cross-links still represent covalent adduct formation, which could still be effective at inhibiting transcription of the target gene.
Characterization of platinated TFO cross-linking reaction
Cyanide, which has been shown to be an exceptionally strong ligand for platinum, is able to displace other platinum ligands, such as N7 of guanine [70-73]. To verify that cross-linking between the trans-platinated N4-(aminopentyl)C-TFO and the target duplex resulted from reaction of the platinum with the pyrimidine strand, we treated the gel-purified cross-linked product with sodium cyanide to reverse the cross-link. The cyanide-treated product was then rerun on a denaturing polyacrylamide gel. As shown in Figure 5, treatment of the cross-linked product with water left the cross-linked product intact. In contrast, treatment with cyanide disrupted the cross-link and caused the labeled pyrimidine strand to revert to its original mobility on the gel. This result is consistent with the notion that the platinum of the N4-(aminopentyl)C reacts with the guanine in the pyrimidine strand of the target.
Figure 5. Reversal of TFO cross-linking with cyanide treatment.
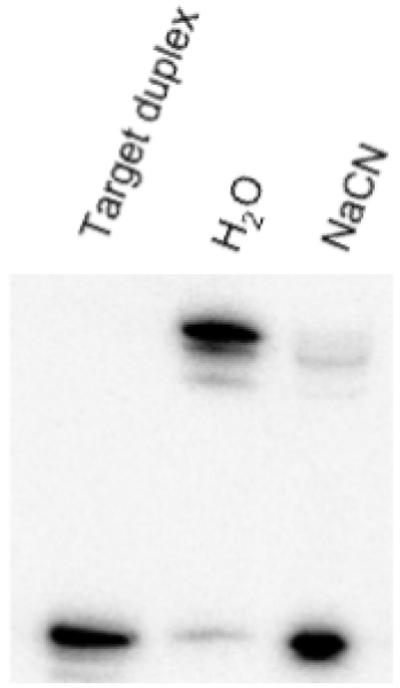
The cross-linked product between the pyrimidine strand of the Pol DNA duplex and platinated N4-(aminopentyl)C-TFO was isolated from a gel and treated with either water or 200 mM sodium cyanide. The samples were then run on a 20% denaturing polyacrylamide gel, shown here.
The kinetics of cross-linking were determined at 37 °C. As shown in Figure 6, the reaction is essentially over after 12 hrs and the half-life of the reaction is approximately 3 hrs. The overall low percentage of cross-linking may be a consequence of reaction of the platinum group with the buffer. We have shown in control experiments that pre-incubation of platinated TFOs with buffer reduces their ability to cross-link with their DNA target (data not shown). In addition or alternatively, intramolecular platination of one of the 5-methylcytosines within the TFO could occur. This type of intramolecular platination has been reported by others and would result in an inactive TFO [27, 28, 74].
Figure 6. Kinetics of cross-linking of trPe-POL to the Pol DNA duplex.
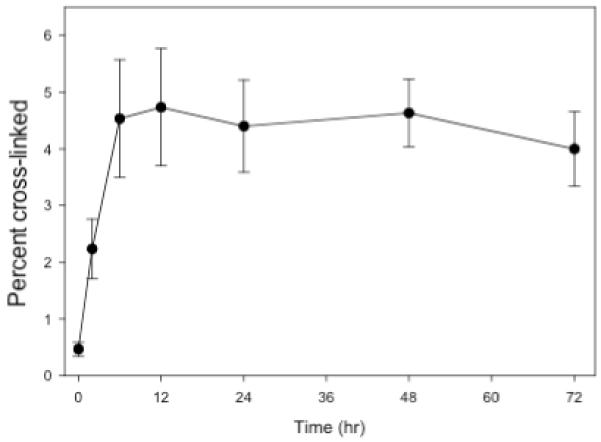
Platinated N4-(aminopentyl)C-TFO, trPe-POL, (20 μM) was incubated with 1 μM target duplex at 37 °C in pH 6.5 MOPS, 100 mM sodium acetate & 2.5 mM magnesium acetate.
Conclusion
Our results show that platinated TFOs can be designed to bind and cross-link to a DNA target that contains a cytosine interruption of the polypurine tract. In addition to expanding the types of sequences that can be targeted by TFOs, the ability to cross-link the TFO to its target could significantly improve the biological activity of the TFO, such as blocking transcription of a targeted gene. This type of modification might be used to design TFOs that can effectively modulate gene expression in cells.
Supplementary Material
Acknowledgements
This research was supported by a grant from the National Institutes of Health, GM 057140 (PSM).
