

Abstract
Closure of the cranial neural tube depends on normal development of the head mesenchyme. Homozygous-mutant embryos for the ENU-induced open mind (opm) mutation exhibit exencephaly associated with defects in head mesenchyme development and dorsal-lateral hinge point formation. The head mesenchyme in opm mutant embryos is denser than in wildtype embryos and displays an abnormal cellular organization. Since cells that originate from both the cephalic paraxial mesoderm and the neural crest populate the head mesenchyme, we explored the origin of the abnormal head mesenchyme. opm mutant embryos show apparently normal development of neural crest-derived structures. Furthermore, the abnormal head mesenchyme in opm mutant embryos is not derived from the neural crest, but instead expresses molecular markers of cephalic mesoderm. We also report the identification of the opm mutation in the ubiquitously expressed Hectd1 E3 ubiquitin ligase. Two different Hectd1 alleles cause incompletely penetrant neural tube defects in heterozygous animals, indicating that Hectd1 function is required at a critical threshold for neural tube closure. This low penetrance of neural tube defects in embryos heterozygous for Hectd1 mutations suggests that Hectd1 should be considered as candidate susceptibility gene in human neural tube defects.
Introduction
The central nervous system of the vertebrate embryo originates during gastrulation with the formation of the neural plate. During subsequent development, the neural plate undergoes extensive morphogenic movements resulting in formation of the neural tube. When the neural tube fails to close completely during its morphogenesis, neural tube defects result. Neural tube defects are one of the most common human congenital malformations occurring in approximately one out of every one thousand live births (Copp et al., 2003; Zohn et al., 2005). Common forms of neural tube defects include spina bifida and exencephaly where the neural tube remains open in the most caudal and rostral aspects of the neural axis, respectively. In humans, neural tube defects represent a complex disease with multiple environmental and genetic contributing factors. Because of the multifaceted etiology of human neural tube defects, identification of causative mutations has been problematic.
Vertebrate model systems have been indispensable for the discovery of the processes required for neural tube closure. The mouse has been particularly useful for identification of genes required for proper morphogenesis of the neural tube and the generation of numerous mouse models for neural tube defects has implicated a long list of candidate genes for human neural tube defects (Copp et al., 2003; Zohn et al., 2005). These genes regulate cell movement, apoptosis, proliferation, patterning and differentiation of not only the neural tissue, but also the surrounding mesenchyme and non-neural ectoderm. Moreover, in some cases, identification of key regulators of neural tube closure in mice has helped to uncover the genetic basis of neural tube defects in humans (Gelineau-van Waes and Finnell, 2001).
Neural tube closure is a complex morphogenic process where the neural plate rolls into a tube forming the central nervous system (Copp et al., 2003; Zohn et al., 2005). The neural folds form at the edges of the neural plate and rise towards the dorsal midline due to forces from both the neural tissue and the surrounding epithelium and mesenchyme. Apical constriction of cells in the midline and in more lateral regions results in the formation of medial and dorsal-lateral hinge points respectively. In the cranial neural tube, neural fold elevation is accompanied by an expansion of the head mesenchyme (Morriss and Solursh, 1978) and reviewed in (Copp, 2005). This expansion is mediated by both increased cell proliferation and an increase in the extracellular space between the mesenchymal cells and is thought to be critical to allow the elevation of the neural folds. The molecular signals regulating these cellular behaviors of the head mesenchyme remain unknown.
Cells that originate from both the cephalic paraxial mesoderm and the neural crest populate the head mesenchyme (Noden and Trainor, 2005). The cephalic mesoderm is derived from the cells in the primitive streak immediately caudal to the node. As gastrulation progresses, cells from the paraxial mesoderm spread medio-laterally from the primitive streak to a position beneath the developing neural plate. In contrast, the cranial neural crest is derived from cells that are located at the junction of the neural and non-neural ectoderm. Once specified, neural crest cells migrate ventral-laterally between the surface ectoderm and the paraxial mesoderm. During later stages of development, the paraxially-derived cephalic mesoderm contributes to multiple structures such as the smooth and skeletal muscles and some of the cartilaginous and bony elements of the skull. The neural crest contributes to cranial nerves, blood vessels and many of the bony elements of the head and face.
Cranial neural tube closure is critically dependent on the proliferation and cellular rearrangement of the head mesenchyme. Mouse models with deletions in the Twist1, Cart1 or Tcfap2a genes exhibit cranial neural tube closure defects linked to defects in development of the head mesenchyme (Chen and Behringer, 1995; Schorle et al., 1996; Zhang et al., 1996; Zhao et al., 1996). Twist1 function is required in the head mesenchyme where it is expressed in both the paraxial mesoderm and the neural crest lineages (Chen and Behringer, 1995). Yet it is unknown if either Twist1 or Cart1 are required in the mesoderm or neural crest or both lineages. Deletion of Tcfap2a specifically in the neural crest results in exencephaly, indicating that AP-2α function in the neural crest is essential for cranial neural tube closure (Brewer et al., 2004).
Here we describe the further characterization of the neural tube defects in the ENU-induced open mind (opm) mutant mouse line. Homozygous opm mutant embryos exhibit severe defects in cranial neural tube closure (Kasarskis et al., 1998). We demonstrate that the neural tube closure defect in opm mutant embryos is associated with a failure of dorsal-lateral hinge point formation and an abnormal organization of the head mesenchyme surrounding the neural tube during closure. In spite of the severe defects in cranial neural tube closure and the abnormal head mesenchyme, opm mutant embryos exhibit apparently normal development of neural crest-derived structures such as facial bones and palate. Neural crest development also appears normal in opm mutant embryos as assayed by both molecular marker and lineage-tracing analysis. In contrast, analyses of the expression pattern of molecular markers that mark the cephalic mesoderm suggest that this tissue is abnormally dense in opm mutants. By positional cloning we identify the opm mutation in an uncharacterized ubiquitin ligase (Hectd1) that is ubiquitously expressed throughout early development of the mouse embryo. Our data also indicate that a critical threshold of Hectd1 function is required for neural tube closure as up to 20% of mutant heterozygotes exhibit neural tube defects depending on the mutant allele. The low penetrance of neural tube defects in heterozygous embryos suggests that Hectd1 should be considered as a candidate susceptibility gene in human neural tube defects.
Materials and Methods
Analysis of mutant phenotype
Whole-mount and section RNA in situs were performed as described (Holmes and Niswander, 2001; Liu et al., 1998) using the following probes: Fgf8 (Crossley and Martin, 1995), BMP4 (Jones et al., 1991), Sox10 (Pusch et al., 1998), AP-2α (Mitchell et al., 1991), Tbx1 (Chapman et al., 1996), Snail (Nieto et al., 1992), Twist (Chen and Behringer, 1995), PDGFRα (Schatteman et al., 1992). The expression pattern of Hectd1 was determined using an anti-sense RNA probe synthesized from IMAGE clone: 3672615 or determining LacZ activity in Hectd1XC/+ embryos. For immunofluorescence experiments, embryos were dissected from the decidua and fixed for 1 hour in 4% paraformaldehyde in PBST (Phosphate-buffered saline (PBS) plus 0.1% Tween-20), washed 3 times in PBST, cryopreserved in 30% sucrose in PBS, embedded in OCT compound (Tissue-Tek) and sectioned at 10 μM. Fixed frozen sections were processed as described (Timmer et al., 2002) using the anti-phospho-Histone H3 Mitosis Marker (1:250; Upstate Biotechnology) for proliferation assays or the cleaved Caspase-3 antibody (1:250; Cell Signaling #9661) for apoptosis assays. Sections were mounted with Vectashield mounting medium with DAPI (Vector Laboratories; H-1200) or stained with Hoechst (10 μg/ml; Sigma) to stain nuclei and allow counts of the total number of cells. For Hematoxylin and Eosin staining, embryos were fixed overnight in 4% paraformaldehyde in PBST and processed for frozen sectioning as described (Timmer et al., 2002). Skeletal and β-galactosidase staining were performed as described (Hogan et al., 1994).
