

Abstract
The protein kinase Chk1 has been implicated as a key regulator of cell cycle progression and DNA repair and inhibitors of Chk1 (e.g., UCN-01, EXEL-9844) potentiate the cytotoxic actions of chemotherapeutic drugs in tumor cells. We have examined the ability of PD-321852, a small-molecule Chk1 inhibitor, to potentiate gemcitabine (Gem)-induced clonogenic death in a panel of pancreatic cancer cell lines, and evaluated the relationship between endpoints associated with Chk1 inhibition and chemosensitization.
Gem chemosensitization by minimally-toxic concentrations of PD-321852 ranged from minimal (< 3 fold change in survival) in Panc1 cells, to over 30-fold in MiaPaCa2 cells. PD-321852 inhibited Chk1 in all cell lines, as evidenced by stabilization of Cdc25A, and, in combination with Gem, a synergistic loss of Chk1 protein was observed in the more sensitized cell lines. Gem chemosensitization, however, did not correlate with abrogation of the S/M or G2/M checkpoint; PD-321852 did not induce premature mitotic entry in Gem-treated BxPC3 or M-Panc96 cells, which were sensitized to Gem 6.2- and 4.6-fold, respectively.
In the more sensitized cells lines, PD-321852 not only inhibited Gem-induced Rad51 focus formation and the recovery from Gem-induced replication stress, as evidenced by persistence of γ-H2AX, but also depleted these cells of Rad51 protein. Our data suggest the inhibition of this Chk1-mediated Rad51 response to Gem-induced replication stress is an important factor in determining Gem chemosensitization by Chk1 inhibition in pancreatic cancer cells.
Keywords: Chk1, checkpoint abrogation, gemcitabine, γ-H2AX, premature mitotic entry, Rad51
Introduction
Most tumor cells exhibit abnormalities in one or more aspects of cell cycle regulation (i.e. checkpoints), and a consensus view has emerged that these abnormalities present an attractive target for therapeutic intervention (reviewed in [1-4]). One member of the checkpoint system that is currently generating intense interest as a therapeutic target is Checkpoint kinase 1 (Chk1), a protein that functions in multiple pathways in response to genotoxic stress. Although interfering with Chk1 function can produce substantial increases in the sensitivity of tumor cells to a variety of chemotherapeutic agents, this effect varies markedly among human tumor cell lines. A better understanding of the mechanistic basis for differential responses to Chk1 inhibitors is therefore necessary in order to rationally translate these agents to the clinic.
One of the best characterized functions of activated Chk1 is its ability to phosphorylate and inactivate Cdc25 phosphatases in response to replication stress [5]. Many chemotherapeutic agents that target DNA synthesis and integrity, including camptothecin [6], 5-fluorodeoxyuridine [7], gemcitabine (Gem; [8-10]) and doxorubicin [6] have been shown to activate Chk1, as indicated by phosphorylation at Ser317 and/or Ser345. This activation is followed by destabilization of Cdc25A, via phosphorylation by Chk1 at multiple sites (reviewed in [11]), which in turn results in inactivation of cyclin-dependent kinase 1 complexes and G2 arrest (the G2/M checkpoint; [6,12]) and/or inactivation of cyclin dependent kinase 2 complexes and intra-S phase arrest [13]. Inhibition of Chk1 with either siRNA [6,10] or chemical inhibitors [8,14,15], prevents drug-induced Cdc25A degradation, leading to abrogation of the intra-S and/or G2/M checkpoint and inappropriate entry into mitosis with either a 4N or a sub-4N DNA content (premature mitotic entry [16]). Abrogation of these checkpoints and premature mitotic entry have previously been correlated with drug sensitization and increased cell death [8,15,17].
In addition to its effects on Cdc25A and cell cycle checkpoints, Chk1 inhibition has been proposed to sensitize cells to DNA damaging agents by destabilizing chemotherapy-induced stalled replication forks and/or inhibiting DNA repair. This hypothesis is supported by studies using histone H2AX phosphorylation at Ser139 (γ-H2AX) as a marker for DNA damage or replication stress, in which inhibition of Chk1 caused an increase in γ-H2AX staining after treatment with Gem [8,18]. Chk1 also functions directly in the homologous recombination repair (HRR) of chemotherapy-induced DNA damage, in part by regulating Rad51, a key protein in HRR [19,20]. Hydroxyurea-induced replication stress induces nuclear accumulation of Rad51 foci in an ATR- and Chk1-dependent manner, and inhibition of Chk1 prevents localization of Rad51 to sites of HRR [19,20].
In preliminary studies using the Chk1 antagonist PD-3218521, we observed that catalytic inhibition of Chk1, stabilization of Cdc25A, and premature mitotic entry were discordant with the degree of Gem chemosensitization in a panel of pancreatic cancer cell lines. These observations suggested to us that other molecular events might better account for potentiation of Gem-induced clonogenic death. In our present study, we determined that molecular events related to replication stress and/or DNA damage and repair, such as inhibition of Rad51 focus formation and persistence of γ-H2AX staining, might be more informative markers for Gem chemosensitization by Chk1 inhibitors in pancreatic cancer cells.
Methods
Cell culture and drug solutions
MiaPaCa2, BxPC3, Panc1 and M-Panc96 cells were grown in DMEM (MiaPaCa2 and Panc1) or RPMI 1640 (BxPC3 and M-Panc96) medium supplemented with 10% fetal bovine serum (Life Technologies, Rockville, MD) and 2 mM L-glutamine (Sigma Chemical, St. Louis, MO). Gemcitabine (Eli Lilly, Indianapolis, IN) was dissolved in PBS and stored in aliquots at -20° C. PD-321852 is a phenyl carbazole (4-(2,6-Dichloro-phenyl)-9-hydroxy-6-(3-methylamino-propyl)-6H-pyrrolo[3,4-c]carbazole-1,3 dione) kinase inhibitor developed in a collaborative effort by Pfizer Global Research and Development and Dr. W.A. Denny, University of Auckland, NZ (Supplementary data, Fig. S1). The IC50 of PD-321852 for inhibition of Chk1 in a cell-free assay system is 5 nM [21]. PD-321852 was dissolved in dimethyl sulfoxide (DMSO; Sigma Chemical) and stored in aliquots at -20° C.
Clonogenic Assay
Cells treated with PD-321852 alone, Gem alone, or Gem + PD-321852 for 24 h were trypsinized and processed for clonogenic survival as previously described [22]. Surviving fractions (SF) for samples treated with a single agent represent the plating efficiency for a given drug-treated sample divided by the plating efficiency for the corresponding control. For samples treated with both Gem and PD-321852, the SF was calculated as the plating efficiency for a given sample treated with both agents divided by the plating efficiency for the corresponding sample treated with PD-321852 alone. Unless noted, cells were treated with a moderately toxic concentration of Gem for 24 h. This concentration was 100 nM in MiaPaCa2 (SF=0.36±0.04) and BxPC3 cells (SF=0.32±0.04), 300 nM in M-Panc96 cells (SF=0.24±0.03) and 1 μM in Panc1 cells (SF=0.45±0.04).