References
- 1.Thuong NT, Helene C. Angew Chem Int Ed Engl. 1993;32:666–690. [Google Scholar]
- 2.Radhakrishnan I, Patel DJ. Biochemistry. 1994;33:11405–11416. doi: 10.1021/bi00204a001. [DOI] [PubMed] [Google Scholar]
- 3.Frank-Kamenetskii MD, Mirkin SM. Annu Rev Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- 4.Chan PP, Glazer PM. J Mol Med. 1997;75:267–282. doi: 10.1007/s001090050112. [DOI] [PubMed] [Google Scholar]
- 5.Fox KR. Curr Med Chem. 2000;7:17–37. doi: 10.2174/0929867003375506. [DOI] [PubMed] [Google Scholar]
- 6.Knauert MP, Glazer PM. Hum Mol Genet. 2001;10:2243–2251. doi: 10.1093/hmg/10.20.2243. [DOI] [PubMed] [Google Scholar]
- 7.Vasquez KM, Glazer PM. Q Rev Biophys. 2002;35:89–107. doi: 10.1017/s0033583502003773. [DOI] [PubMed] [Google Scholar]
- 8.Postel EH, Flint SJ, Kessler DJ, Hogan ME. Proc Natl Acad Sci USA. 1991;88:8227–8231. doi: 10.1073/pnas.88.18.8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young SL, Krawczyk SH, Matteucci MD, Toole JJ. Proc Natl Acad Sci USA. 1991;88:10023–10026. doi: 10.1073/pnas.88.22.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duval-Valentin G, Thuong NT, Helene C. Proc Natl Acad Sci USA. 1992;89:504–508. doi: 10.1073/pnas.89.2.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaggiante B, Morassutti C, Tolazzi G, Michelutti A, Baccarani M, Quadrifoglio F. FEBS Lett. 1994;352:380–384. doi: 10.1016/0014-5793(94)00995-3. [DOI] [PubMed] [Google Scholar]
- 12.Carbone GM, McGuffie E, Napoli S, Flanagan CE, Dembech C, Negri U, Arcamone F, Capobianco ML, Catapano CV. Nucleic Acids Res. 2004;32:2396–2410. doi: 10.1093/nar/gkh527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang G, Levy DD, Seidman MM, Glazer PM. Mol Cell Biol. 1995;15:1759–1768. doi: 10.1128/mcb.15.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang G, Seidman MM, Glazer PM. Science. 1996;271:802–805. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- 15.Vasquez KM, Narayanan L, Glazer PM. Science. 2000;290:530–533. doi: 10.1126/science.290.5491.530. [DOI] [PubMed] [Google Scholar]
- 16.Faruqi AF, Datta HJ, Carroll D, Seidman MM, Glazer PM. Mol Cell Biol. 2000;20:990–1000. doi: 10.1128/mcb.20.3.990-1000.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor MJ, Dervan PB. Bioconjug Chem. 1997;8:354–364. doi: 10.1021/bc970035x. [DOI] [PubMed] [Google Scholar]
- 18.Grant KB, Dervan PB. Biochemistry. 1996;35:12313–12319. doi: 10.1021/bi9608469. [DOI] [PubMed] [Google Scholar]
- 19.Reed MW, Lukhtanov EA, Gorn V, Kutyavin IV, Gall AA, Wald A, Meyer RB. Bioconjug Chem. 1998;9:64–71. doi: 10.1021/bc970134a. [DOI] [PubMed] [Google Scholar]
- 20.Kutyavin IV, Gamper HB, Gall AA, Meyer RB. J Am Chem Soc. 1993;115:9303–9304. [Google Scholar]
- 21.Giovannangeli C, Thuong NT, Helene C. Nucleic Acids Res. 1992;20:4275–4281. doi: 10.1093/nar/20.16.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takasugi M, Guendouz A, Chassignol M, Decout JL, Lhomme J, Thuong NT, Helene C. Proc Natl Acad Sci USA. 1991;88:5602–5606. doi: 10.1073/pnas.88.13.5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Havre PA, Gunther EJ, Gasparro FP, Glazer PM. Proc Natl Acad Sci USA. 1993;90:7879–7883. doi: 10.1073/pnas.90.16.7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ping YH, Rana TM. Biochemistry. 2005;44:2501–2509. doi: 10.1021/bi0488707. [DOI] [PubMed] [Google Scholar]
- 25.Guieysse AL, Praseuth D, Giovannangeli C, Asseline U, Helene C. J Mol Biol. 2000;296:373–383. doi: 10.1006/jmbi.1999.3466. [DOI] [PubMed] [Google Scholar]