Mouse Strains and Genotyping
The opm mouse line was identified in a screen for recessive ENU-induced mutations that cause defects in neural tube closure at E9.5 (Garcia-Garcia et al., 2005; Kasarskis et al., 1998; Zohn et al., 2005). The opm mutation was generated on a C57BL/6J genetic background and backcrossed to C3H/HeJ for at least 10 generations to obtain a congenic line. The mapping of the opm mutation was done during the creation of the congenic line, while the high resolution mapping data was generated once the congenic line (e.g. >N10) was established. Approximately 5% of opm/+ embryos exhibit defects in cranial neural tube closure (see Table 1). However, the neural tube defect in these heterozygous embryos is less severe (only mid- and hindbrain exencephaly) and can be easily distinguished at E9.5 and E10.5 from opm/opm embryos (exencephaly from the forebrain to the hindbrain). For this reason, mapping of the opm mutation was done using only E9.5 and E10.5 embryos from crosses of opm/+ females mated to opm/+ males. In a mapping cross of 590 opportunities for recombination, opm was mapped between the Massachusetts Institute of Technology (MIT) simple sequence length polymorphism (SSLP) markers D12mit64 and D12mit54 with no recombination with D12mit110. For high-resolution mapping, additional polymorphic DNA markers were generated based on nucleotide repeat sequences and include: D12ski18, D12ski16 and D12ski12 (see http://mouse.ski.mskcc.org/ for sequence of primers).
Table 1.
Mendelian rations of opm/opm embryos are found upon dissection at E9.5-12.5 and a small percentage of opm/+ embryos exhibit exencephaly
| Number of embryos | ||||||
|---|---|---|---|---|---|---|
| Age | +/+ | opm/+ | opm/opm | Number of embryos with exencephaly | Number of opm/+ embryos with exencephaly | Total number of embryos |
| E9.5 | 81 | 178 | 76 | 86 | 10 | 335 |
| E10.5 | 50 | 83 | 43 | 45 | 2 | 176 |
| E11.5 | 11 | 31 | 13 | 13 | 0 | 55 |
| E12.5 | 4 | 9 | 9 | 11 | 3 | 22 |
| Total: | 146 | 301 | 141 | 165 | 15 | 588 |
| 25% | 51% | 24% | 28% | 5% | ||
The entire Hectd1 transcript was sequenced by RT-PCR (Superscript One-Step RT-PCR, Invitrogen) using RNA isolated from E10.5 opm/opm and C57BL/6 control embryos. At position 430, a T to A transversion was discovered in the 7833 base pair open reading frame that encodes Hectd1. This mutation results in a nonsense mutation changing a Leucine to a stop codon at amino acid 145 in the predicted 2611 amino acid Hectd1 protein.
A mouse embryonic stem (ES) cell line carrying an insertion in the Hectd1 gene (XC266) was identified from a blast search using the Hectd1-coding sequence against the BayGenomics (http://baygenomics.ucsf.edu/) database (Skarnes et al., 2004). Mice carrying the Hectd1 insertion were generated by standard blastocyst injection followed by breeding for at least two generations into a C3H background and named Hectd1XC. Mice carrying the genetrap allele were genotyped using LacZ-specific primers (Liu et al., 1998). Other mouse strains used were Wnt1-Cre (Danielian et al., 1998) and the R26R conditional reporter allele (Soriano, 1999).
Results
opm mutant embryos exhibit exencephaly
In our ongoing ENU mutagenesis screens to identify genes that are required for neural tube closure (Zohn et al., 2005), we isolated the open mind (opm) mutant mouse line (Kasarskis et al., 1998). This mouse mutant is distinct from the ENU-induced openmind (om) mouse mutant, which harbors a mutation in the Rere/atrophin-2 gene (Zoltewicz et al., 2004). As summarized in Table 1, Mendelian ratios of opm/opm mutant embryos are recovered with exencephaly upon dissection at E9.5-E12.5. Additionally, approximately 5% of heterozygous embryos show a less severe exencephaly affecting only the midbrain and hindbrain (Table 1 and data not shown). Homozygous opm mutant embryos display exencephaly characterized by a failure of neural tube closure from the hindbrain to the forebrain. The closure point at the most anterior aspect of the forebrain is typically closed, but in some embryos this closure point remains open resulting in a split of the facial primordia down the midline at E9.5 although this appears to resolve as we do not detect cranial-facial defects at later stages (Figure 1A-L; see below and data not shown (Kasarskis et al., 1998). Eye morphogenesis is also affected as opm mutant eyes are malformed, smaller and are rotated in the head (Figure 1O, P).
Figure 1. opm mutant embryos exhibit defects in neural tube closure without other significant defects in craniofacial development.
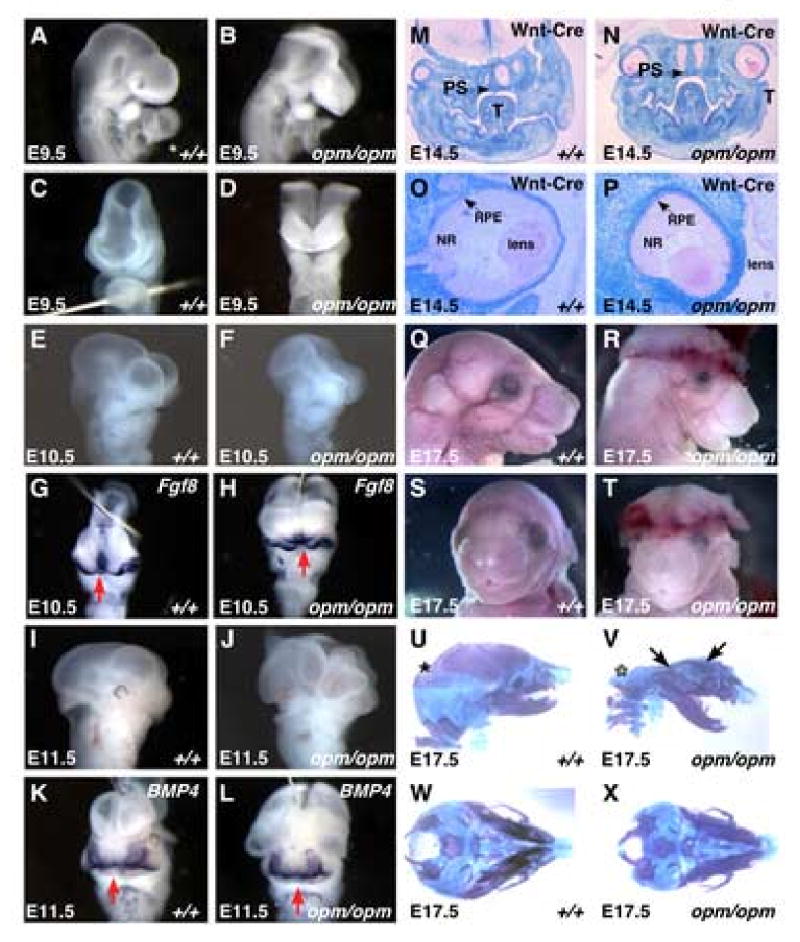
Panels A-L: lateral (A, B, E, F, I and J) and frontal (C, D, G, H, K and L) views of wildtype (A, C, E, G, I and K) and opm mutant (B, D, F, H, J, L) heads demonstrating neural tube closure defect from the forebrain to the hindbrain and normal development of the face (A-D, E9.5; E-H, E10.5; I-L, E11.5). Frontal views of wildtype and opm mutant embryos at E10.5 and E11.5 were labeled by in situ hybridization using probes against Fgf8 and BMP4 respectively to highlight the developing frontal nasal processes. Fate of the neural crest as shown in coronal sections of E14.5 Wnt1-Cre/R26R wildtype (M,O) and opm mutant (N,P) embryos stained for β-galactosidase activity and counterstained with eosin. Arrow points to the properly fused secondary palatal shelves (PS; T=Tongue) in wildtype (M) and opm mutant (N) heads. Eye development is abnormal in opm mutant embryos (P). The retinal pigmented epithelium (RPE, arrow), neural retina (NR) and lens appear histologically normal, but eyes are rotated in the head of opm mutant embryos. Profile (Q,R) and frontal (S,T) views of E17.5 wildtype (Q,S) and opm mutant (R,T) heads demonstrating normal craniofacial development in spite of the severe neural tube closure defect. Lateral (U,V) and basal (W,X) views of E17.5 skeletal preparations stained with Alcian blue and Alizarin Red in wildtype (U,W) and opm mutant (V,X) skulls. Asterisk (*) denotes exoccipital, petrosal and interpariatal bones that are missing in opm mutant skulls. Arrows point to frontal and pariatal bones that are malformed in opm mutant skulls.
