

Abstract
Quantum dots (Qdots) are now used extensively for labeling in biomedical research, and this use is predicted to grow because of their many advantages over alternative labeling methods. Uncoated Qdots made of core/shell CdSe/ZnS are toxic to cells because of the release of Cd2+ ions into the cellular environment. This problem has been partially overcome by coating Qdots with polymers, poly(ethylene glycol) (PEG), or other inert molecules. The most promising coating to date, for reducing toxicity, appears to be PEG. When PEG-coated silanized Qdots (PEG-silane-Qdots) are used to treat cells, toxicity is not observed, even at dosages above 10–20 nM, a concentration inducing death when cells are treated with polymer or mercaptoacid coated Qdots. Because of the importance of Qdots in current and future biomedical and clinical applications, we believe it is essential to more completely understand and verify this negative global response from cells treated with PEG-silane-Qdots. Consequently, we examined the molecular and cellular response of cells treated with two different dosages of PEG-silane-Qdots. Human fibroblasts were exposed to 8 and 80 nM of these Qdots, and both phenotypic as well as whole genome expression measurements were made. PEG-silane-Qdots did not induce any statistically significant cell cycle changes and minimal apoptosis/necrosis in lung fibroblasts (IMR-90) as measured by high content image analysis, regardless of the treatment dosage. A slight increase in apoptosis/necrosis was observed in treated human skin fibroblasts (HSF-42) at both the low and the high dosages. We performed genome-wide expression array analysis of HSF-42 exposed to doses 8 and 80 nM to link the global cell response to a molecular and genetic phenotype. We used a gene array containing ~22,000 total probe sets, containing 18,400 probe sets from known genes. Only ~50 genes (~0.2% of all the genes tested) exhibited a statistically significant change in expression level of greater than 2-fold. Genes activated in treated cells included those involved in carbohydrate binding, intracellular vesicle formation, and cellular response to stress. Conversely, PEG-silane-Qdots induce a down-regulation of genes involved in controlling the M-phase progression of mitosis, spindle formation, and cytokinesis. Promoter analysis of these results reveals that expression changes may be attributed to the down-regulation of FOXM and BHLB2 transcription factors. Remarkably, PEG-silane-Qdots, unlike carbon nanotubes, do not activate genes indicative of a strong immune and inflammatory response or heavy-metal-related toxicity. The experimental evidence shows that CdSe/ZnS Qdots, if appropriately protected, induce negligible toxicity to the model cell system studied here, even when exposed to high dosages. This study indicates that PEG-coated silanized Qdots pose minimal impact to cells and are a very promising alternative to uncoated Qdots.
Introduction
Toxicity of nanomaterials is a major healthcare concern that may impact the nanotechnology industry.1–3 Concern has been rising following studies on the toxicity of carbon nanophase materials, some of which are found in flames, welding fumes, diesel exhausts, and other petrol byproducts.4–7 There is evidence for the contribution of many factors to the toxicity of these organic nanostructures including their size, shape, and surface functionalization. Assuming an equivalent mass of carbon, cytotoxicity grows in the following order: fullerene (C60) < multiwall carbon nanotube (MWCNT) < single-wall carbon nanotube (SWCNT).8 For example, C60, with a well-defined surface and no available dangling bonds, is harmful to cells even at low doses.9–14 C60 is an excellent electron acceptor that can readily react with available oxygen and water to generate free radicals leading to oxidative damage of the cellular membrane. Derivatized fullerenes are less efficient in producing oxygen radicals,14 therefore C60 derivatized with hydroxyl groups is much less toxic. Less is known about the toxicity of fluorescent semiconductor quantum dots, or Qdots. Qdots are CdSe/ZnS core/shell nanocrystals15 and the heavy elements that make up the core may induce a more pronounced and acute cytotoxic response than carbon nanostructures. It has been reported that Cd2+ is released from CdSe through oxidative attack.16,17 This released cadmium can bind to the sulfohydryl groups of critical mitochondria proteins leading to mitochondria dysfunction and ultimately cell poisoning.18
Qdots are small fluorescent tags that have tremendous potential for advancing knowledge in biology because of their unique characteristics.15 Because of their large extinction coefficient, they can be excited at much lower power than organic dyes, in a range of energies not absorbed by the cells. They also exhibit intense light emission with negligible photobleaching over minutes or hours. This offers a tremendous advantage over organic dyes and engineered fluorescent proteins that photobleach in seconds when they are used to label single molecules in living cells. Photobleaching causes the formation of reactive oxygen radicals and further triggers a cascade of chemical reactions resulting in the poisoning and death of cells. Therefore, the detrimental effects of radiation exposure are minimized for Qdot-labeled cells. These properties may allow the observation of long-lasting chemical or biological processes within or around the cell, which includes information on cell communication.19,20 For example, such long-lasting probes would allow the multiplexed tracking of signaling biomolecular events in live cells for hours or provide a method to encode particular cells with colored tags to study cell–cell interactions from days to months (C. Larabell, private communication). Because of the tenability, stability, and brightness of Qdots, several studies have explored in vivo labeling in live animals for optical imaging of cancer providing greater insight into tumorigenesis.21,22 The use of these molecules for labeling has provided methodology for in vivo experiments that previously had been impossible.