- 26.Besch R, Marschall C, Schuh T, Giovannangeli C, Kammerbauer C, Degitz K. J Invest Dermatol. 2004;122:1114–1120. doi: 10.1111/j.0022-202X.2004.22521.x. [DOI] [PubMed] [Google Scholar]
- 27.Colombier C, Lippert B, Leng M. Nucleic Acids Res. 1996;24:4519–4524. doi: 10.1093/nar/24.22.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernal-Mendez E, Sun J-S, Gonzalez-Vilchez F, Leng M. New J Chem. 1998;22:1479–1483. [Google Scholar]
- 29.Muller J, Drumm M, Boudvillain M, Leng M, Sletten E, Lippert B. J Biol Inorg Chem. 2000;5:603–11. doi: 10.1007/s007750000143. [DOI] [PubMed] [Google Scholar]
- 30.Dieter-Wurm I, Sabat M, Lippert B. J Am Chem Soc. 1992;114:357–359. [Google Scholar]
- 31.Sharma SK, McLaughlin LW. J Am Chem Soc. 2002;124:9658–9659. doi: 10.1021/ja020500n. [DOI] [PubMed] [Google Scholar]
- 32.Sharma SK, McLaughlin LW. J Inorg Biochem. 2004;98:1570–1577. doi: 10.1016/j.jinorgbio.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Macaya RF, Gilbert DE, Malek S, Sinsheimer JS, Feigon J. Science. 1991;254:270–274. doi: 10.1126/science.254.5029.270. [DOI] [PubMed] [Google Scholar]
- 34.Sun JS, Mergny JL, Lavery R, Montenay-Garestier T, Helene C. J Biomol Struct Dyn. 1991;9:411–424. doi: 10.1080/07391102.1991.10507925. [DOI] [PubMed] [Google Scholar]
- 35.Mergny JL, Sun JS, Rougee M, Montenay-Garestier T, Barcelo F, Chomilier J, Helene C. Biochemistry. 1991;30:9791–9798. doi: 10.1021/bi00104a031. [DOI] [PubMed] [Google Scholar]
- 36.Roberts RW, Crothers DM. Proc Natl Acad Sci USA. 1991;88:9397–9401. doi: 10.1073/pnas.88.21.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoon K, Hobbs CA, Koch J, Sardaro M, Kutny R, Weis AL. Proc Natl Acad Sci USA. 1992;89:3840–3844. doi: 10.1073/pnas.89.9.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller PS, Cushman CD. Biochemistry. 1993;32:2999–3004. doi: 10.1021/bi00063a010. [DOI] [PubMed] [Google Scholar]
- 39.Kiessling LL, Griffin LC, Dervan PB. Biochemistry. 1992;31:2829–2834. doi: 10.1021/bi00125a026. [DOI] [PubMed] [Google Scholar]
- 40.Stilz HU, Dervan PB. Biochemistry. 1993;32:2177–2185. doi: 10.1021/bi00060a008. [DOI] [PubMed] [Google Scholar]
- 41.Huang CY, Cushman CD, Miller PS. J Org Chem. 1993;58:5048–5049. [Google Scholar]
- 42.Verma S, Miller PS. Bioconjug Chem. 1996;7:600–605. doi: 10.1021/bc960049n. [DOI] [PubMed] [Google Scholar]
- 43.Huang CY, Bi G, Miller PS. Nucleic Acids Res. 1996;24:2606–2613. doi: 10.1093/nar/24.13.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kukreti S, Sun JS, Garestier T, Helene C. Nucleic Acids Res. 1997;25:4264–4270. doi: 10.1093/nar/25.21.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gowers DM, Fox KR. Nucleic Acids Res. 1999;27:1569–1577. doi: 10.1093/nar/27.7.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prevot-Halter I, Leumann CJ. Bioorg Med Chem Lett. 1999;9:2657–2660. doi: 10.1016/s0960-894x(99)00451-5. [DOI] [PubMed] [Google Scholar]
- 47.Loakes D. Nucleic Acids Res. 2001;29:2437–2447. doi: 10.1093/nar/29.12.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guianvarc’h D, Benhida R, Fourrey JL, Maurisse R, Sun JS. Chem Commun. 2001:1814–1815. doi: 10.1039/b103743a. [DOI] [PubMed] [Google Scholar]
- 49.Parel SP, Marfurt J, Leumann CJ. Nucleosides Nucleotides Nucleic Acids. 2001;20:411–417. doi: 10.1081/NCN-100002315. [DOI] [PubMed] [Google Scholar]
- 50.Obika S, Hari Y, Sekiguchi M, Imanishi T. Chem Eur J. 2002;8:4796–4802. doi: 10.1002/1521-3765(20021018)8:20<4796::AID-CHEM4796>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 51.Torigoe H, Hari Y, Obika S, Imanishi T. Nucleosides Nucleotides Nucleic Acids. 2003;22:1097–1099. doi: 10.1081/NCN-120022745. [DOI] [PubMed] [Google Scholar]