Western Blot Analysis
Whole cell lysates were prepared in buffer containing 10 mM Tris (pH 7.4), 2% sodium dodecyl sulfate (SDS), 1× Complete Protease Inhibitor Cocktail (Roche, Mannheim, Germany), 1 mM sodium fluoride, 2 mM sodium orthovanadate, and 1 mM sodium pyrophosphate, diluted in 2× loading buffer (0.32 M Tris-HCl, 10% glycerol, 2% SDS, 0.2% bromophenol blue, 4% 2-mercaptoethanol, pH 6.8) and resolved on 10% polyacrylamide gels. Separated proteins were then transferred to Immobilon PVDF membranes (Millipore Corporation, Billerica, MA) and incubated overnight at 4°C with one the following antibodies: anti-Chk1 (G-4), anti-Cdc25A (F-6) and anti-Rad51 (H-92) (Santa Cruz Biotechnology, Santa Cruz, CA); anti-Chk2 (clone 7) (Upstate Biotechnology, Lake Placid, NY) or anti-β-actin (Sigma Chemical). For Chk1 quantitation, autoradiograms were scanned for densitometric analysis and normalized first to actin loading control, then to the effects of Gem alone. Data were analyzed with AlphaEase FC software (Alpha Innotech, San Leandro, CA). For experiments with the proteasome inhibitor calpain inhibitor I N-Acetyl-l-leucinyl-l-leucinyl-l-norleucinol (LLnL; Sigma Chemical), 25 μg/mL LLnL or vehicle control (0.1% DMSO) was added to the drug media for the last 3 h of treatment.
Flow Cytometry
Exponentially growing cells were treated as indicated, trypsinized, washed with ice-cold PBS, and fixed at a concentration of 2 × 106 cells/mL in ice-cold 70% ethanol. For phospho-histone H3 analysis, washed, fixed cells were incubated with a rabbit anti-phospho-histone H3-specific antibody (no. 06-570; Upstate Cell Signaling Solutions, Lake Placid, NY), followed by a FITC-conjugated anti-rabbit secondary antibody (F-0382; Sigma Chemical) as previously described [7]. Trout erythrocyte nuclei (BioSure, Grass Valley, CA) were included as internal standards. Samples were then stained with propidium iodide to measure total DNA content and analyzed on a FACScan flow cytometer (Becton-Dickinson, Palo Alto, CA) with FlowJo software (Tree Star, Inc., Ashland, OR). Normal and premature mitoses were separated by first defining normal mitosis under control conditions as phospho-histone H3 positive-cells with 4N DNA content, and then applying those parameters to treated samples. Premature mitosis was defined as phospho-histone H3 positive cells with sub-4N DNA content.
For γ-H2AX analysis, samples were processed as above, then incubated with a mouse anti-γ-H2AX specific antibody (clone JBW301, no. 05-636; Upstate) overnight at 4°C prior to incubation with a FITC-conjugated anti-mouse secondary antibody (F-0257; Sigma Biochemical) as previously described [23]. For each cell line, a gate was set arbitrarily to define a region of positive staining for γ-H2AX (approximately 5%). This gate was then overlaid upon the drug-treated samples.
Immunofluorescence
Cells were subcultured onto coverslips in 12-well dishes. Forty-eight h later, cells were treated with either Gem alone, PD-321852 or Gem + PD-321852 for 24 h. Alternatively, cells were irradiated with 7.5 Gy. Immediately after drug treatment, or 5 hours post-irradiation, cells were fixed for 15 min in ice-cold 3.7% paraformaldehyde, 2% sucrose, 0.5% Triton X-100 solution, washed 3 × 5 min with PBS, permeabilized for 10 min in PBS with 0.5% Triton X-100, and blocked for at least 30 min in PBS containing 5% non-fat milk, 1% goat serum and 0.05% Triton X-100. Samples were then incubated in blocking buffer with both a polyclonal rabbit anti-Rad51 antibody (1:400 dilution; (H-92) Santa Cruz Biotechnology) and a mouse anti-γ-H2AX antibody (1:2000 dilution; (clone JBW301) Upstate Biotechnology) for 1 hour at room temperature. Samples were then washed 3 × 5 min with PBS and incubated with both Alexa 488-conjugated goat anti-rabbit secondary antibody and Alexa 594-conjugated goat anti-mouse secondary antibody (1:1000 dilutions; Molecular Probes, Eugene, OR) for 30 min at room temperature. Cells were then washed 3 × 5 min with PBS and stained with DAPI (0.1 mg/ml, 4,6 diamidino-2-phenylindole, Roche) for 5 min. After extensive washing, cover slips were mounted on slides with anti-fade reagent (Prolong Gold, Molecular Probes). Samples were imaged with an Olympus FV500 confocal microscope (Olympus America Inc., Center Valley, PA) with a 60× objective. Fields were chosen at random based on DAPI staining. For quantitation of Rad51 foci, at least 100 cells from each of 3 independent experiments were visually scored for each condition. Cells with 10 or more Rad51 foci were scored as positive (+).
Statistics
Statistically significant differences were determined using one-way analysis of variance (ANOVA) with the Newman-Keuls post-comparison test using GraphPad PRISM v3 (GraphPad Software, Inc., San Diego, CA).[24]
Results
Effect of PD-321852 on clonogenic survival
We first assessed the toxicity of PD-321852 (for structure, see Supplementary Data, Fig. S1) both by itself and in combination with Gem, in a panel of pancreatic cancer cell lines. The four cell lines studied were similarly sensitive to PD-321852-induced clonogenic death, with a “threshold” toxic (i.e., highest non-toxic) concentration of 0.1 μM, and minimal toxicity at 0.3 μM (see Supplementary Data, Fig. S2A). We next assessed the ability of PD-321852 to sensitize these cell lines to Gem (Fig. 1 and Supplementary Data Fig. S2B). MiaPaCa2, M-Panc96 and BxPC3 cells were each sensitized to Gem by PD-321852, with a 13-, 17-, or 6-fold shift in the Gem IC50, respectively (Table 1), and a significant increase in Gem-induced clonogenic death. In each of these sensitized cell lines, the effects of PD-321852 on Gem-induced clonogenic death were most pronounced at minimally- or, in MiaPaCa2 cells, even non-toxic concentrations of Gem. Although PD-321852 also caused a significant 4-fold shift in the Gem IC50 in Panc1 cells, the differences in clonogenic survival between Panc1 cells treated with Gem alone compared to Gem + PD-321852 were minimal (less than 3-fold), compared to the other cell lines.