Because of the potential for quantum dots to provide additional information about biological process in vivo, it is important to understand the toxicology of these foreign nanomaterials. Studies so far have been limited to determining the survival rate of cells exposed to Qdots for less than 48 h.23–26 These studies have demonstrated that the surface functionalization plays the key role in nanoparticle toxicity.24–26 For example, CdSe/ZnS solubilized by a simple ligand exchange with a mercaptoacid, are less soluble and are toxic to breast cancer cells above a threshold concentration in the nanomoles per liter range.26 This is caused by the release of Cd2+ ions into solution because of the weak and dynamic bond between the Qdot surface and the mercapto surfactant. In contrast, Cd2+ release is noticeably slowed if Qdots are embedded in a cross-linked shell reducing toxicity. For example, cells treated with CdSe/ZnS nanoparticles embedded in a silica shell do not show signs of toxicity, even when treated with dosages 6–12 times higher than the toxicity-inducing dosage of mercaptoacid coated CdSe/ZnS Qdots.26 These experiments have helped to illuminate some of the reasons for the toxic effects of Qdots to live organisms and have provided guidance on how to modify the Qdots to negate these concerns.
One question that remains to be answered is how cells respond at the molecular level after treatment with nano-materials below the dosage causing high percentage cell death. Even minute changes may have profound effects on the integrity and viability of the cells over multiple cellular divisions. To address this question we examined the impact of the treatment of both human lung and skin epithelial cells to two dosages of poly(ethylene glycol) silanized quantum dots (PEG-silane-Qdots). Two dosages were selected, one reported to be nontoxic to breast cancer cells and a 10-fold higher dosage. Human skin (HSF-42) and lung fibroblasts (IMR-90) were selected because skin and the respiratory track are the most likely route of human exposure. Furthermore, molecular and genetic data addressing the cytotoxicity of carbon nanostructures for these cells lines is available.27 Presented here are measurements of phenotypic changes in large populations of cells combined with expression array analysis of exposed cells. Our results indicate that both high and low doses of PEG-silane-Qdots present a similar average response from the cells. We do not see an adverse effect in lung epithelial cells, while in the case of skin epithelial cells, PEG-silane-Qdot treatment exerts a slight repression of genes regulating cell cycle progression. In general though, only <50 genes (equivalent to ~0.2% of total genes) out of more than 22,000 probed show significant changes in the expression level due to the presence of the PEG-silane-Qdots. Detailed analysis allows the classification of these genes into functional categories, and promoter analysis reveals affected regulatory pathways. As expected, we observe minor involvement of cell endocytosis and intracellular transport pathways. Remarkably, the global picture emerging from our study is that PEG-silica-Qdots have a negligible toxicological effect on these two cell lines. This study is a critical first step to characterize the toxicity of coated Qdots at the molecular level in an in vitro culture system.
Materials and Methods
A detailed description of the experimental procedures and of the materials used can be found in the Supporting Information. Here we describe only some salient features of the experimental protocol. We used silica-coated CdSe/ZnS Qdots terminated with both thiol and PEG functional groups.28 While the core/shell Qdots are only ~4–5 nm in size, the silane shell adds ~2–3 nm in thickness and thus silanized Qdots are ~8–10 nm in diameter.29 Such Qdot chemistry was observed to pose minimal toxicity to breast cancer cells when the cells were exposed to a solution containing 2–10 nM of PEG-silane-Qdots.26 Human lung (IMR-90) and skin epithelial (HSF-42) cells were exposed for 48 h to a medium containing 8 or 80 nM Qdots or to an equivalent amount of 10 mM phosphate buffer as a control.
Phenotypical measurements of cell proliferation, apoptosis, necrosis, and cell cycle distribution were performed using a high content image analyzer (HCA, Cellomics KineticScan HCS Reader). Cells were detected and counted by staining their nucleus with Hoechst dye. Further distinction between apoptotic and necrotic cells was done using DNA dyes that transverse the membrane of apoptotic and necrotic cells, respectively. For instance YO-PRO-1, a green dye, can cross the slightly impermeable membranes of apoptotic cells while propidium iodide (PI), a red dye, crosses the membrane of necrotic cells due to their greater permeability. Cell cycle distribution was performed by adding bromo-deoxyuridine (BrdUrd) to the cell medium and subsequently staining the cells using anti-BrdUrd antibody labeled with AlexaFluor 488 and PI to obtain DNA content information. After images from stained culture plates were obtained, intensity measurements for both BrdUrd and DNA staining were made for each identified cell to generate a scatter plot with BrdUrd intensity on the Y-axis and PI intensity on the X-axis. Analysis of these scatter plots allows estimation of the percentages of these cells in G0/G1, S, and G2/M phases.
Gene expression profiling was obtained with an Affymetrix high throughput analysis automated Genechip system. Target preparation, washing, and staining were carried out on a Affymetrix/Caliper robotic system, and scanning was performed on a CCD-based high throughput scanner. The chip contains ~22,000 probe set, among which 18,400 are known genes or probe sets. Data analysis has been performed using Genesping, Bioconductor, GeneTraffic, Cluster 3.0, PAINT, GoMiner, and Pathway Assist. More details and the software and the protocol of analysis can be found in the Supporting Information.