In spite of the severe defects in neural tube closure in opm mutant embryos, other aspects of cranial-facial development appear to be unaffected. The facial primordial consists of the frontonasal prominence and the first branchial arch. Around day 10, the nasal placodes in the frontonasal prominence invaginate resulting in formation of the medial and lateral nasal prominences (Kaufman and Bard, 1999). As shown in Figure 1G-H, Fgf8 is expressed in the ectoderm of the nasal pits and the medial nasal process and is expressed in a similar domain in both opm mutant and wildtype embryos. By E11.5, the medial and lateral prominences fuse to form the nasal pits as highlighted by Bmp4 expression (Figure 1K). In the developing face of opm mutant embryos, Bmp4 is expressed in a similar domain as in wildtype (Figure 1L). The secondary palate forms from outgrowths of the medial walls of the maxillary prominence, which fuse in the midline with the nasal septum. Coronal sections of E14.5 wildtype and mutant heads reveals that the secondary palatal shelves fuse normally in opm mutant embryos (Figure 1M, N). These results indicate that unlike other mouse mutants which exhibit similar neural tube closure defects from the forebrain to the hindbrain such as Twist1, Cart1 or Tcfap2a (Chen and Behringer, 1995; Schorle et al., 1996; Zhang et al., 1996; Zhao et al., 1996), the facial primordia are able to undergo proper morphogenesis in opm mutant embryos.
To further examine the effect of the opm mutation on cranial-facial growth, development of the facial skeleton was examined later in development (E17.5). As shown by the gross morphology of the face of opm mutant embryos (Figure 1Q-T), the face and snout of mutant embryos develops normally. Skeletal staining reveals that opm mutant embryos exhibit some malformations in the growth of the skull vault such as abnormally shaped frontal and parietal bones (arrowheads in Figure 1V) and the absence of the exoccipital, petrosal and interpariatal bones (* in Figure 1V); however, these malformations are likely a secondary result of the cranial neural tube closure defect (Copp, 2005). Aside from these malformations, this analysis does not reveal any other gross morphological defects in development of the cranial-facial skeleton (Figure 1U-X). Therefore, unlike other mutants with similar neural tube defects such as Twist, CART1 and Tcfap2a, craniofacial development of structures rostral to the open neural tube is normal in opm mutants.
Cranial neural tube closure defects in opm mutant embryos are associated with defects in development of the head mesenchyme
The failure of neural fold elevation and the open neural tube from the forebrain to the hindbrain is strikingly similar to the neural tube defects observed in CART1 and Twist mutant embryos (Chen and Behringer, 1995; Zhao et al., 1996). Previous studies have implicated the involvement of these genes in proper development of the head mesenchyme. Therefore, histological analysis was used to investigate whether the head mesenchyme is affected in opm mutants. Coronal sections of wildtype and opm mutant heads were stained with hematoxylin and eosin to reveal the cellular organization of the head mesenchyme surrounding the neural tube (Figure 2A-D). This analysis demonstrates that the head mesenchyme in opm mutants is abnormal and appears to be denser around the open neural tube (Figure 2B,D). To further quantify the density of the head mesenchyme cells, the number of head mesenchyme cells in identical areas of wildtype and mutant heads in many embryos were counted to obtain cell densities. Since an increased cell density could result from a failure of neural fold elevation, areas of head mesenchyme were selected ventral to the dorsal-lateral hinge points such as those in the boxed regions of Figure 2A and C. The density of cells in the head mesenchyme of E9.5 mutant embryos were significantly (p <0.0001) greater than in wildtype embryos as determined by the Mann-Whitney U test (Figure 2I). Furthermore, the increased cell density is observed early as the neural folds are beginning to elevate (e.g. at E8.5). This is in contrast to our earlier findings suggesting that the mesenchyme was less dense (Kasarskis et al., 1998). After our current careful analysis, our previous observation now appears to be an artifact of the plane of few sagittal sections. We also observed an increase in the thickness of the head mesenchyme around the dorsal and lateral aspects of the neural tube in mutant versus wildtype embryos (Figure 2A,C,E,F brackets). This expansion of head mesenchyme is not seen in other mouse mutant lines with exencephaly from our screens. For example, neural tube defects in the humdy mutant mouse line are associated with an increase in the proliferation of neural tissue (Taehee Kim and Lee Niswander, submitted). Furthermore, the head mesenchyme is not denser in humdy mutants indicating that abnormal head mesenchyme is not always secondary to failure of neural tube closure. These findings indicate that the opm phenotype is associated with an abnormal organization of the head mesenchyme around the cranial neural tube.
Figure 2. Abnormal head mesenchyme development in opm mutant embryos is associated with neural tube closure defects.
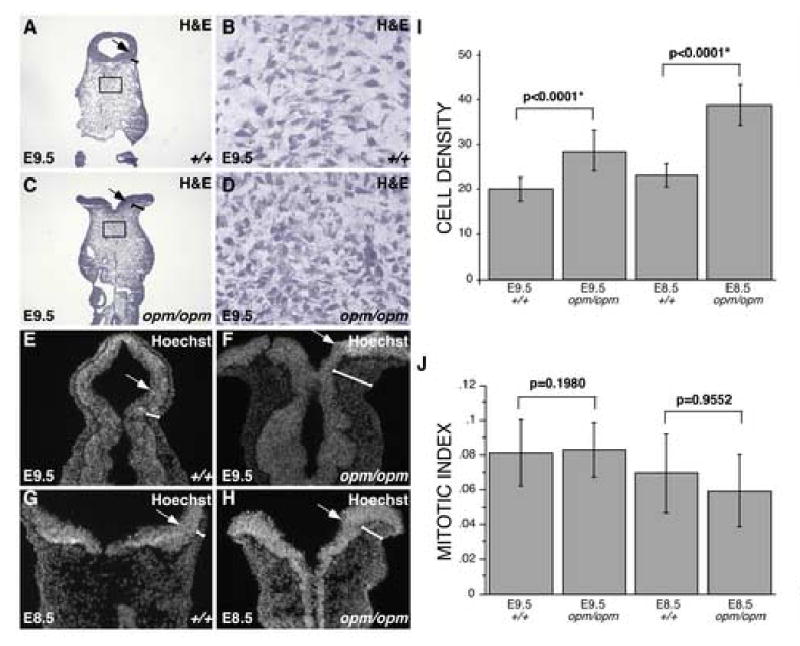
A-D. Hematoxylin and Eosin (H&E) staining of coronal sections of wildtype (A,B) and opm mutant (C,D) heads at E9.5. Brackets highlight the region of head mesenchyme that are expanded around the opm neural tube. Panels B&D show magnified views of the boxes drawn in panels A&C, demonstrating the abnormally dense cellular organization of opm mutant head mesenchyme. E-H. Nuclei were visualized by staining with Hoechst in coronal sections of wildtype (E,G) and opm mutant (F,H) at E9.5 (E,F) and E8.5 (G,H) demonstrating that the head mesenchyme is denser around the dorsal neural tube of opm mutant embryos during neurulation. There is also a failure of dorsal-lateral hinge point formation leading to a flat or convex curvature of the opm neural tube compared to the concave neural tube in wildtype embryos at both stages. Arrows point to dorsal-lateral hinge points in A-H. I. Cell density was determined by counting the number of nuclei in 1-2 defined areas on many sections from many embryos (E9.5: wildtype n=39 areas, opm mutant n=41 areas; E8.5: wildtype n=58 areas, opm mutant n=63 areas). Error bars represent one standard deviation from the mean. The density of cells between wildtype and opm mutant samples are significantly different as calculated by the Mann-Whitney U test (E9.5: P-value<0.0001*; E8.5: P-value<0.0001*). J. Mitotic indexes were calculated by dividing the number of P-H3 positive cells by the number of nuclei in a given area for wildtype and opm mutant in two to three sections from many embryos (E9.5: wildtype n=19 sections, opm mutant n=20 sections; E8.5: wildtype n=17 sections, opm mutant n=18 sections). Error bars represent one standard deviation from the mean. Samples are not significantly different as calculated by the Mann-Whitney U test (E9.5: P-value=0.9552; E8.5: P-value=0.1980).
In order to determine if the abnormal organization of head mesenchyme in opm mutant embryos occurs before cranial neural tube closure, the head mesenchyme was examined at a stage that precedes this event. Coronal sections of embryos at E8.5 (12 somites; Figure 2G,H) were stained with Hoechst to highlight the morphology of the tissue. As indicated by brackets in Figure 2G,H, these defects precede cranial neural tube closure as the thickness of the head mesenchyme around the neural tube is already increased at this stage. Additionally, quantification of cell density in the head mesenchyme of E8.5 embryos indicates that there is a significant difference (p<0.0001; Mann-Whitney U test) between the density of cells in wildtype and mutant embryos even before neural tube closure (Figure 2I). Since, the increase in cell density precedes neural tube closure, it is unlikely due to the failure of neural tube closure. Taken together, these results suggest that the neural tube defect in opm mutant embryos could be due (at least in part) to the abnormal organization and increased density of the head mesenchyme surrounding the neural tube during neural fold elevation and neural tube closure.