Figure 1. The effect of PD-321852 on Gem-induced clonogenic death in pancreatic cancer cell lines.
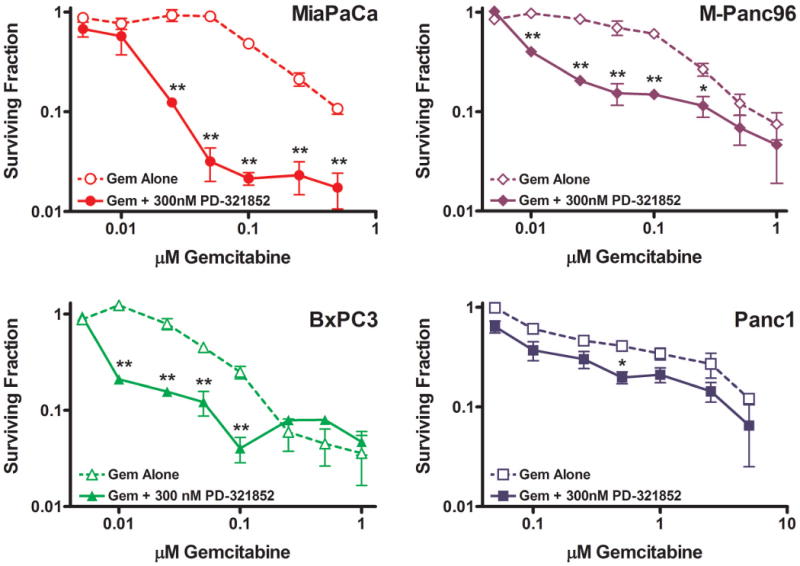
MiaPaCa2, BxPC3, M-Panc96 and Panc1 cells were treated with Gem alone or Gem + 0.3 μM PD-321852 for 24 h and assayed for clonogenic death. Data plotted are the mean ± SEM (n=3-7), *p<0.05 or **p<0.01, one-way ANOVA.
Table 1.
Gem IC50 values for pancreatic cancer cells treated with Gem ± 0.3 μM PD-321852 for 24 h and assayed for clonogenic survival. Data are presented as the mean ± SEM (n=3-7; *p<0.05, one-way ANOVA).
| Cell Line | Gem IC50 (nM) (Gem Alone) |
Gem IC50 (nM) (Gem + PD-321852) |
|---|---|---|
| MiaPaCa2 | 160±19 | 12±4.6* |
| M-Panc96 | 150±21 | 9.2±0.16* |
| BxPC3 | 50±8.5 | 8.0±0.05* |
| Panc1 | 480±63 | 110±29* |
Catalytic inhibition and degradation of Chk1 induced by PD-321852
To test the hypothesis that differences in the ability of PD-321852 to sensitize different pancreatic cancer cell lines to Gem-induced clonogenic death result from differences in Chk1 inhibition, we examined Cdc25A protein levels in cells treated with either PD-321852 alone, Gem alone or Gem and PD-321852 combined. The generally accepted model of Cdc25A regulation by Chk1 predicts that inhibition of Chk1 should be reflected by abrogation of Gem-induced Cdc25A degradation [17,25]. Although each of the cell lines examined expressed similar levels of Chk1, and PD-321852 stabilized Cdc25A protein levels in each, both basal Cdc25A levels and the effects of PD-321852 on Gem-induced Cdc25A degradation varied (Fig. 2A&B). While MiaPaCa2 and BxPC3 cells were sensitized to Gem by 0.03 μM PD-321852 (Supplementary Data, Fig. S2B), there was no measurable effect on Cdc25A protein levels until higher concentrations of PD-321852 were used (0.1-0.3 μM) and the effects of PD-321852 on Cdc25A stability were minimal in BxPC3 cells. Inhibition of Gem-induced Cdc25A degradation was most effective in the Panc1 cells, the cell line least sensitized to Gem by PD-321852. Finally, although Cdc25A levels were stabilized by PD-321852 in M-Panc96 cells, these cells did not exhibit Gem-induced Cdc25A degradation.2 Collectively, these results suggest that while PD-321852 inhibits Chk1 in each of these cell lines, as indicted by attenuated Cdc25A degradation under basal conditions, Gem sensitization by PD-321852 did not require abrogation of Gem-induced Cdc25A degradation.
Figure 2. The effects of PD-321852, Gem or Gem + PD-321852 on checkpoint protein levels.

Basal levels of Cdc25A, Chk1, Chk2 and Rad51 in pancreatic cancer cell lines (A). Image shown is representative of 2 independent experiments. In (B), cells treated with either Gem alone (100 nM for MiaPaCa and BxPC3, 300 nM for M-Panc96 and 1 μM for Panc1), 0.3 μM PD-321852 or Gem + PD-321852 for 24 h were assayed for Cdc25A, Chk1 and Chk2 protein levels. Breaks indicate non-contiguous lanes from a single membrane. Images shown are representative of 3-5 independent experiments. For quantitation of Chk1 depletion in cells treated with Gem + PD-321852 (C), autoradiograms were scanned for densitometric analysis and normalized first to actin loading control, then to the effects of Gem alone. Data are given as the mean ± SEM (n=3-5), *p<0.05 or **p<0.01, one-way ANOVA. In (D), MiaPaCa2 and BxPC3 cells were treated with Gem + 0.3 μM PD-321852 for 24 h, with no other addition, or with 25 μg/ml LLnL added for the final 3 h of drug treatment. Images shown are representative of two independent experiments.
Surprisingly, PD-321852 caused a synergistic loss of Chk1 protein in Gem-treated cells. This effect was most pronounced in the three more sensitized cell lines, MiaPaCa2, BxPC3 and M-Panc96 (Fig. 2C), and may be attributed to the activation-mediated degradation of Chk1 [26]. To test whether PD-321852 accelerated Chk1 degradation in Gem-treated cells, we added the proteasome inhibitor, LLnL, during the last 3 h of drug treatment and measured Chk1 protein levels by western blot. As shown in Fig. 2D, Chk1 protein levels were restored by LLnL, suggesting the loss of Chk1 in cells treated with both Gem and PD-321852 is at least partly due to proteasome-mediated degradation.