Results
Cellomics
PEG-coated silanized Qdots (Qdots), schematically represented in Figure 1A, were added to the cell culture medium. Human skin fibroblasts and lung fibroblasts were selected as model systems because entry of nanomaterials through the skin and respiratory track is the most likely route of human exposure to nanomaterials. In addition, genotoxicity data of carbon nanotubes and nano-onions are available for these cells and can be used for comparison purposes.27 Human skin fibroblast and lung fibroblast cells exposed to 8 or 80 nM PEG-silane-Qdots for 48 h internalize them (Figure 1B). As shown in Figure 1B, all cells are labeled by Qdots. The entry mechanism is likely endocytosis, as observed previously by Jaiswal et al. for Hela and D. discoideum cells.30,31 The nanoparticles are stored in the perinuclear region, as most studies report,23,26 but we also observed PEG-silane-Qdots dispersed in the cytoplasm (Figure 1B). A careful look at Figure 1 and comparable images indicate a slightly elevated number of labeled cells are in the cytokinesis stage of mitotic cell cycle. This warrants further quantitative analysis of the cell cycle profile.
Figure 1.

(A) Schematics of the PEG-silica embedded Qdots. The silica shell is functionalized with –SH groups and with PEG groups. The latter provide additional stability and reduced nonspecific bindings. The scale bar (~3 nm) provides a qualitative comparison between the overall size of the silanized dot (~8 nm) and the size of the semiconductor core (~3 nm). (B) Qdots localization in HSF42 cells, after 48 h of incubation. The nuclei are stained with DAPI, a blue dye. Yellow Qdots are localized either in the cytoplasm or in the perinuclear region. Notice that about half of the cells in the image are in the postmitotic stage.
Cell Proliferation
Forty-eight hours after transfection, proliferation of cells labeled by PEG-silane-Qdots was evaluated through an automated counting method. Cells were labeled with Hoechst dye and counted with the Cellomics KineticScan HCS Reader (KSR). Figure 2.I shows the average results from 10 independent runs, with error bars representing the standard deviation. For both cell lines after 48 h, statistically significant differences in the number of cells, exposed to either PEG-silane-Qdots or the unexposed control, are not observed regardless of the PEG-silane-Qdot dosage used for treatment. This indicates a neutral effect of treatment both on the cell proliferation rate and on the cell death rate over a period of 48 h, i.e., encompassing about two cellular division cycles.
Figure 2.

(I) Cell counts for IMR-90 and HSF-42 cells after treatment with silanized Qdots in various doses. When treated with PEG-silane-Qdots, the survival rate of both cell lines is mostly unaffected. The statistically insignificant reduction in the cell number may be explained by a mild block of the G2/M phase (see Figure 2III). This contrasts with the marked effect that organic nanostructures (carbon nanotube and nano-onions) have on IMR-90 and HSF42 cells.27 (II.a) There’s no statistically significant change in apoptotic/necrotic profile for PEG-silane-Qdot-treated IMR-90 cells, with either high or low dosage of Qdots. PEG-silanized Qdots appear much less detrimental than organic nano-structures. (II.b) Treating HSF42 cells with Qdots causes a slight increase in apoptotic/necrotic cells for both dosages. (III) The distribution of PEG-silane-Qdot treated HSF42 cells in different phases of cell cycle. The baseline of 1 is equivalent to the control distribution. There are slightly fewer PEG-silane-Qdot treated cells in the S phase and slightly more (~10% more) in the M phase than control cells.
Apoptosis/Necrosis
Quantifying apoptotic or necrotic cells generated further information on cell cytotoxicity. Live cells are impermeable to YO-PRO 1 and PI, two DNA staining dyes, but apoptotic cells are permeable to YO-PRO 1 (a green dye), and necrotic cells are permeable to PI (a red dye). Thus, we could count and differentiate cells undergoing apoptosis or necrosis. The results of large scale analysis over more than 20,000 cells, replicated 10 times, are reported in Figure 2II as the percentage of all cells exhibiting apoptosis or necrosis. Exposing human lung fibroblasts to either high or low dosages of PEG-silane-Qdots does not significantly increase the percentage of cells in an apoptotic or necrotic state compared to the control (~1.8–2% vs ~1.2–1.5%, Figure 2II.A). In contrast, a slight increase in both apoptosis and necrosis, from ~1–1.2% to ~2.7–2.8% is observed in human skin fibroblasts (HSF-42, Figure 2II.B). The increase is very modest and is independent of the dosage of PEG-silane-Qdots in the medium.