Another mechanistic defect that can lead to neural tube defects is a failure to form the hinge points, which facilitate the convergence of the neural folds in the dorsal midline. The medial hinge point forms normally in opm mutant embryos (Figure 2A-H), consistent with normal dorsal-ventral patterning of the neural tissue (data not shown), both processes which are known to be regulated by Shh signaling (Caspary and Anderson, 2003; Ybot-Gonzalez et al., 2002). In contrast, the dorsal-lateral hinge points completely fail to form in opm mutants (arrows; Figure 2A-H), resulting in a flat or convex curvature of the neural tissue rather than the concave and closed neural tube seen in wildtype embryos (Figure 1C,D and Figure 2A-F).
To determine if the dense head mesenchyme results from changes in cell proliferation or apoptosis, these processes were examined in E8.5 and E9.5 embryos, prior to and during the time of cranial neural closure. Apoptosis was examined using the cleaved caspase-3 antibody, which marks cells that are initiating apoptosis. In wildtype and mutant embryos, very few cells were undergoing apoptosis in the head mesenchyme surrounding the neural tube (data not shown). Therefore it is unlikely that differences in apoptosis are responsible for the dramatic differences in the number of cells populating the head mesenchyme of wildtype and opm mutant embryos. Proliferation was measured by examining expression of phosphorylated-Histone-3 (P-H3), which increases during mitosis. To assess the rate of proliferation, the number of P-H3 positive cells were counted and divided by the number of cells in a given area of the head mesenchyme to give a mitotic index as shown in Figure 2J. To determine if mitotic indexes are significantly different in opm mutant versus wildtype head mesenchyme, the Mann-Whitney U test was used. For E8.5 and E9.5 wildtype versus opm mutants the p-value was 0.1980 and 0.9552, respectively. These results indicate that there are no significant differences in the mitotic indexes of the abnormal head mesenchyme surrounding the neural tube in opm mutant embryos as compared to control littermates.
opm mutant embryos do not exhibit defects in neural crest cell development
Our data indicate that neural tube defects in opm mutant embryos are associated with defects in development of the head mesenchyme. Additionally, the open neural tube phenotype of opm mutants is reminiscent of the neural tube defects exhibited by Twist and AP-2α mutants (Chen and Behringer, 1995; Schorle et al., 1996; Zhang et al., 1996; Zhao et al., 1996), which also exhibit severe defects in the development of the head mesenchyme. Since the head mesenchyme is composed of cells that are of both neural crest and mesoderm origin (Noden and Trainor, 2005), the head mesenchyme defect in opm mutant embryos could originate from either source. Therefore, we sought to determine the origin of the abnormal head mesenchyme using a molecular marker analysis. If the abnormal head mesenchyme is due to defects in neural crest cell development, this should be evident by examination of neural crest specific markers at early developmental stages. Examination of Sox10 and AP-2α expression at E8.5 indicated that neural crest induction and its initial migration are normal in opm mutant embryos (Figure 3A-D). Additionally, expression of Sox10 and AP-2α at E9.5 indicates that the neural crest migrates and undergoes its initial differentiation normally (Figure 3E-H). Lastly, the expression of AP-2α at E9.5 indicates that the neural crest does not contribute to the regions of abnormal mesenchyme of opm mutants (* in Figure 3H). This normal initial development of neural crest derived structures in opm mutant embryos agrees with our observations that later in development, neural crest-derived structures such as the facial bones and palate develop normally in opm mutants (Figure 1M-X).
Figure 3. Neural crest cells are not present in the abnormal opm mutant head mesenchyme.
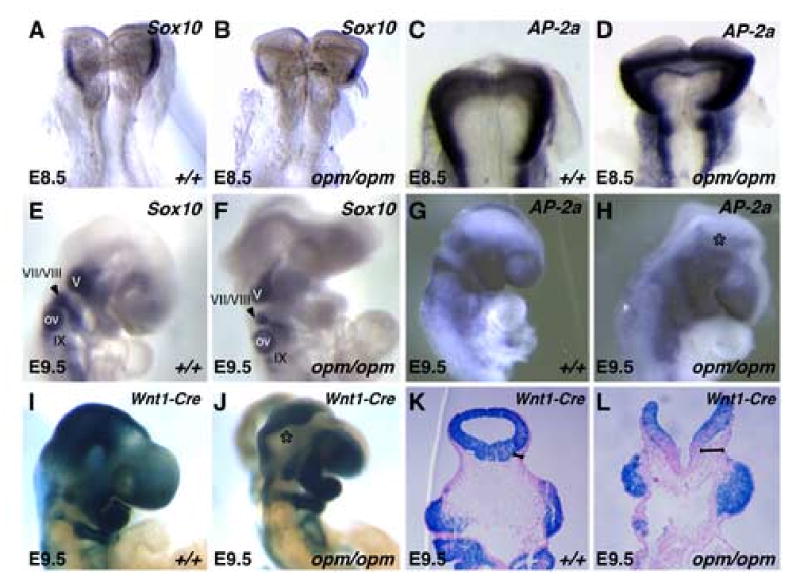
E8.5 (A-D) and E9.5 (F-H) wildtype (A,C,E,G) and opm mutant (B,D,F,H) embryos were analyzed by whole-mount in situ hybridization analysis for expression of Sox10 (A,B,E,F) and AP-2α (C,D,G,H). Asterisk (*) in panel H denotes the lack of AP-2α expression in the abnormal head mesenchyme surrounding the open neural tube in the opm mutant head. V, VII/VIII and IX denote the Sox10-expressing cranial nerves. OV = otic vesicle. Fate of the neural crest in E9.5 Wnt1-Cre/R26R wildtype (I) and opm mutant (J) embryos that were stained in whole-mount for β-galactosidase activity. Asterisk (*) highlights the abnormal head mesenchyme in opm mutant embryos that is negative for β-galactosidase activity. Coronal sections of E9.5 Wnt1-Cre/R26R wildtype (K) and opm mutant (L) head stained for β-galactosidase and counterstained with eosin. Brackets highlight the abnormally expanded head mesenchyme in opm mutant embryos that is negative for β-galactosidase activity.
To further explore a potential contribution of neural crest cells to the abnormal head mesenchyme in opm mutants, genetic fate-mapping experiments were performed. For these experiments, the opm mutation was crossed into the Wnt1-Cre and R26R transgenic mouse strains (Danielian et al., 1998; Soriano, 1999). As shown in Figure 3I,J, E9.5 opm mutant embryos exhibit normal development of Wnt1-lineage traced cells in the head mesenchyme. In fact, the abnormally dense tissue surrounding the neural tube in opm mutant embryos is LacZ-negative (* in Figure 3J). This is better visualized in coronal sections where it is evident that cells of neural crest origin do not populate the abnormal head mesenchyme surrounding the open neural tube and migrate to the correct position in the developing heads of opm mutant embryos (Figure 3K,L). Thus, this lineage tracing experiment clearly indicates that it is not of neural crest origin (brackets in Figure 3K,L).
Molecular markers of cephalic mesoderm
The results described above indicate that the abnormal head mesenchyme surrounding the open neural tube in opm mutants is not derived from the neural crest. Therefore, we next sought to determine if this head mesenchyme is derived from the cephalic mesoderm. To do so, the expression of a number of molecular markers of cephalic mesoderm was examined. Tbx1 is expressed in the caudal cephalic mesoderm and the mesoderm that populates the branchial arches. Examination of Tbx1 in wildtype and opm mutant embryos at E9.5 and E8.5 indicates that its expression domain is not expanded in opm mutant embryos (Figure 4A-D). In contrast, in situ hybridization analysis at E9.5 using antisense probes against Snail, Twist and PDGFRα, molecular markers of both cranial neural crest and mesoderm, indicates that the abnormal head mesenchyme in opm mutant embryos expresses these markers (Figure 4E-J). Since the head mesenchyme is partially obscured by exencephalic neural tissue in the whole mount in situs, expression of Twist and PDGFRα was also visualized in coronal sections. Examination of section in situs clearly shows that the expression domain of both Twist and PDGFRα is expanded in opm mutant heads (Figure 4K-N). This is not simply due to the exencephaly, as mouse mutants that have exencephaly associated with, for example, defects in the organization of actin in the neural tissue do not show this expansion of Twist surrounding the neural tube (Hildebrand and Soriano, 1999). At E8.5, the expression of Snail in the abnormal head mesenchyme in opm mutant embryos is less evident. However, Twist appears to be expressed in an expanded domain at this stage (Figure 4O-R, *). These results combined with our analysis of neural crest cell fate indicates that the abnormal head mesenchyme associated with the cranial neural tube closure defects in opm mutant embryos is derived from the cephalic mesoderm rather than the neural crest and that this defect occurs as early as E8.5.