The relationship between the effects of PD-321852 on cell cycle progression and chemosensitization
Previous studies have linked aberrant Cdc25A expression to premature induction of mitosis (in cells with less than 4N DNA content) and cell death [7,8,17]. Based on these observations, we examined whether PD-321852 would promote premature mitotic entry, or S/M checkpoint abrogation, in Gem-treated cells, and whether these cell cycle effects were associated with chemosensitization. Cells treated with either PD-321852, Gem, or Gem and PD-321852 were analyzed by flow cytometry for expression of the mitotic marker, phospho-Histone H3 [27] (Fig. 3). Despite the elevated levels of Cdc25A, Chk1 inhibition with PD-321852 alone (0.3 μM) did not cause premature mitotic entry. It did, however, lead to an enrichment of cells in late S and G2/M (see also Supplementary Data, Fig. S3). In each of the cell lines examined, Gem alone caused an early S-phase arrest which is accompanied by activation of Chk1 [10]. Consistent with our previous Chk1 siRNA data, Chk1 inhibition with PD-321852 partially abrogated the G2/M and/or S/M checkpoints in Gem-treated Panc1 cells, with phospho-Histone H3-postive cells equally distributed between normal mitosis (4.8±0.6%) and premature mitosis (4.4±0.7%) (Fig. 3B; [17]). Despite this checkpoint abrogation, Panc1 cells were minimally sensitized to Gem under these conditions (Fig. 1D). Surprisingly, M-Panc96 cells appeared to be resistant to PD-321852-induced premature mitotic entry, or abrogation of the Gem-induced S/M checkpoint, even though PD-321852 attenuated Cdc25A degradation in these cells (Fig. 2B). It should also be noted that, despite a failure to degrade Cdc25A, each of these cell lines still arrested in late G1 or early S-phase when treated with Gem and PD-321852, likely as a consequence of Gem-induced nucleotide pool depletion. Only in MiaPaCa2 cells did the effects of Chk1 inhibition on Cdc25A correlate with premature mitotic entry, S/M checkpoint abrogation and sensitization to Gem. These data suggest aberrant Cdc25A regulation and premature mitotic entry, or abrogation of the S/M checkpoint, are neither required nor sufficient for sensitization of pancreatic cancer cells to Gem.
Figure 3. Phosphorylation of Histone H3 (Ser10) following treatment with Gem ± PD-321852.
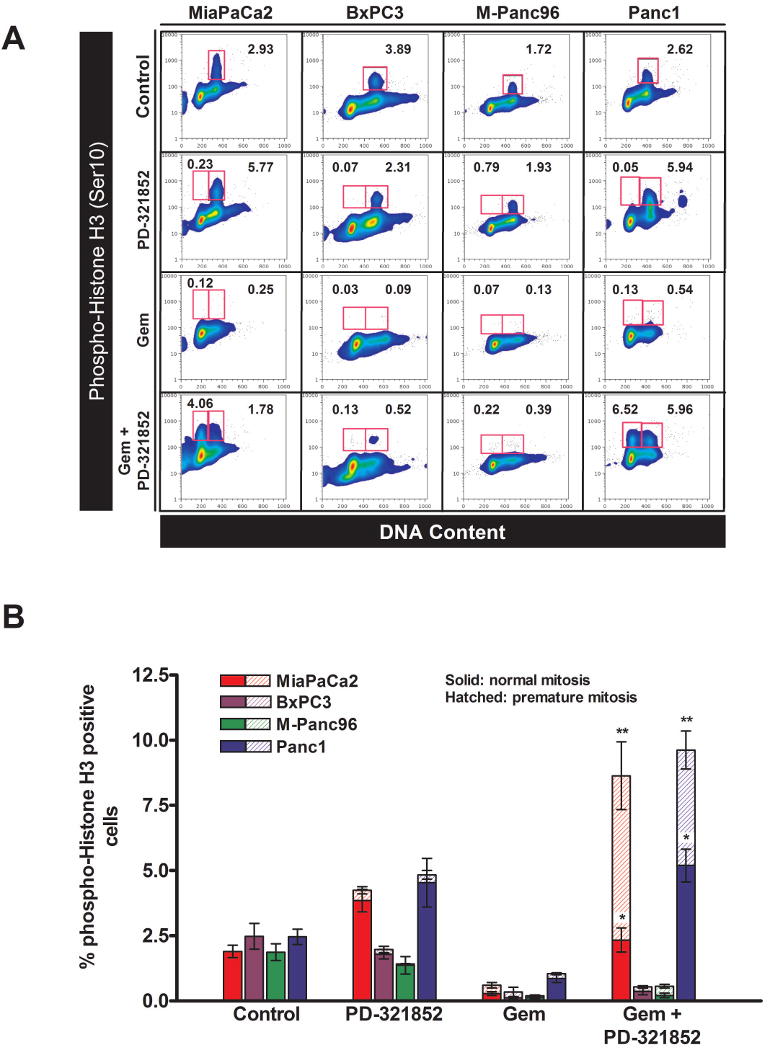
Cells treated with either 100 nM (MiaPaCa2 and BxPC3) 300 nM (M-Panc96) or 1 μM Gem (Panc1) ± 0.3 μM PD-321852 for 24 h were analyzed for phospho-Histone H3 (Ser10) expression, as a function of DNA content, by flow cytometry. Samples shown in (A) are representative of 3-4 individual experiments. Numbers given are the percentage of cells staining positive for phospho-Histone H3 (gated areas). Data presented in (B) are the mean ± SEM (n=3-4; *p<0.05, **p<0.001, compared to Gem alone, one-way ANOVA) of the percentage of phospho-Histone H3-positive cells with either a 4N (normal mitosis – solid bars) or sub-4N DNA content (premature mitosis – hatched bars) for each cell line.
The relationship between the effects of PD-321852 on Gem-induced Rad51 focus formation, γ-H2AX expression and chemosensitization
We next considered that the ability of PD-321852 to sensitize pancreatic cancer cells to Gem may correlate with inhibition of the DNA damage response and recovery from Gem-induced replication stress, rather than S/M or G2/M checkpoint abrogation. Chk1 participates in the cell's response to DNA damage in part through regulation of Rad51, a critical protein that functions in homologous recombination repair (HRR) [19]. To assess the effects of Chk1 inhibition on the DNA damage response, we tested the ability of PD-321852 to abrogate Gem-induced Rad51 focus formation (Fig. 4). While PD-321852 alone (0.3 μM) did not significantly alter the percentage of BxPC3 or Panc1 cells staining positive for Rad51 foci (10 or more foci/cell), it did attenuate Rad51 focus formation in MiaPaCa2 and M-Panc96 cells (Fig. 4C). There were also differences in PD-321852-induced γ-H2AX staining between the cell lines; 0.3 μM PD-321852 had minimal effects on γ-H2AX in Panc1 cells, but induced strong, pan-nuclear γ-H2AX staining in the other cell lines (see also Supplementary data Fig. S4). In most cases, Gem alone induced Rad51 focus formation and γ-H2AX staining, and in each cell line PD-321852 significantly blocked formation of Rad51 foci in Gem-treated cells. The magnitude of this effect, however, varied. While PD-321852 caused a greater than 10-fold decrease in the percentage of Gem-treated BxPC3, MiaPaCa2 and M-Panc96 cells staining positive for Rad51 foci, the percentage of Panc1 cells staining positive only decreased 2.4-fold, from 46.4 ± 2.57% to 19.6 ± 5.5%. Surprisingly, PD-321852 not only blocked Rad51 focus formation in the sensitized cell lines, but also caused a decrease in Rad51 protein levels, a result confirmed by western blot (Fig. 4D). Therefore, as with Chk1 protein depletion, drug-induced loss of Rad51 protein is associated with Gem sensitization in these cell lines.