Cell Cycle Profile
Because lung fibroblasts IMR-90 do not show marked signs of cytotoxicity, we focused on the response of skin fibroblasts HSF-42 to the presence of PEG-silane-Qdots in all subsequent analyses. First, we studied the proliferation profile of this cell line by incorporating BrdUrd into replicating DNA and counterstaining with PI to determine total DNA content.27 For each individual HSF-42 cell, the ratio of the signal intensity from antibody staining of incorporated BrdUrd versus total DNA content measured by PI staining is plotted in a scatter plot. We analyzed the cell cycle status of more than 20,000 cells and then classified them into G0/G1, S, or G2/M phases. Figure 2.III shows the relative percentage of treated cells compared to control cells in each of the three phases of the cell cycle. The ratio of PEG-silane-Qdot treated cells to control cells in G0/G1 is close to 1, indicating that PEG-silane-Qdot treatment does not induce a block in G1. Similarly, the ratio of cells in the S-phase of treated to control is ~0.94, with a Student t-test demonstrating only borderline statistical significance. The largest difference in ratio occurs at the G2/M phase, where the ratio of cells treated with PEG-silane-Qdot vs control is ~1.1, possibly indicating a block in G2/M. However, because only two cell divisions have occurred in 24 h, this ratio suggests either no significant G2/M block or that it may only become apparent after multiples cell division cycles. An important observation based on these data is that the effect of PEG-silane-Qdots on the cell proliferation, cell death, and cell cycle regulation is much more subtle than the marked cytotoxic effects induced by treatment with carbon-based nanostructures, i.e. nanotubes and nanoonions, in these cell lines.27 These observations are also consistent with gene expression results presented below.
Gene Expression
The Affymetrix high throughput array (HTA) GeneChip system was used to profile gene expression changes in human skin fibroblasts labeled with PEG-silane-Qdots. The results are plotted in a two-dimensional diagram in Figure 3 where each gene is represented by an (X,Y) value in a log scale. The Affymetrix HG-U133Av2.0AofA Gene-Chip contains 25 mer oligoprobes, in sets for identification of transcripts from ~22,000 genes and ESTs in the human genome. Each dot on the graph represents a gene where the X-value corresponds to the level of expression in control cells, while the Y-value corresponds to the level of expression of that same gene in the PEG-silane-Qdots labeled cells. A dot that lands on the graph where the slope is 1 (red line) indicates no difference between the gene expression level of the treated and control samples. The two dotted lines flanking the central line indicate the cutoff for 2-fold up-regulation (top line) or down-regulation (bottom line) of the sample vs the control. Dots above or below the 2-fold box lines represent genes with a greater than 2-fold change in gene expression and are discussed below.
Figure 3.

(A) Scatter plots for the two doses of PEG-silane-Qdot treatment, in a log10 scale. The Y axis represents treated cells; the X axis represents the control. The line X = Y correspond to no difference in gene expression between the treated and control sample. The dashed lines correspond to changes of level of expression by a factor of 2. The tightness of the plot indicates that most of the genes do not change significantly after PEG-silane-Qdots treatment. (B) The functional categories of the genes affected by low and high doses of PEG-silane-Qdots. All functional categories affected by high doses are also affected by a low dose treatment. A significant portion of the down-regulated genes are related to the M phase of the mitotic cell cycle, especially the spindle assembly and cytokinesis. The up-regulated genes include those for carbohydrate binding proteins (possibly in recognition of the PEG coating of Qdots), intracellular organelle (especially vacuole and intracelluar vesicle) related proteins (possibly involved in intracellular transport of Qdots), and stress-response genes (possibly due to the slight stress induced by treatment).
In Figure 3A it is remarkably clear that most of the dots lie close to where the slope = 1, with minimal dispersion up or down, indicating minimal changes in the gene expression in the PEG-silane-Qdot treated cells compared to untreated. This is in strong contrast to treatment of these same cells with carbon nanotubes and nano-onions.27 For instance, the MWCNT at a concentration of 0.6 mg/L induced significant changes in 216 genes, while the PEG-silane-Qdot induced changes in 20 times fewer genes at a much higher concentration of 40 mg/L (80 nM, with molecular weight approximately 500 KDa). Because there is little to no toxicity for these PEG-silane-Qdots compared to other types of nanoparticles, this provides evidence that the composition and surface functionalization of the particle are the most important determinants of toxicity.
Thirty-eight genes were identified as being differentially expressed by more than 2-fold in the cells treated with a low dosage of Qdots, while only 12 were identified for the higher dosage. Among these genes, 4 are shared both by low and high dosage experiments, representing approximately 20–30% of the genes analyzed. The combined number of genes demonstrating significant changes with these two treatments totals 46 genes, ~0.2% of the total number probed. We classified the genes into functional categories using the GoMiner program.32 Figure 3B lists the categories. The functional categories of the changed genes are consistent across the two different dosages. Genes overexpressed are mostly related to carbohydrate binding (CHI3L1, GPNMB, PRELP, TNXB), intracellular vesicle localization (CTSF, CTSH, GPNMB, PTGIS/CYP8A1), and cell-membrane-associated and intracellular vesicular proteins involved in cellular response to stress (CLU, MAP2K6/MKK6, FST). Interestingly, both MAP2K6 and CLU are both implicated in the inhibition of apoptosis33,34 and induction of senescence,35–38 while CLU is a sulfated glycoprotein on the cell surface.39 While there was some phenotypic evidence of some apoptosis, it was minimal. The four common genes between the low-dose-induced group and the high-dose-induced group are CORIN, BUB1, CHI3L1, and CLU, suggesting the interaction of PEG-silane-Qdots with cell surface binding proteins (CHI3L1 and CLU).