Figure 4. The abnormal head mesenchyme in opm mutant embryos expresses molecular markers of cephalic mesoderm.
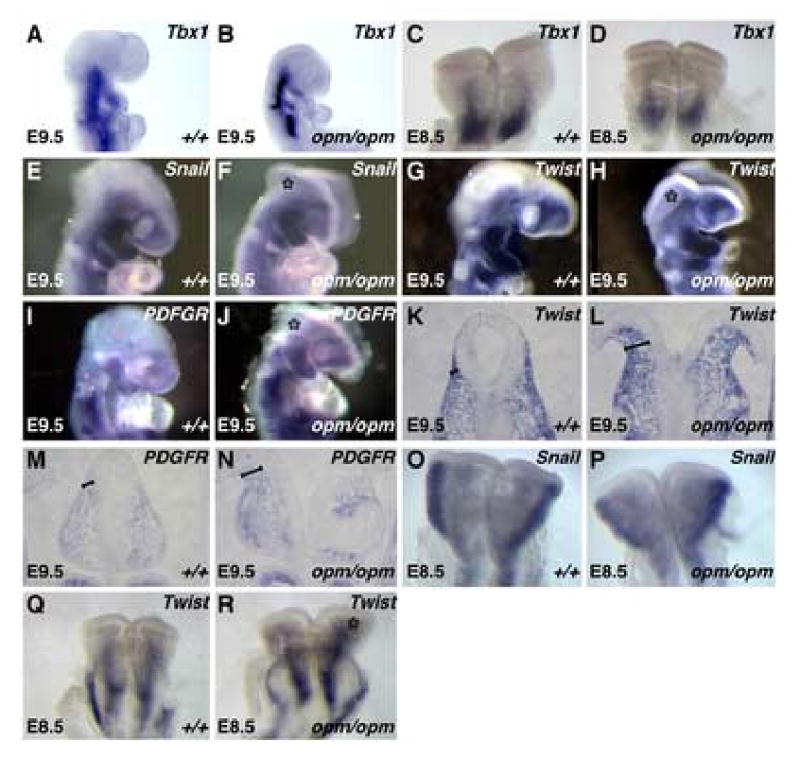
E9.5 (A,B,E,F,G,H,I,J) and E8.5 (C,D,O,P,Q,R) wildtype (A,C,E,G,I,O,Q) and opm mutant (B,D,F,H,J,P,R) heads were analyzed by whole-mount in situ hybridization analysis for expression of Tbx1 (A-D), Snail (E,F,O,P), Twist (G,H,Q,R) and PDGFRα (I,J). Asterisk (*) denotes staining in the abnormal head mesenchyme surrounding the open neural tube in opm mutant embryos. Coronal sections of wildtype (K,M) and opm mutant (L,N) heads stained by in situ hybridization for expression of Twist (K,L) and PDGFRα (M,N). Brackets highlight the abnormal head mesenchyme surrounding the open neural tube in opm mutant embryos.
The opm mutation disrupts a HECT domain ubiquitin ligase
Using positional cloning techniques, the opm mutation was mapped to a 1.7 MB interval on mouse chromosome 12 that contains 12 genes (Figure 5A). One interesting candidate was a novel ubiquitin ligase (Hectd1) with a Hect-domain (Homologous to E6-AP Carboxy Terminal). Hect-domain ubiquitin ligases mediate transfer of ubiquitin and target substrate proteins for either degradation or modification of activity (Kerscher et al., 2006). Upon sequencing, a point mutation was identified in the Hectd1 gene (Figure 5B). The mutation results in a T to A transversion and a change of a Leucine to a stop codon after amino acid 144 in the predicted 2611 amino acid Hectd1 protein (predicted molecular weight of 289 KD). The Hectd1 protein contains a number of conserved protein domains (Figure 5G): an ankyrin repeat in the N-terminal portion of the protein likely mediates protein-protein interactions; a central mind bomb (mib) domain and a C-terminal HECT domain. These domains are also present in a number of E3 ubiquitin ligases. The opm mutation results in truncation of the Hectd1 protein before any of these conserved domains and likely represents a null allele (Figure 5G).
Figure 5. The opm mutation disrupts the uncharacterized Hectd1 ubiquitin ligase.
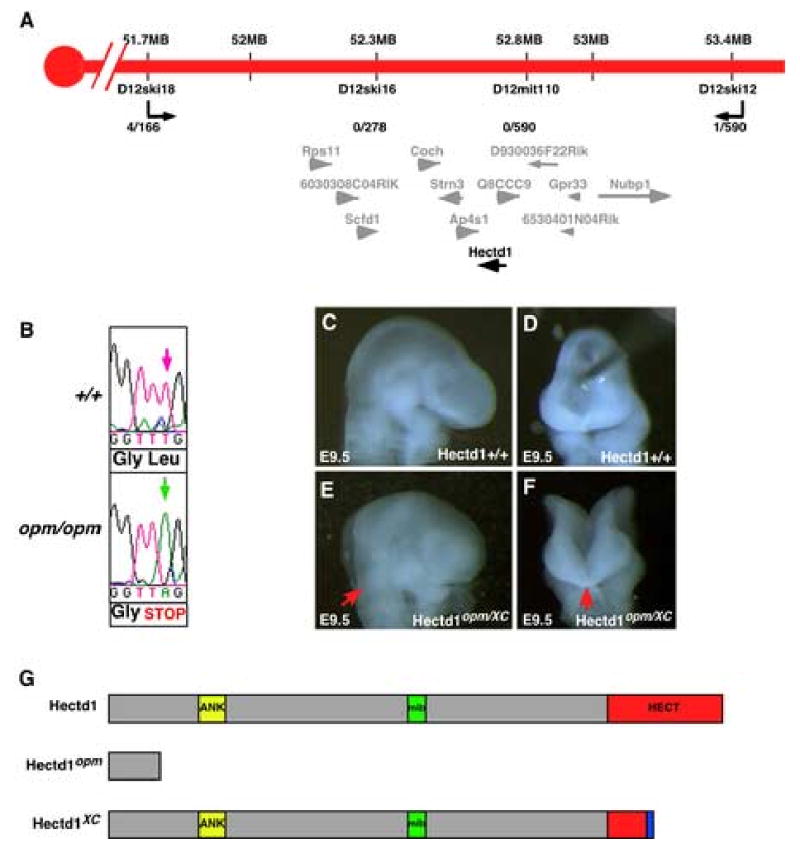
A. Genetic map of opm interval on mouse chromosome 12. The number of recombination events over the number of opportunities for recombination is indicated for each polymorphic marker. Markers D12ski16 and D12MIT110 never separated from the opm phenotype. Within this interval are twelve transcription units: Rps11 (ribosomal protein S11), 6030308C04RIK (RIKEN cDNA 6030408C04 gene), Scfd1 (sec1 family domain containing 1), Coch (coagulation factor C homolog), Strn3 (striatin, calmodulin binding protein 3), Ap4s1 (adaptor-related protein complex AP-4, sigma 1), Hectd1 (HECT domain containing 1), Q8CCC9 (PREDICTED: hypothetical protein), D930036F22Rik (RIKEN cDNA D930036F22 gene), 6530401N04Rik (RIKEN cDNA 6530401N04 gene), Gpr33 (G protein-coupled receptor 33) and Nubp1 (nucleotide binding protein-like). The transcripts in the opm interval and physical map position (mb) is from the Ensemble mouse genome assembly release #40. B. The opm ENU-induced mutation results in a T to A transversion (green arrow) at position 430 in the Hectd1 coding sequence. This mutation results in a nonsense mutation changing a Leucine to a stop codon. C-F. The Hectd1XC gene trap allele fails to complement Hectd1opm as embryos at E9.5 exhibit exencephaly from the hindbrain to the forebrain (E,F). Panels C-F show lateral (C,E) and frontal (D,F) views of wildtype (C,D) and Hectd1opm/XC mutant (E,F) E9.5 embryos. G. Predicted protein motifs in Hectd1: an ankyrin domain (ANK), MIB-HERC2 domain (mib) and a C-terminal Homologous to the E6-AP Carboxyl Terminus domain (HECT). The Hectd1opm mutation results in a truncated protein at amino acid 145. The Hectd1XC mutation results in an insertion and truncation of the HECT domain.