Figure 4. Immunofluorescent analysis of Gem-induced Rad51 and γ-H2AX focus formation.
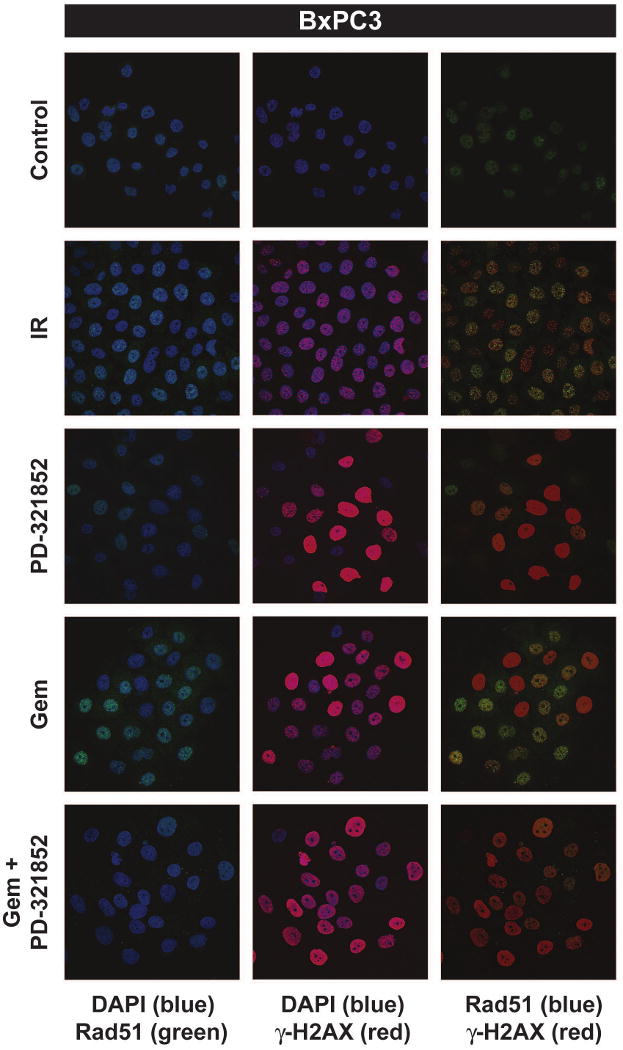

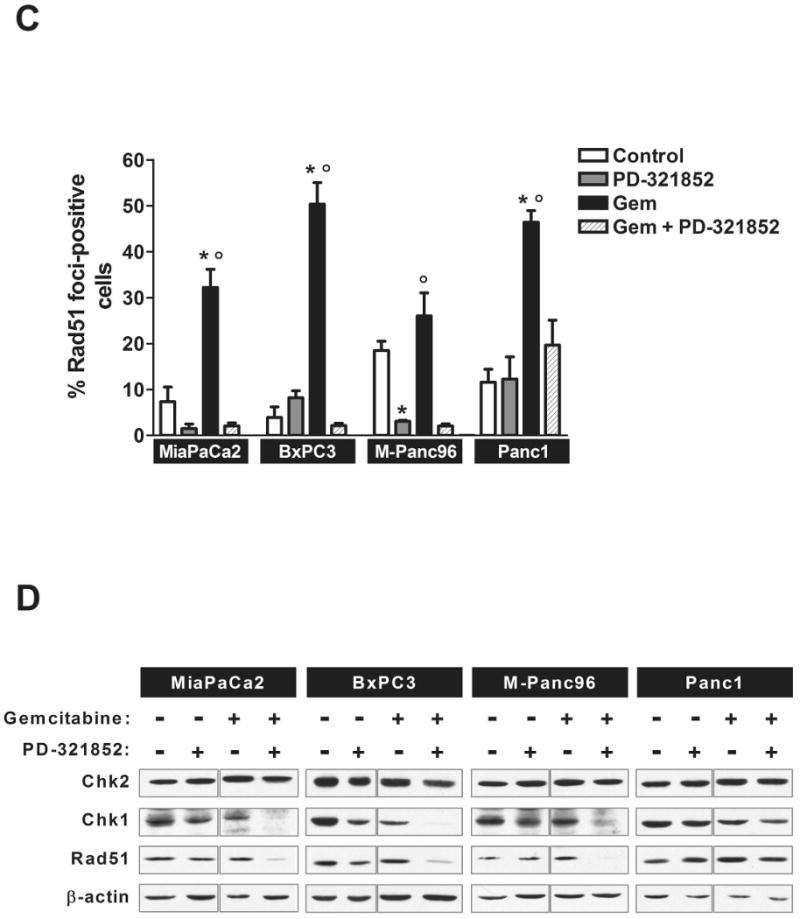
BxPC3 (A) and Panc1 (B) cells were either irradiated with 7.5 Gy and fixed 5 h post-irradiation or treated with Gem ± 0.3 μM PD-321852 for 24 h and stained for both Rad51 (green) and γ-H2AX (red). Samples shown are representative of 3 independent experiments. In (C), the percentage of cells with 10 or more Rad51 foci after treatment with either 100 nM (MiaPaCa2), 300 nM (BxPC3 and M-Panc96) or 3 μM Gem (Panc1) ± 0.3 μM PD-321852 for 24 h was determined. Data presented are the mean ± SEM (*p<0.01, compared to control, °p<0.01 compared to Gem + PD-321852, one-way ANOVA). Whole cell lysates from cells treated with either Gem alone, PD-321852 or Gem + PD-321852 for 24 h were assayed for Chk1, Chk2 and Rad51 protein levels (D). Blots shown are representative of 2-3 independent experiments.