There are far more genes observed to be down-regulated in the treated cells as compared to upregulated genes. The majority of the down-regulated genes fall into the functional categories controlling the M-phase progression in mitosis, spindle formation, and cytokinesis (BUB1, CyclinA2/CCNA1, CDC20, KIF2A, KIF2C, NEK2, PLK1, PTTG, TACC3 for low dose and BUB1, MPHOSPH1 for high dose),40–52 indicating that these proteins might account for the limited perturbation of M-phase progression by PEG-silane-Qdots. In addition, the expression of the transcription factors FOXM1 and BHLHB2/Dec1 is also down-regulated in low-dose-treated cells. Interestingly, PEG-silane-Qdots treatment does not seem to illicit any genes involved in wound healing or the immune response, contrary to both responses we observed in human skin fibroblasts treated with carbon nanotubes27 and the response of dendritic cells to nanosphere treatment by others.53 The lack of induction of these genes may underscore the negligible toxic effects of PEG-silane-Qdot treatment in this cell line. This observation also counters a widely held preconception that Qdots are toxic to cells because of the presence of Cd in the nano-crystal.
One important discovery of this study was that genes associated with heavy metal exposure were not induced by PEG-silane-Qdot treatment. The gene expression changes revealed by gene expression profiling can be mostly attributed to the interaction between the cellular machinery to the PEG coating of the Qdot. The PEG silica coating is very robust under the biological conditions used in this study, greatly reducing or even eliminating the concern of Cd poisoning. Table 1 shows a comprehensive list of significantly changed genes.
Table 1.
Significantly Changed Genes after Treatment with PEG-silane-Qdotsa
| gene symbol | gene name | fold changes of gene expression in log2 |
|---|---|---|
| 8 nM Qdots | ||
| CHI3L1 | chitinase 3-like 1 (cartilage glycoprotein-39) | 4.79653 |
| CLU | clusterin (complement lysis inhibitor, SP-40,40, sulfated glycoprotein 2, testosterone-rep) | 3.23731 |
| IGFBP2 | insulin-like growth factor binding protein 2, 36 kDa | 2.87535 |
| PTGIS | prostaglandin I2 (prostacyclin) synthase///prostaglandin I2 (prostacyclin) synthase | 2.63813 |
| CCND2 | cyclin D2 | 2.03349 |
| PRELP | proline arginine-rich end leucine-rich repeat protein | 1.9751 |
| MAP2K6 | mitogen-activated protein kinase kinase | 1.975 |
| TNXB | tenascin XB | 1.93589 |
| FMOD | fibromodulin | 1.72529 |
| FST | follistatin | 1.70054 |
| HCA112 | hepatocellular carcinoma-associated antigen 112 | 1.69136 |
| CTSH | cathepsin H | 1.58153 |
| GPNMB | glycoprotein (transmembrane) nmb | 1.50888 |
| GLTSCR2 | glioma tumor suppressor candidate region gene 2 | 1.31639 |
| CTSF | cathepsin F | 1.23353 |
| PTTG1 | pituitary tumor-transforming 1 | −1.05541 |
| TBPIP | TBP-1 interacting protein | −1.08364 |
| DDA3 | differential display and activated by p53 | −1.15054 |
| TK1 | thymidine kinase 1, soluble | −1.22634 |
| POSTN | periostin, osteoblast specific factor | −1.30694 |
| ITGA6 | integrin, alpha 6 | −1.34346 |
| TACC3 | transforming, acidic coiled-coil containing protein 3 | −1.35367 |
| FOXM1 | forkhead box M1 | −1.36322 |
| HCAP-G | chromosome condensation protein G | −1.40545 |
| KIF2C | kinesin family member 2C | −1.48635 |
| NEK2 | NIMA (never in mitosis gene a)-related kinase 2///NIMA (never in mitosis gene a)-relate | −1.52081 |
| CDCA3 | cell division cycle associated 3///cell division cycle associated 3 | −1.5265 |
| KIF4A | kinesin family member 4A | −1.58766 |
| CDC20 | CDC20 cell division cycle 20 homologue (S. cerevisiae) | −1.59272 |
| CCNA2 | cyclin A2 | −1.60045 |
| PLK1 | polo-like kinase 1 (Drosophila) | −1.64545 |
| BM039 | uncharacterized bone marrow protein BM039 | −1.66054 |
| SLC4A4 | solute carrier family 4, sodium bicarbonate cotransporter, member 4 | −1.80529 |
| BUB1 | BUB1 budding uninhibited by benzimidazoles 1 homologue (yeast) | −1.81555 |
| SERPINB7 | serine (or cysteine) proteinase inhibitor, clade B (ovalbumin), member 7 | −1.8309 |
| BHLHB2 | basic helix-loop-helix domain containing, class B, 2 | −1.9595 |
| KISS1 | KiSS-1 metastasis-suppressor | −2.45044 |
| CORIN | corin, serine protease | −3.4274 |
| 80 nM Qdots | ||
| EGFL6 | EGF-like-domain, multiple 6 | 4.28111 |
| CHI3L1 | chitinase 3-like 1 (cartilage glycoprotein-39) | 4.04108 |
| CLU | clusterin (complement lysis inhibitor, SP-40,40, sulfated glycoprotein 2, testosterone-rep) | 3.26671 |
| RARRES3 | retinoic acid receptor responder (tazarotene induced) 3 | 2.38346 |
| HIST1H2BD | histone 1, H2bd | 1.73794 |
| MPHOSPH1 | M-phase phosphoprotein 1 | −1.21531 |
| SPHK1 | sphingosine kinase 1 | −1.23641 |
| KLF6 | Kruppel-like factor 6 | −1.62409 |
| FLNB | filamin B, beta (actin binding protein 278) | −1.64077 |
| KLF6 | Kruppel-like factor 6 | −1.6771 |
| BUB1 | BUB1 budding uninhibited by benzimidazoles 1 homologue (yeast) | −1.71227 |
| CORIN | corin, serine protease | −3.41574 |
The genes presented in the table are the ones with fold change more than 2, and P value less than 0.05.