To confirm that this mutation in Hectd1 is responsible for the developmental defects observed in the opm mutant line, a second allele was obtained from the BayGenomics genetrap resource (Skarnes et al., 2004). The genetrap allele, Hectd1XC, consists of a LacZ-Stop cassette inserted into the HECT domain (Figure 5G). A complementation test was done to confirm that the Hectd1 mutation in the opm mouse line is responsible for the observed phenotypes. Embryos were generated that consist of the genetrap allele over the opm allele (Hectd1XC/opm) and the phenotype in mutant embryos was examined at E9.5. Hectd1XC/opm embryos exhibit exencephaly that is identical to that observed in opm homozygous mutant embryos (Figure 5C-F). Moreover, the phenotype of Hectd1XC/XC mutant embryos was identical to that of Hectd1opm/opm mutants (data not shown and Table 2). Interestingly, embryos heterozygous for the Hectd1XC allele exhibited exencephaly at a much higher frequency (8/40 = 20% of embryos) than Hectd1opm/+ embryos, which exhibit exencephaly in only 5% of cases, indicating a weak dominant-negative effect of the Hectd1XC allele (compare Tables 1 and 2). These results confirm that the point mutation identified in the Hectd1 gene is responsible for the phenotypes observed in opm mutants. Furthermore, the observation that disruption of the HECT domain produces the same neural tube closure defect phenotype as the null allele indicates that the ubiquitin ligase activity of Hectd1 is essential for its biological function.
Table 2.
Hectd1XC/+ embryos exhibit exencephaly at a higher frequency than Hecd1opm/+ embryos
| Number of embryos | ||||||
|---|---|---|---|---|---|---|
| Age | +/+ | XC/+ | XC/XC | Number of embryos with exencephaly | Number of XC/+ embryos with exencephaly | Total number of embryos |
| E9.5 | 9 | 22 | 14 | 18 | 4 | 45 |
| E10.5 | 1 | 5 | 0 | 1 | 1 | 6 |
| E11.5 | 2 | 10 | 5 | 8 | 3 | 17 |
| E12.5 | 2 | 3 | 2 | 2 | 0 | 7 |
| Total: | 17 | 40 | 22 | 30 | 8 | 79 |
| 22% | 50% | 28% | 38% | 20% | ||
Expression of the opm gene during embryogenesis
We examined the expression pattern of Hectd1 during neurulation in Hectd1XC embryos that expresses the βgeo reporter under the endogenous Hectd1 promoter. In phenotypically normal Hectd1XC/+ embryos, Hectd1:βgeo was expressed ubiquitously during gastrulation and neurulation (Figure 6). Importantly, LacZ activity was detected in the developing head mesenchyme, consistent with a role for Hectd1 in development of this tissue (Figure 6C-F). Additionally, LacZ activity was detected in the placenta (Figure 6G), and at higher levels in other developing tissues such as the lens of the eye (Figure 6H), the differentiated neurons in the spinal neural tube (Figure 6I) and the atrium of the heart (Figure 6J).
Figure 6. Hectd1 is ubiquitously expressed during development of the mouse embryo.
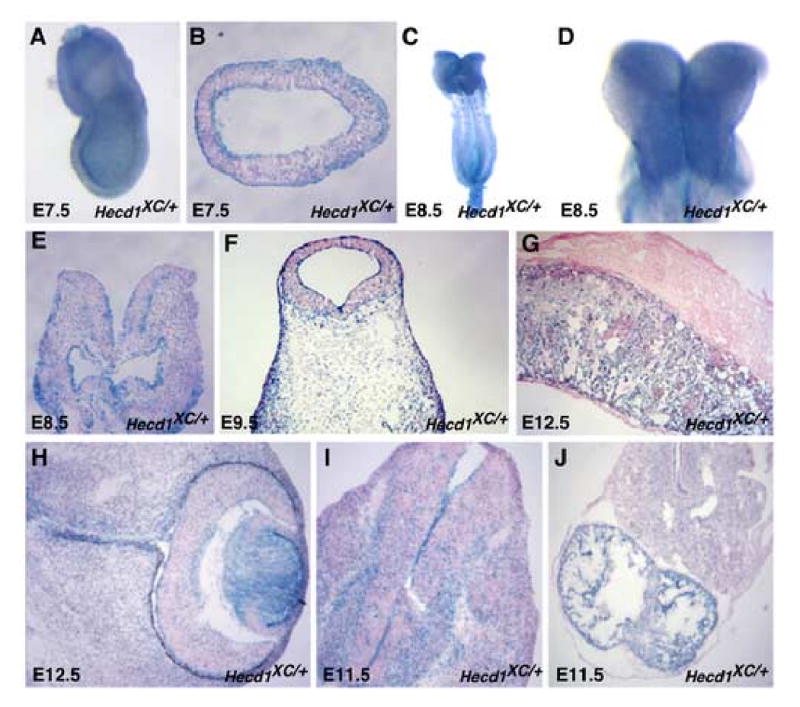
Hectd1 expression was monitored by staining for β-galactosidase activity in whole mount (A,C,D) or in section and counterstained with eosin (B,E-J) in Hectd1XC/+ embryos. Hectd1 is expressed during development of the head mesenchyme at E7.5 (A,B), E8.5 (C-E) E9.5 (F). Hectd1 is also expressed in the placenta (G) and eye (H) at E12.5. Hectd1 is expressed at E11.5 at higher levels in differentiated neurons of the developing spinal cord (I) and the atrium of the heart (J).
Discussion
Neural tube closure defects are common developmental malformations that result in death or long-term physical disabilities in humans. Here we show that an ENU-induced mutation in the open mind (opm) mutant mouse line disrupts the gene encoding the uncharacterized Hectd1 ubiquitin ligase. The cranial neural tube of opm mutant embryos fails to undergo proper morphogenesis and is open from the forebrain to the hindbrain. This neural tube defect is accompanied by an increase in the density and abnormal organization of the head mesenchyme surrounding the open neural tube, as well as failure to form the dorsal-lateral hinge points. Neural crest development in opm mutant embryos is apparently normal as determined by development of neural crest-derived structures. Additionally, molecular marker and lineage-tracing analysis during the induction and migration of the neural crest indicates that these processes occur normally in opm mutants. In contrast, the abnormal head mesenchyme expresses molecular markers of cephalic mesoderm indicating that Hectd1 is required for normal development of this lineage. Finally, the low penetrance of neural tube defects in Hectd1 mutant heterozygotes indicates that the level of Hectd1 gene activity is critical for proper closure of the neural tube.
Mechanism of cranial neural tube closure defects in opm mutant embryos
During cranial neural tube closure, the head mesenchyme undergoes expansion by increasing both cell proliferation and the extracellular space between mesenchymal cells (Morriss and Solursh, 1978). This expansion is thought to be required for elevation of the neural folds (Copp, 2005; Copp et al., 2003). Neural fold elevation is also mediated by bending of the neuroepithelium through apical constriction resulting in the formation of the medial and dorsal-lateral hinge points (Copp, 2005; Copp et al., 2003). Together these forces convert the flat neural plate to a concave morphology contributing to the convergence of the neural folds in the dorsal midline and their subsequent fusion. The cranial neural tube in opm mutant embryos exhibits a complete failure of dorsal-lateral hinge point formation and neural fold elevation. Furthermore, expanded and denser head mesenchyme around the cranial neural tube during closure accompanies this failure in neural fold elevation.