To further test the hypothesis that sensitization to Gem correlates with inhibition of a Chk1 mediated DNA damage response, BxPC3 and Panc1 cells treated with PD-321852, Gem alone or Gem and PD-321852 were assayed for γ-H2AX by flow cytometry (Fig. 5). γ-H2AX is a commonly used marker for DNA damage [28] and/or replication stress [29] that has previously been used to assay for Gem-induced stalled replication forks [9,18]. In these experiments, we used 0.1 μM PD-321852 as this concentration did not induce γ-H2AX staining. In BxPC3 cells, treatment with 100 nM Gem for 24 h induced γ-H2AX in 34.4±5.4% of cells (Fig. 5C). After removal of Gem, cells resumed progression through S-phase over the next 48 h, and γ-H2AX staining fell to near control levels, suggestive of either DNA repair or recovery from Gem-induced replication arrest. Seventy-two h after the removal of Gem, we observed an increase in γ-H2AX positive cells that was accompanied by the accumulation of cells with sub-G1 DNA content. While PD-321852 alone (0.1 μM) had minimal effects on γ-H2AX staining in BxPC3 cells, it enhanced induction of γ-H2AX when combined with Gem, and significantly attenuated the loss of γ-H2AX after removal of both drugs (*p<0.05, one-way ANOVA). This persistence of γ-H2AX suggests PD-321852 inhibits recovery from Gem-induced replication stress, in BxPC3 cells. Similar results were observed in the other cell lines sensitized to Gem by Chk1 inhibition, MiaPaCa2 and M-Panc96 (data not shown). In Panc1 cells, a similarly toxic concentration of Gem (1 μM; SF=0.45±0.04) induced γ-H2AX in 74.9±3.8% of cells, and this level was maintained for 72 h after removal of drug (Fig. 5D). This persistence of γ-H2AX is consistent with the persistence of the Gem-induced S-phase arrest [18]. Panc1 cells treated with a lower, minimally-toxic concentration of Gem (100 nM; SF=0.76±0.09) also retained a high level of γ-H2AX staining after drug removal (Fig. 5D). In contrast to BxPC3 cells, PD-321852 did not enhance the induction of γ-H2AX in Panc1 cells. Due to the result that Panc1 cells accumulated in S-phase, and γ-H2AX intensity did not decrease over the course of this experiment, we were unable to determine the effects of Chk1 inhibition on recovery from Gem-induced replication arrest in Panc1 cells with this assay. Collectively, the Rad51 and γ-H2AX results suggest that Rad51 plays a significant role in the recovery from Gem-induced replication stress in BxPC3 cells, but not in Panc1 cells, and that inhibition of this response contributes to Gem chemosensitization.
Figure 5. Phosphorylation of Histone H2AX (γ-H2AX) in BxPC3 and Panc1 cells following treatment with Gem ± PD-321852.
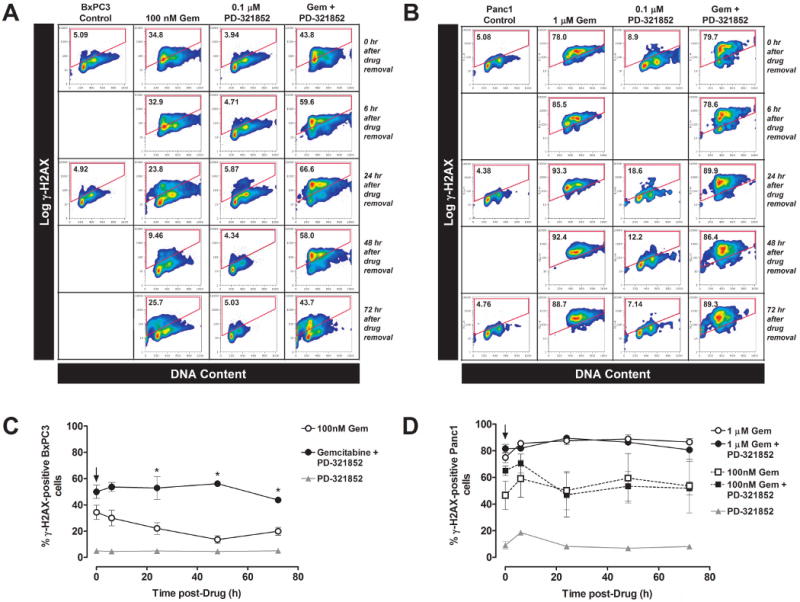
BxPC3 cells were treated with 100 nM Gem ± 0.1 μM PD-321852 for 24 h. The agents were then washed out (as indicated by the arrow in (C)), and cells were collected either immediately, or after incubation in control medium for an additional 6, 24, 48 or 72 h. Cells were analyzed by flow cytometry for the presence of γ-H2AX staining, as a function of DNA content (A & C). The number inside the gate is the percentage of the population staining positive for γ-H2AX. Panc1 cells were treated with either 1 μM Gem ± 0.1 μM PD-321852 (B & C) or 100 nM Gem ± 0.1 μM PD-321852 (C) for 24 h and analyzed as above. Data presented in (C) are the mean ± SEM of 3-5 independent experiments (*p<0.05; one-way ANOVA).
Discussion
In this study of pancreatic cancer cells, we found evidence supporting the hypothesis that Gem chemosensitization by Chk1 inhibition correlates well with inhibition of the Rad51 DNA damage response. Our data further suggest that the significance of the replication stress or DNA damage marker γ-H2AX depends upon the cell line examined, and may not always be a reliable indicator of Gem-induced DNA damage and/or repair.
It had previously been reported that Chk1 directly activates Rad51 and HRR in response to replication stress [19]. Consistent with previous reports [30], we found that Gem treatment caused the accumulation of Rad51 nuclear foci. It is unclear whether these foci result from Gem-induced stalled replication forks (replication stress) and/or Gem-induced accumulation of cells in S-phase. This distinction may be important in some cell lines as Tarsounas et al. have suggested that the mechanism for DNA damage-induced Rad51 focus formation, which is BRCA2-dependent, differs from the BRCA2-independent formation of Rad51 foci in unperturbed S-phase cells [31].
It has been reported that Rad51 overexpression may be a common event in pancreatic cancer that contributes to tumor resistance to DNA damaging agents [32]. PD-321852 may therefore sensitize cells to Gem by inhibiting Rad51 localization in response to Gem-induced replication arrest. Indeed, we found that PD-321852 attenuated Gem-induced Rad51 focus formation in each cell line examined. The magnitude of this effect, however, was much greater in the more sensitized cell lines where PD-321852 not only inhibited Gem-induced Rad51 focus formation, but was associated with depletion of Rad51 protein from whole cell lysates. This result is consistent with previous reports from Yao et al., who reported that depletion of Chk1 with siRNA leads to a loss of Rad51 protein in human leukemia cells [33], and Bahassi et al. who reported that Chk1 siRNA treatment prevented UV-induced Rad51 focus formation [20]. The ability of PD-321852 to block induction of Gem-induced Rad51 foci is further evidence that Chk1 is inhibited by this compound.