Promoter Analysis
By identifying and analyzing the enriched cis-regulatory transcription regulatory elements (TRE) on the promoters of differentially expressed genes in this experiment (Figure 4), we were able to identify putative trans-regulating transcription factors. Data from HSF cells treated at high and low dosages of PEG-silane-Qdots were included. Promoter analysis of the predominantly down-regulated genes at the higher dosage of PEG-silane-Qdots suggests the enrichment of FOXO family transcription regulatory elements. Even though we did not see the under-expression of FOXM1 in the high-dose-treated cells in contrast to low-dose-treated cells, the over all transcriptional profile points to the down regulation of its activity. FOXM1 has been shown to activate the transcription of genes essential for mitotic progression.54
Figure 4.
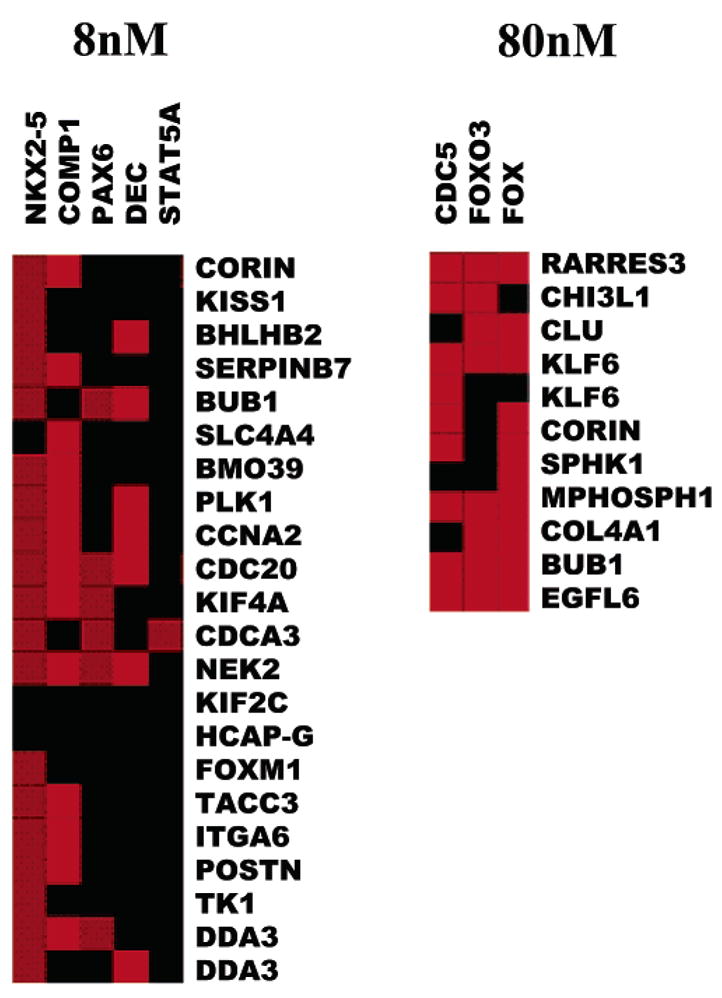
Analysis of transcription regulatory elements (TREs) in the promoters of the altered genes. The TREs for different transcription factors on the promoter regions of the altered genes are analyzed for over-/under-representation relative to all promoters in the PAINT database. The relationships of TREs and input genes are represented as an image of the interaction matrix: the columns of the interaction matrix correspond to the enriched TREs and each row corresponds to a gene from the input list. Individual elements of the matrix are colored by the significance of the p-values: over-representation in the matrix is indicated in red. There is an enrichment of FOX transcription binding elements on the high dose responsive genes. In low dose responsive genes that are down-regulated, there is enrichment of DEC/BHLHB2 and COMP1 (cooperates with myogenic protein 1).
The promoter analysis of the down-regulated genes at the lower dosage points to the enrichment of two transcriptional regulatory elements: DEC and COMP1. Genes under-expressed in response to low-dose PEG-silane-Qdot treatment include BHLHB2/DEC1/STRA13. This gene is involved in transcriptional repression, differentiation, hypoxia-induced stress response, and circadian clock regulation. It was recently proposed to have a role in differentiation by promoting cell cycle exit.55–58 There is not enough information about COMP1 to deduce its putative role in PEG-silane-Qdot response. The limited number of TREs identified by promoter analysis from the expression information from PEG-silane-Qdot treated cells contrasts with the large number of promoter regulatory pathways perturbed by other nano-materials or environmental factors,27,53,59 providing additional evidence for the minimal impact of PEG-silane-Qdots on cells.