Genetic evidence that cranial neural tube closure is dependent on the proper expansion of the head mesenchyme is supported by studies of the Twist and CART1 mutant phenotypes (Chen and Behringer, 1995; Zhao et al., 1996). Like opm mutant embryos, Twist and CART1 mutants exhibit failures of cranial neural fold elevation. However, rather than the increase density of head mesenchyme observed in opm mutant embryos, Twist and CART1 mutants exhibit a decrease in the density of head mesenchyme cells due to decreased cell proliferation and increased apoptosis, respectively (Chen and Behringer, 1995; Zhao et al., 1996). In contrast, our data do not indicate a statistically significant change in proliferation or apoptosis of the head mesenchyme during neurulation in opm mutants. Furthermore, we show through both lineage tracing and molecular marker analysis that the abnormal head mesenchyme is not derived from the cranial neural crest but instead from the cephalic mesoderm. Below we suggest possible molecular and cellular mechanisms by which loss of Hectd1 ubiquitin ligase function could lead to the increased density of the cephalic mesoderm observed in opm mutant embryos. Together these data indicate that neural fold elevation requires that the amount of head mesenchyme be carefully balanced; either too much (opm mutants) or too little (Twist and CART1 mutants) head mesenchyme can lead to a failure of neural fold elevation and neural tube closure.
opm mutant embryos also display a lack of dorsal-lateral hinge point formation in the cranial neural tube, although formation of the medial hinge point appears normal. With respect to the cranial neural tube, it is known that dorsal-lateral hinge point formation is essential for neural fold elevation; however the mechanisms that regulate dorsal-lateral hinge point formation are unknown. Some potential mechanisms could be extrapolated from our knowledge of dorsal-lateral hinge point formation in the spinal neural tube; however these mechanisms have not been determined experimentally for cranial neurulation. In the spinal neural tube there are two described mechanisms that contribute to formation of the dorsal-lateral hinge points. First, Shh signaling inhibits formation of the dorsal-lateral hinge points (Ybot-Gonzalez et al., 2002). In the anterior regions of the spinal neural tube, dorsal-lateral hinge point formation is inhibited by robust Shh signals from the notochord and floorplate. In contrast, in the more caudal regions of the spinal neural tube, Shh signals from the notochord and floorplate diminish allowing the formation of dorsal-lateral hinge points. Failure of this mechanism does not likely play a role in opm mutants as our data indicate that there are no obvious changes in dorsal-ventral patterning of the anterior neural tissue in opm mutants (data not shown), consistent with normal levels of Shh signaling. Second, in the spinal neural tube, contact with the surface ectoderm is required for induction of the dorsal-lateral hinge points (Hackett et al., 1997). If this mechanism also plays a role in induction of the dorsal-lateral hinge points in the cranial neural tube, then it is possible that the overexpansion of head mesenchyme in opm mutant embryos around the neural tube could interfere with this inductive interaction. Indeed, in opm mutants the surface ectoderm is greatly separated from the cranial neural tube (Figure 2), suggesting that contact between these two tissues could be required for dorsal-lateral hinge point formation in the cranial regions of the neural tube. Alternatively, it is possible that the denser head mesenchyme simply causes mechanical inhibition of neural fold elevation.
While we detected a defect in the development of the head mesenchyme associated with cranial neural tube closure defects in opm mutant embryos it is still a distinct possibility that Hectd1 is also required in other cell lineages for neural tube closure. Our analysis of the expression of Hectd1 indicates that the transcript is also expressed in the neural and non-neural ectoderm during neural tube closure. Therefore, further experiments such as tissue specific deletion of Hectd1 or chimeric analysis are needed to determine if expression of Hectd1 in these tissues also contributes to the lack of dorsal-lateral hinge point formation and the subsequent failure to close the cranial neural tube in opm mutant embryos.
Ubiquitination and regulation of development
In recent years, the importance of posttranslational modification of proteins by ubiquitin during embryonic development has become increasingly apparent. Ubiquitination results in the conjugation of a 76 amino acid polypeptide (ubiquitin) to substrate proteins (Kerscher et al., 2006). Polyubiquitination targets proteins for destruction in the proteasome, while monoubiquitination results in the modification of protein function and/or processing. Ubiquitination is a multistep process where an Ubiquitin-activating enzyme (E1) activates ubiquitin and transfers it to an ubiquitin-conjugating enzyme (E2). The E3 ubiquitin ligase associates with both the E2 enzyme and the target substrate through protein-protein interaction domains. There are two main types of ubiquitin ligases: ones that contain a Ring domain and ones that contain a C-terminal HECT (homologous to E6-AP carboxy terminal) domain, the latter type includes Hectd1. The substrate specificity of the ubiquitin pathway arises from the interaction of the E3 ligases with their substrates and the HECT domain ligases interact directly with their substrates.
The opm mutation occurs in a gene encoding a novel E3 ubiquitin ligase, Hectd1. Gene identification was confirmed by failure of complementation with the genetrap allele (Hectd1XC) in which the HECT domain was disrupted. Additionally, the identical phenotypes observed in mutant embryos homozygous for the Hectd1opm (presumed null) and Hecd1XC alleles indicate that ubiquitin ligase activity is essential for the function of Hectd1 in vivo. The observation that mutation of Hectd1 results in the opm phenotype suggests that posttranslational modification of an unknown substrate protein(s) is required for normal development of the head mesenchyme. Since we find that the head mesenchyme around the neural tube is denser as early as E8.5, and that no significant difference in proliferation rates in the head mesenchyme are observed between wildtype and mutant embryos at this stage, expansion of the head mesenchyme could be due to misregulation of the numbers of progenitor cells specified in the primitive streak, the origin of the cephalic mesoderm. As an example, it is possible that Hectd1 normally regulates a pathway that is required for patterning of the primitive streak, for instance by regulating the strength and/or duration of a patterning signal. Therefore, in this situation, loss of ubiquitin modification by Hectd1 in opm mutants could result in abnormal signaling and an increase in the number of cephalic mesoderm precursors set aside in the primitive streak. Alternatively, it is possible that the apparent expansion of the head mesenchyme is due to a failure to reorganize during neural tube closure. Since the increased density of head mesenchyme indicates that the extracellular space between the mesenchymal cells is also not increased appropriately, it is possible that Hectd1 could regulate the quantity or quality of the extracellular matrix required for reorganization of the head mesenchyme.
Determination of the substrate(s) of the Hectd1 ubiquitin ligase will also lead to a greater understanding of the mechanisms underlying the neural tube closure defects in opm mutants. Currently the substrate(s) of Hectd1 are unknown; however the human orthologue of Hectd1, KIAA1131 was recently identified in yeast two-hybrid screens (Nakayama et al., 2002) and (NCBI accession: AAP13073). In both of these screens, Hectd1 was found to bind Immunoglobulin-like domain containing 1 protein (Igsf1), implicating Igsf1 as a potential Hectd1 substrate. Originally, Igsf1 was identified in a screen for proteins that bound the TGFβ antagonist Inhibin and was thus also named InhBP/p120 (Chong et al., 2000); however, subsequent studies have questioned the ability of InhBP/p120 to modulate Inhibin signaling (Bernard et al., 2003; Chapman et al., 2002). The future identification of Hectd1 substrates and their modification by ubiquitination will increase our understanding of the molecular pathways that regulate the normal (and abnormal) development of the cephalic mesoderm and neural tube closure and provide further molecular insight into the etiology of the opm phenotype.
Variable expressivity of neural tube defects in Hectd1 heterozygotes suggests a candidate susceptibility gene locus
Embryos heterozygous for the opm allele (presumed null) display a low frequency (∼5%) neural tube defects. This suggests that the level of Hectd1 function is critical and that loss of one allele can decrease protein function below a critical threshold. Interestingly, embryos heterozygous for the gene trap allele (Hectd1XC) in which the C-terminal HECT domain is disrupted display a higher frequency (∼20%) of neural tube defects. It has been shown that mutation of a conserved cysteine in the HECT domain results in a dominant-negative protein by maintaining the interaction of the ligase with the substrate but preventing ubiquitination (Huibregtse et al., 1995; Talis et al., 1998). Thus, the Hectd1XC allele could create a dominant-negative protein that prevents interaction of the wildtype protein with the substrate, potentially explaining the higher frequency of neural tube defects in the XC heterozygotes relative to the opm allele.
Neural tube closure defects are one of the most common birth defects in humans with approximately one in one thousand live births affected. It is likely that in some cases the genetic factors contributing to human neural tube defects result from a low penetrance of neural tube defects in heterozygous individuals with null or dominant negative mutations in genes that are required for neural tube closure (Harris and Juriloff, 2006). Therefore, it is of interest that Hectd1 heterozygotes display variable expressivity of neural tube defects in heterozygous individuals. Our observations that Hectd1 function is required at a critical threshold level suggest Hectd1 would be a good candidate for a susceptibility gene contributing to neural tube closure defects in humans.