Recent studies have demonstrated that inhibition of Chk1 sensitizes tumor cells to a variety of chemotherapeutic agents [8,9,15,17]. As a result, this protein has become a promising target for therapeutic intervention, with at least three Chk1 inhibitors entering clinical trials recently, and others poised to follow [34]. A major issue in clinical development of molecularly targeted agents, including Chk1 inhibitors, is how to assess pharmacodynamic endpoints that will reflect the variation in efficacy among individual patients. It has been widely assumed that accelerated cell cycle progression resulting from increased Cdc25A activity is pivotal in mediating the cytotoxic consequences of Chk1 inhibition, and that the molecular markers for these events should be useful correlates for therapeutic effectiveness. Perez et al observed G2/M checkpoint abrogation in tumor biopsy specimens from patients treated with a Chk1 inhibitor (UCN-01) in combination with cis-platinum, demonstrating this effect in tumor tissue for the first time in a clinical trial [35].
Our results indicate, however, that Cdc25A stabilization and S/M or G2/M checkpoint abrogation are not necessarily reliable correlates of the chemosensitizing effects of a Chk1 inhibitor. For example, PD-321852 did not cause premature mitotic entry in either Gem-treated BxPC3 or M-Panc96 cells, both of which were chemosensitized to Gem by this compound. This result was unexpected, as we and others have previously found sensitization to Gem by Chk1 inhibition with either siRNA [17] or the checkpoint inhibitor EXEL-9844, which inhibits both Chk1 and Chk2 [8], was associated with premature mitotic entry. Only in MiaPaCa2 cells did our results fit the prevailing model: inhibition of Chk1 led to abrogation of Gem-induced Cdc25A degradation, premature mitotic entry and sensitization to Gem. While these results do not mean that Cdc25A upregulation and S/M or G2/M checkpoint abrogation are not important players in mediating the sensitizing effects of Chk1 inhibitors, they indicate that endpoints reflecting other molecular events need to be considered in order to assess the efficacy of treatment with a Chk1 antagonist.
While catalytic inhibition of Chk1, as indicated by Cdc25A stabilization, was not strictly associated with Gem chemosensitization, depletion of Chk1 protein occurred in each of the sensitized cell lines after drug treatment. This result is consistent with our previous siRNA studies, where we found depletion of Chk1 with siRNA resulted in modest chemosensitization of Panc1 cells [17]. Zhang et al. have suggested that, in response to replication stress, the proteasome-mediated degradation of activated Chk1 functions to terminate this cell cycle checkpoint, and that the loss of Chk1 protein in chronically-stressed tumor cells may be a determinant of sensitivity to topoisomerase inhibitors such as camptothecin [26]. Our data suggest there may be an added benefit, in terms of chemosensitization, with stimulation of the Chk1 degradation pathway, in addition to catalytic inhibition of Chk1 activity.
Although our original intent was to use γ-H2AX as a marker of Gem-induced DNA damage and repair, it has become increasing clear that γ-H2AX also accumulates in response to drug-induced stalled replication forks in S-phase cells [18,29], and, in our model, most likely represents an inability to recover from Gem-induced replication stress. This interpretation is consistent with the recovery from Gem-induced S-phase arrest and accompanied loss of γ-H2AX we observed in BxPC3 cells, the persistence of γ-H2AX in Gem-treated Panc1 cells, which remained S-phase arrested 72 h after removal of drug, and the ability of PD-321852 to inhibit Rad51 focus formation, recovery of replication stress and loss of γ-H2AX in BxPC3 cells. Another possible interpretation of our results is that the high level of γ-H2AX induction in Panc1 cells, at relative non-toxic drug concentrations, reflects a more robust response to Gem-induced replication stress that is actually protective, in terms of drug-induced loss of clonogenicity.
One limitation of the current work is that, while PD-321852 is reasonably selective for Chk1 [20], it may inhibit other kinases as well, and these activities may contribute to the chemosensitization patterns observed. Furthermore, each of the three Chk1 inhibitors that are now in clinical trials has a distinctive selectivity profile (for example, two of the three also inhibit Chk2), and results obtained with PD-321852, or any particular Chk1 inhibitor, may not necessarily extrapolate to others.
Finally, while the four cell lines used in this study clearly represent some extent of the biological diversity of human pancreatic cancer, it is unclear whether any of the phenotypes observed are more representative of a “typical” tumor than the others. A limitation shared by all in vitro studies is that established cell lines are frequently seen to have diverged substantially from the tumors from which they were derived. It would therefore be useful to interrogate the endpoints used in this study with a model system that is closer to primary tumors, such as early passage xenografts [36].
The results of our studies suggest that although stabilization of Cdc25A and S/M or G2/M checkpoint abrogation may be good surrogate markers for validating catalytic inhibition of Chk1, abrogation of drug-induced Rad51 focus formation and depletion of Rad51 protein, accompanied by inhibitor-induced persistence of γ-H2AX, may be more reliable markers of the chemosensitizing effects of a Chk1 inhibitor, in pancreatic cancer cells.
Supplementary Material
Acknowledgments
The authors would like to thanks Drs. Mary A. Davis and Sheryl A. Flanagan for their critical reviews of this manuscript.
Abbreviations used
- ATR
ATM-Rad3-related kinase
- Chk1
checkpoint kinase 1
- Chk2
checkpoint kinase 2
- DAPI
4,6 diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- γ-H2AX
Ser139-phosphorylated histone H2AX variant
- Gem
gemcitabine
- HRR
homologous recombination repair
- LLnL
N-Acetyl-l-leucinyl-l-leucinyl-l-norleucinol
- PBS
phosphate buffered saline
- SF
surviving fraction
Footnotes
Supported by 1 R01-CA078554 and the University of Michigan Medical School Endowment for the Basic Sciences.
LA Parsels, JP Parsels, RJ Booth, WA Denny, AJ Kraker, J Maybaum. The small-molecule chk1 inhibitor, PD-321852, causes synergistic depletion of chk1 protein and clonogenic death when combined with gemcitabine in colorectal and pancreatic cancer tumor cells. AACR Meeting Abstracts 2006, Abstract #4911, unpublished.
We previously described a similar phenotype in HT29 cells, where, despite activation of both Chk1 and Chk2, Cdc25A was induced, rather than degraded, after treatment with the antimetabolite 5-fluorodeoxyuridine [7]. HT29 cells, like M-Panc96 cells, express relatively low levels of Cdc25A (LA Parsels, J Maybaum, unpublished results).