Discussion
Because of the increasing use of Qdots in biomedical research, it has become extremely important to understand the impact and toxicity of Qdots on cells and ultimately living organisms. Data obtained from our studies predict that silanized CdSe/ZnS nanocrystals will have minimal, if any, impact on cellular functions. Even for the highest dosage we used, negligible phenotypic response of cells to PEG-silane-Qdots and minimal global gene expression changes were observed. In fact, concentrations of 80 nM of PEG-silane-Qdots (i.e., ~5 × 1010 particles/mm3) in lungs or skin fibroblast cells represent a dosage that would be extreme and unlikely in cases of an accidental inhalation or exposure to Qdots. Qdots solutions are typically stored in micromolar concentrations and if inhaled will be spontaneously diluted below toxic concentrations.
When lung or skin fibroblast cells are treated with PEG-coated silanized Qdots, the nanoparticles stay in vesicles in the perinuclear region or in the cytoplasm, as previously observed.23,30 In contrast to Qdots with a nuclear localization sequence on the surface, PEG-silane-Qdots are unable to cross the nuclear membrane,23 preventing their direct interaction with the genetic machinery in the cell nucleus. This precludes studies requiring the labeling of nuclear materials, creating a definite disadvantage.
Our data uncover a surprising observation that low or high dosages of Qdots during the incubation step do not induce a marked difference in the phenotypic response of cells. The higher dosage of Qdots during incubation does however result in a higher degree of particle uptake as measured by a stronger fluorescent signal. It is unclear, however, if the 10-fold increase of PEG-silane-Qdot used for the incubation period results in a 10-fold increase of particle uptake. Of importance, the high concentration of Qdots used in this study corresponds to an approximately 5-fold greater concentration than reported previously in toxicity studies using non-PEGalated Qdots.26 Despite this high concentration, skin HSF-42 and lung IMR-90 cells only show a mild phenotypic response to PEG-silane-Qdots, as measured by changes in cell proliferation, cell cycle regulation, and cell death. Whether the same conclusion will hold true for treatment times covering multiple (>10) cell division cycles remains an open question. Similarly, it may seem counterintuitive that lung fibroblast cells are less susceptible to PEG-silane-Qdot exposure than skin fibroblast cells. It is possible that the tissue-different gene expression pattern contributes to this effect. It is expected that organ-specific toxicological profiles will emerge if the Qdots are administered to whole organism, and there will be issues such as clearance, transport, retention, and degradation of the PEG coating. However, the cellular-level molecular and cellomic profiling is an important first step for understanding the nanotoxicology of Qdots, and the data here strongly indicate that in vitro cell imaging study can benefit from the nontoxicity of Qdots.
The stimuli induced by the presence of Qdots in human skin fibroblasts can be readout at the genetic level by monitoring gene expression changes in the cells. By applying significance analysis with Bonferroni multitesting correction, we found that only a minute number of genes exhibit statistically significant expression level changes. Out of more than 22,000 genes probed on the array, only ~50 (i.e., ~0.2%) show more than a 2-fold expression change. Such mild change contrasts sharply with the much larger number of genes affected when HSF-42 are exposed to carbon nanoparticles.27
A careful analysis of the genes affected by the dosage of Qdots reveals that 20–30% of genes affected at high dosage are also affected at low dosage. This may indicate a similar gene expression profile. In fact all functional categories of genes affected at high dosage are also affected at low dosage. The observed response in HSF-42 to Qdots seems to consist of several aspects: reduced expression of genes involved in M-phase exit, including spindle checkpoint and cytokinesis, and increased expression of genes involved in vesicle transport and apoptosis avoidance. From the promoter analysis, we identified FOXM1 and BHLHB2 as the transcription factors responsible for the reduced expression, with minor biological significance. One of the main concerns in using Qdots is the potential cytotoxicity generated by exposure to cadmium. In this study, we found no evidence for altered expression of any genes involved in cadmium (Cd2+) and Se toxicity during the treatment. This is strong evidence for a resilient silica shell that restrains the leakage of CdSe. The altered expression of a few cytoskeletal proteins suggests that Qdots may interact with the intracellular trafficking system during endocytosis and intracellular movement. This is a common mechanism used by intracellular labels that enter the cell through endocytosis. Of importance is that PEG-silane-Qdots do not significantly impact cellular functions through these possible interactions. Both the high content imaging analysis (Cellomics) and high throughput gene expression profiling showed a consistent result for the PEG-silane-Qdots.
In conclusion, results from both high content cellomics analysis and comprehensive analysis of expression over the entire genome of cells treated with PEG-silica-coated Qdots indicate minimal impact on cell health and molecular response of exposed cells. This provides evidence that proper coating and passivation of Qdots allows their safe use for in vivo applications. This contradicts the commonly held belief that CdSe nanocrystals are poisonous due to Cd leakage and may have widespread implications on the use of these particles in biomedical studies in living cells and organisms. Qdots are much less toxic than carbon nanoparticles when used to treat skin fibroblasts, as observed by us27 and others.53 These studies can now be extended to determine if there are any long term effects of Qdots on skin or lung cells and finally extended to animal studies. Using cells has provided us the opportunity to very carefully control our experimental conditions to obtain valid comparisons between treatment and control cells. These types of initial studies are required before moving to more complex biological systems, such as more detailed studies in small animals and, eventually, preclinical and clinical tests to provide a baseline for further studies. On the other hand, longer-term fate studies in a living system, including degradation and clearance, are necessary before full clinical usage of Qdots becomes a reality. In addition, previous studies have been done using these types of model systems, allowing us to directly compare our results to results from these studies.27 These results demonstrate that the surface chemistry of Qdots is very important for determining toxicity and further open the field for long-term labeling of live cells and in vivo clinical imaging applications. This should provide guidance for any future improvements upon surface chemistry to reduce or eliminate the toxicity of other nanomaterials as well.