Acknowledgments
We are grateful to Trevor Williams and members of the Niswander and Anderson laboratories for helpful discussions and suggestions, Karen Sears for statistical analysis and Trevor Williams for critical reading of the manuscript. We also thank Lori Bulwith and Andrew Pollock for technical assistance. ES cells used to generate Hectd1XC mutant mice were from BayGenomics. This work was supported by NIH grants F32-HD08605 to IEZ, U01-HD43478 and R01-HD035455 to KVA. LAN is a HHMI investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernard DJ, Burns KH, Haupt B, Matzuk MM, Woodruff TK. Normal reproductive function in InhBP/p120-deficient mice. Mol Cell Biol. 2003;23:4882–91. doi: 10.1128/MCB.23.14.4882-4891.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer S, Feng W, Huang J, Sullivan S, Williams T. Wnt1-Cre-mediated deletion of AP-2alpha causes multiple neural crest-related defects. Dev Biol. 2004;267:135–52. doi: 10.1016/j.ydbio.2003.10.039. [DOI] [PubMed] [Google Scholar]
- Caspary T, Anderson KV. Patterning cell types in the dorsal spinal cord: what the mouse mutants say. Nat Rev Neurosci. 2003;4:289–97. doi: 10.1038/nrn1073. [DOI] [PubMed] [Google Scholar]
- Chapman DL, Garvey N, Hancock S, Alexiou M, Agulnik SI, Gibson-Brown JJ, Cebra-Thomas J, Bollag RJ, Silver LM, Papaioannou VE. Expression of the T-box family genes, Tbx1-Tbx5, during early mouse development. Dev Dyn. 1996;206:379–90. doi: 10.1002/(SICI)1097-0177(199608)206:4<379::AID-AJA4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Chapman SC, Bernard DJ, Jelen J, Woodruff TK. Properties of inhibin binding to betaglycan, InhBP/p120 and the activin type II receptors. Mol Cell Endocrinol. 2002;196:79–93. doi: 10.1016/s0303-7207(02)00227-7. [DOI] [PubMed] [Google Scholar]
- Chen ZF, Behringer RR. Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9:686–99. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Chong H, Pangas SA, Bernard DJ, Wang E, Gitch J, Chen W, Draper LB, Cox ET, Woodruff TK. Structure and expression of a membrane component of the inhibin receptor system. Endocrinology. 2000;141:2600–7. doi: 10.1210/endo.141.7.7540. [DOI] [PubMed] [Google Scholar]
- Copp AJ. Neurulation in the cranial region--normal and abnormal. J Anat. 2005;207:623–35. doi: 10.1111/j.1469-7580.2005.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Greene ND, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev Genet. 2003;4:784–93. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121:439–51. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–6. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Garcia-Garcia MJ, Eggenschwiler JT, Caspary T, Alcorn HL, Wyler MR, Huangfu D, Rakeman AS, Lee JD, Feinberg EH, Timmer JR, Anderson KV. Analysis of mouse embryonic patterning and morphogenesis by forward genetics. Proc Natl Acad Sci U S A. 2005;102:5913–9. doi: 10.1073/pnas.0501071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelineau-van Waes J, Finnell RH. Genetics of neural tube defects. Semin Pediatr Neurol. 2001;8:160–4. doi: 10.1053/spen.2001.26449. [DOI] [PubMed] [Google Scholar]
- Hackett DA, Smith JL, Schoenwolf GC. Epidermal ectoderm is required for full elevation and for convergence during bending of the avian neural plate. Dev Dyn. 1997;210:397–406. doi: 10.1002/(SICI)1097-0177(199712)210:4<397::AID-AJA4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res A Clin Mol Teratol. 2006 doi: 10.1002/bdra.20333. [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Soriano P. Shroom, a PDZ domain-containing actin-binding protein, is required for neural tube morphogenesis in mice. Cell. 1999;99:485–97. doi: 10.1016/s0092-8674(00)81537-8. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington RS, Costantini F, Lacy E. Manipulating the Mouse Embryo, a Laboratory Manual. Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Holmes G, Niswander L. Expression of slit-2 and slit-3 during chick development. Dev Dyn. 2001;222:301–7. doi: 10.1002/dvdy.1182. [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci U S A. 1995;92:5249. doi: 10.1073/pnas.92.11.5249-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CM, Lyons KM, Hogan BL. Involvement of Bone Morphogenetic Protein-4 (BMP-4) and Vgr-1 in morphogenesis and neurogenesis in the mouse. Development. 1991;111:531–42. doi: 10.1242/dev.111.2.531. [DOI] [PubMed] [Google Scholar]
- Kasarskis A, Manova K, Anderson KV. A phenotype-based screen for embryonic lethal mutations in the mouse. Proc Natl Acad Sci U S A. 1998;95:7485–90. doi: 10.1073/pnas.95.13.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman MH, Bard JBL. The Anatomical Basis of Mouse Development. Academic Press; San Diego: 1999. [Google Scholar]
- Kerscher O, Felberbaum R, Hochstrasser M. Modification of Proteins by Ubiquitin and Ubiquitin-Like Proteins. Annu Rev Cell Dev Biol. 2006 doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- Liu A, Joyner AL, Turnbull DH. Alteration of limb and brain patterning in early mouse embryos by ultrasound-guided injection of Shh-expressing cells. Mech Dev. 1998;75:107–15. doi: 10.1016/s0925-4773(98)00090-2. [DOI] [PubMed] [Google Scholar]
- Mitchell PJ, Timmons PM, Hebert JM, Rigby PW, Tjian R. Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev. 1991;5:105–19. doi: 10.1101/gad.5.1.105. [DOI] [PubMed] [Google Scholar]
- Morriss GM, Solursh M. Regional differences in mesenchymal cell morphology and glycosaminoglycans in early neural-fold stage rat embryos. J Embryol Exp Morphol. 1978;46:37–52. [PubMed] [Google Scholar]
- Nakayama M, Kikuno R, Ohara O. Protein-protein interactions between large proteins: two-hybrid screening using a functionally classified library composed of long cDNAs. Genome Res. 2002;12:1773–84. doi: 10.1101/gr.406902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Bennett MF, Sargent MG, Wilkinson DG. Cloning and developmental expression of Sna, a murine homologue of the Drosophila snail gene. Development. 1992;116:227–37. doi: 10.1242/dev.116.1.227. [DOI] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207:575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch C, Hustert E, Pfeifer D, Sudbeck P, Kist R, Roe B, Wang Z, Balling R, Blin N, Scherer G. The SOX10/Sox10 gene from human and mouse: sequence, expression, and transactivation by the encoded HMG domain transcription factor. Hum Genet. 1998;103:115–23. doi: 10.1007/s004390050793. [DOI] [PubMed] [Google Scholar]
- Schatteman GC, Morrison-Graham K, van Koppen A, Weston JA, Bowen-Pope DF. Regulation and role of PDGF receptor alpha-subunit expression during embryogenesis. Development. 1992;115:123–31. doi: 10.1242/dev.115.1.123. [DOI] [PubMed] [Google Scholar]
- Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235–8. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, von Melchner H, Wurst W, Hicks G, Nord AS, Cox T, Young SG, Ruiz P, Soriano P, Tessier-Lavigne M, Conklin BR, Stanford WL, Rossant J. A public gene trap resource for mouse functional genomics. Nat Genet. 2004;36:543–4. doi: 10.1038/ng0604-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Talis AL, Huibregtse JM, Howley PM. The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)-positive and HPV-negative cells. J Biol Chem. 1998;273:6439–45. doi: 10.1074/jbc.273.11.6439. [DOI] [PubMed] [Google Scholar]
- Timmer JR, Wang C, Niswander L. BMP signaling patterns the dorsal and intermediate neural tube via regulation of homeobox and helix-loop-helix transcription factors. Development. 2002;129:2459–72. doi: 10.1242/dev.129.10.2459. [DOI] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Cogram P, Gerrelli D, Copp AJ. Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development. 2002;129:2507–17. doi: 10.1242/dev.129.10.2507. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hagopian-Donaldson S, Serbedzija G, Elsemore J, Plehn-Dujowich D, McMahon AP, Flavell RA, Williams T. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature. 1996;381:238–41. doi: 10.1038/381238a0. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Behringer RR, de Crombrugghe B. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat Genet. 1996;13:275–83. doi: 10.1038/ng0796-275. [DOI] [PubMed] [Google Scholar]
- Zohn IE, Anderson KV, Niswander L. Using genomewide mutagenesis screens to identify the genes required for neural tube closure in the mouse. Birth Defects Res A Clin Mol Teratol. 2005 doi: 10.1002/bdra.20164. [DOI] [PubMed] [Google Scholar]
- Zoltewicz JS, Stewart NJ, Leung R, Peterson AS. Atrophin 2 recruits histone deacetylase and is required for the function of multiple signaling centers during mouse embryogenesis. Development. 2004;131:3–14. doi: 10.1242/dev.00908. [DOI] [PubMed] [Google Scholar]