References
- 1.Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther. 2004;3:513–9. [PubMed] [Google Scholar]
- 2.Eastman A. Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem. 2004;91:223–31. doi: 10.1002/jcb.10699. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz GK. Development of cell cycle active drugs for the treatment of gastrointestinal cancers: a new approach to cancer therapy. J Clin Oncol. 2005;23:4499–4508. doi: 10.1200/JCO.2005.18.341. [DOI] [PubMed] [Google Scholar]
- 4.Tao ZF, Lin NH. Chk1 inhibitors for novel cancer treatment. Anticancer Agents Med Chem. 2006;6:377–88. doi: 10.2174/187152006777698132. [DOI] [PubMed] [Google Scholar]
- 5.Sorensen CS, Syljuasen RG, Falck J, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–58. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 6.Xiao Z, Chen Z, Gunasekera AH, et al. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278:21767–73. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- 7.Parsels LA, Parsels JD, Tai DC, Coughlin DJ, Maybaum J. 5-fluoro-2′-deoxyuridine-induced cdc25A accumulation correlates with premature mitotic entry and clonogenic death in human colon cancer cells. Cancer Res. 2004;64:6588–94. doi: 10.1158/0008-5472.CAN-03-3040. [DOI] [PubMed] [Google Scholar]
- 8.Matthews DJ, Yakes FM, Chen J, et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle. 2007;6:104–10. doi: 10.4161/cc.6.1.3699. [DOI] [PubMed] [Google Scholar]
- 9.Karnitz LM, Flatten KS, Wagner JM, et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68:1636–44. doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- 10.Morgan MA, Parsels LA, Parsels JD, Mesiwala AK, Maybaum J, Lawrence TS. Role of checkpoint kinase 1 in preventing premature mitosis in response to gemcitabine. Cancer Res. 2005;65:6835–42. doi: 10.1158/0008-5472.CAN-04-2246. [DOI] [PubMed] [Google Scholar]
- 11.Busino L, Chiesa M, Draetta GF, Donzelli M. Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene. 2004;23:2050–56. doi: 10.1038/sj.onc.1207394. [DOI] [PubMed] [Google Scholar]
- 12.Chen MS, Ryan CE, Piwnica-Worms H. Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14-3-3 binding. Mol Cell Biol. 2003;23:7488–97. doi: 10.1128/MCB.23.21.7488-7497.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci U S A. 2002;99:14795–800. doi: 10.1073/pnas.182557299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampath D, Shi Z, Plunkett W. Inhibition of cyclin-dependent kinase 2 by the Chk1-Cdc25A pathway during the S-phase checkpoint activated by fludarabine: dysregulation by 7-hydroxystaurosporine. Mol Pharmacol. 2002;62:680–8. doi: 10.1124/mol.62.3.680. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Xiao Z, Gu WZ, et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int J Cancer. 2006;119:2784–94. doi: 10.1002/ijc.22198. [DOI] [PubMed] [Google Scholar]
- 16.Zachos G, Rainey MD, Gillespie DA. Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol Cell Biol. 2005;25:563–74. doi: 10.1128/MCB.25.2.563-574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan MA, Parsels LA, Parsels JD, Lawrence TS, Maybaum J. The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle. 2006;5:1983–8. doi: 10.4161/cc.5.17.3184. [DOI] [PubMed] [Google Scholar]
- 18.Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007;6:1239–48. doi: 10.1158/1535-7163.MCT-06-0633. [DOI] [PubMed] [Google Scholar]
- 19.Sorensen CS, Hansen LT, Dziegielewski J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 20.Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 2008;27:3977–85. doi: 10.1038/onc.2008.17. [DOI] [PubMed] [Google Scholar]
- 21.Palmer BD, Boushelle S, Bridges AJ, et al. Pyrrolo[3,4-c]carbazole-1,3-dione inhibitors of the G2/M checkpoint kinase Wee1. European Journal of Cancer. 2004:20. [Google Scholar]
- 22.Lawrence TS, Davis MA, Maybaum J, et al. The potential superiority of bromodeoxyuridine to iododeoxyuridine as a radiation sensitizer in the treatment of colorectal cancer. Cancer Res. 1992;52:3698–704. [PubMed] [Google Scholar]
- 23.Huang X, Kurose A, Tanaka T, Traganos F, Dai W, Darzynkiewicz Z. Sequential phosphorylation of Ser-10 on histone H3 and ser-139 on histone H2AX and ATM activation during premature chromosome condensation: relationship to cell-cycle phase and apoptosis. Cytometry A. 2006;69:222–9. doi: 10.1002/cyto.a.20257. [DOI] [PubMed] [Google Scholar]
- 24.Motulsky HJ. Prism 4 Statistics Guide - Statistical analysis for laboratory and clinical researchers. San Diego: GraphPad Software Inc; 2003. pp. 61–5. [Google Scholar]
- 25.Syljuasen RG, Sorensen CS, Hansen LT, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–62. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang YW, Otterness DM, Chiang GG, et al. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–18. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 27.Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001;21:3445–50. doi: 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 29.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 30.Wachters FM, van Putten JW, Maring JG, Zdzienicka MZ, Groen HJ, Kampinga HH. Selective targeting of homologous DNA recombination repair by gemcitabine. Int J Radiat Oncol Biol Phys. 2003;57:553–62. doi: 10.1016/s0360-3016(03)00503-0. [DOI] [PubMed] [Google Scholar]
- 31.Tarsounas M, Davies D, West SC. BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene. 2003;22:1115–23. doi: 10.1038/sj.onc.1206263. [DOI] [PubMed] [Google Scholar]
- 32.Maacke H, Jost K, Opitz S, et al. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19:2791–95. doi: 10.1038/sj.onc.1203578. [DOI] [PubMed] [Google Scholar]
- 33.Yao Q, Weigel B, Kersey J. Synergism between etoposide and 17-AAG in leukemia cells: critical roles for Hsp90, FLT3, topoisomerase II, Chk1, and Rad51. Clin Cancer Res. 2007;13:1591–1600. doi: 10.1158/1078-0432.CCR-06-1750. [DOI] [PubMed] [Google Scholar]
- 34.Janetka JW, Ashwell S, Zabludoff S, Lyne P. Inhibitors of checkpoint kinases: from discovery to the clinic. Curr Opin Drug Discov Devel. 2007;10:473–86. [PubMed] [Google Scholar]
- 35.Perez RP, Lewis LD, Beelen AP, et al. Modulation of cell cycle progression in human tumors: a pharmacokinetic and tumor molecular pharmacodynamic study of cisplatin plus the Chk1 inhibitor UCN-01 (NSC 638850) Clin Cancer Res. 2006;12:7079–85. doi: 10.1158/1078-0432.CCR-06-0197. [DOI] [PubMed] [Google Scholar]
- 36.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–37. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.