- 52.Torigoe H, Hari Y, Obika S, Imanishi T. Nucleosides Nucleotides Nucleic Acids. 2003;22:1571–1573. doi: 10.1081/NCN-120023036. [DOI] [PubMed] [Google Scholar]
- 53.Jazouli M, Guianvarc’h D, Bougrin K, Soufiaoui M, Vierling P, Benhida R. Nucleosides Nucleotides Nucleic Acids. 2003;22:1277–1280. doi: 10.1081/NCN-120022945. [DOI] [PubMed] [Google Scholar]
- 54.Buchini S, Leumann CJ. Nucleosides Nucleotides Nucleic Acids. 2003;22:1199–1201. doi: 10.1081/NCN-120022835. [DOI] [PubMed] [Google Scholar]
- 55.Osborne SD, Powers VE, Rusling DA, Lack O, Fox KR, Brown T. Nucleic Acids Res. 2004;32:4439–4447. doi: 10.1093/nar/gkh776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sasaki S, Taniguchi Y, Takahashi R, Senko Y, Kodama K, Nagatsugi F, Maeda M. J Am Chem Soc. 2004;126:516–528. doi: 10.1021/ja037211z. [DOI] [PubMed] [Google Scholar]
- 57.Taniguchi Y, Nakamura A, Senko Y, Kodama K, Nagatsugi F, Sasaki S. Nucleosides Nucleotides Nucleic Acids. 2005;24:823–827. doi: 10.1081/ncn-200060309. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Rusling DA, Powers VE, Lack O, Osborne SD, Fox KR, Brown T. Biochemistry. 2005;44:5884–5892. doi: 10.1021/bi050013v. [DOI] [PubMed] [Google Scholar]
- 59.Fox KR, Brown T. Q Rev Biophys. 2005;38:311–320. doi: 10.1017/S0033583506004197. [DOI] [PubMed] [Google Scholar]
- 60.Taniguchi Y, Nakamura A, Senko Y, Nagatsugi F, Sasaki S. J Org Chem. 2006;71:2115–2122. doi: 10.1021/jo052413u. [DOI] [PubMed] [Google Scholar]
- 61.Buchini S, Leumann CJ. Angew Chem Int Ed Engl. 2004;43:3925–3928. doi: 10.1002/anie.200460159. [DOI] [PubMed] [Google Scholar]
- 62.Ranasinghe RT, Rusling DA, Powers VE, Fox KR, Brown T. Chem Commun (Camb) 2005:2555–2557. doi: 10.1039/b502325d. [DOI] [PubMed] [Google Scholar]
- 63.Rusling DA, Powers VE, Ranasinghe RT, Wang Y, Osborne SD, Brown T, Fox KR. Nucleic Acids Res. 2005;33:3025–3032. doi: 10.1093/nar/gki625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li JS, Chen FX, Shikiya R, Marky LA, Gold B. J Am Chem Soc. 2005;127:12657–12665. doi: 10.1021/ja0530218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brennan L, Peng G, Srinivasan N, Fox KR, Brown T. Nucleosides Nucleotides Nucleic Acids. 2007;26:1283–1286. doi: 10.1080/15257770701530525. [DOI] [PubMed] [Google Scholar]
- 66.Gerrard SR, Srinivasan N, Fox KR, Brown T. Nucleosides Nucleotides Nucleic Acids. 2007;26:1363–1367. doi: 10.1080/15257770701533958. [DOI] [PubMed] [Google Scholar]
- 67.Puglisi JD, Tinoco I., Jr. Methods Enzymol. 1989;180:304–325. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- 68.Campbell MA, Mason TM, Miller PS. Can J Chem. 2007;85:241–248. [Google Scholar]
- 69.Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, et al. Nature. 1985;313:277–284. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- 70.Lippard SJ, Ushay HM, Merkel CM, Poirier MC. Biochemistry. 1983;22:5165–5168. [Google Scholar]
- 71.Raudaschl-Sieber G, Lippert B. Inorg Chem. 1985;24:2426–2432. [Google Scholar]
- 72.Blommaert FA, Saris CP. Nucleic Acids Res. 1995;23:1300–1306. doi: 10.1093/nar/23.8.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anin MF, Leng M. Nucleic Acids Res. 1990;18:4395–4400. doi: 10.1093/nar/18.15.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Comess KM, Costello CE, Lippard SJ. Biochemistry. 1990;29:2102–2110. doi: 10.1021/bi00460a020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.