Acknowledgments
We thank Ms. Lonnette Robinson for administrative support. We thank Professors Marc Shuman, Luke P. Lee, and Song Li for support. F. Chen was supported by NIH Grant R1CA95393-01, DOD, DARPA, UCSF Prostate Cancer SPORE award (NIH Grant P50 CA89520), by DOE LDRD grant to A. P. Alivisatos/J. W. Gray, and by NIH P50 Grant CA112970 to J. W. Gray. This work was performed under the auspices of the U.S. Department of Energy, at the University of California/Lawrence Berkeley National Laboratory under Contract No. DE-AC03-76SF00098, and at the University of California/Lawrence Livermore National Laboratory under Contract No. W-7405-Eng-48.
Footnotes
Supporting Information Available: A detailed description of the experimental procedures and of the materials used. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Colvin VL. Scientist. 2004;18:26–27. [Google Scholar]
- 2.Colvin VL. Nat Biotechnol. 2004;22:760–760. [Google Scholar]
- 3.Service RF. Science. 2004;304:1732–1734. doi: 10.1126/science.304.5678.1732. [DOI] [PubMed] [Google Scholar]
- 4.Maynard AD, Baron PA, Foley M, Shvedova AA, Kisin ER, Castranova V. J Toxicol Environ Health, Part A. 2004;67:87–107. doi: 10.1080/15287390490253688. [DOI] [PubMed] [Google Scholar]
- 5.Silva VM, Corson N, Elder A, Oberdorster G. Toxicol Sci. 2005 doi: 10.1093/toxsci/kfi142. [DOI] [PubMed] [Google Scholar]
- 6.Frampton MW, Utell MJ, Zareba W, Oberdorster G, Cox C, Huang LS, Morrow PE, Lee FE, Chalupa D, Frasier LM, Speers DM, Stewart J. Res Rep—Health Eff Inst. 2004:1–47. discussion 49–63. [PubMed] [Google Scholar]
- 7.Block ML, Wu X, Pei Z, Li G, Wang T, Qin L, Wilson B, Yang J, Hong JS, Veronesi B. FASEB J. 2004;18:1618–1620. doi: 10.1096/fj.04-1945fje. [DOI] [PubMed] [Google Scholar]
- 8.Jia G, Wang H, Yan L, Wang X, Pei R, Yan T, Zhao Y, Guo X. 2005;39:1378–1383. doi: 10.1021/es048729l. [DOI] [PubMed] [Google Scholar]
- 9.Ali SS, Hardt JI, Quick KL, Kim-Han JS, Erlanger BF, Huang TT, Epstein CJ, Dugan LL. Free Radicals Biol Med. 2004;37:1191–1202. doi: 10.1016/j.freeradbiomed.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Burlaka AP, Sidorik YP, Prylutska SV, Matyshevska OP, Golub OA, Prylutskyy YI, Scharff P. Exp Oncol. 2004;26:326–327. [PubMed] [Google Scholar]
- 11.Nelson MA, Domann FE, Bowden GT, Hooser SB, Fernando Q, Carter DE. Toxicol Ind Health. 1993;9:623–630. doi: 10.1177/074823379300900405. [DOI] [PubMed] [Google Scholar]
- 12.Oberdorster E. Environ Health Perspect. 2004;112:1058–1062. doi: 10.1289/ehp.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rancan F, Rosan S, Boehm F, Cantrell A, Brellreich M, Schoenberger H, Hirsch A, Moussa FJ. Photochem Photobiol, B. 2002;67:157–162. doi: 10.1016/s1011-1344(02)00320-2. [DOI] [PubMed] [Google Scholar]
- 14.Sayes CM, Fortner JD, Guo W, Lyon D, Boyd AM, Ausman KD, Tao YJ, Sitharaman B, Wilson LJ, Hughes JB, West JL, Colvin VL. Nano Lett. 2004;4:1881–1887. [Google Scholar]
- 15.Alivisatos AP, Gu W, Larabell C. Annu Rev Biomed Eng. 2004 doi: 10.1146/annurev.bioeng.7.060804.100432. [DOI] [PubMed] [Google Scholar]
- 16.Tang Z, Wang Y, Sun K, Kotov NA. Nano Lett. 2005;17:358–363. [Google Scholar]
- 17.Zaitseva N, Manna L, Gerion D, Saw CK. Adv Mater. 2005;17:1321–1324. doi: 10.1002/adma.200401597. [DOI] [PubMed] [Google Scholar]
- 18.Rikans LE, Yamano T. J Biochem Mol Toxicol. 2000;14:110–117. doi: 10.1002/(sici)1099-0461(2000)14:2<110::aid-jbt7>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 19.Tsien RY. Nat Cell Biol. 2003:SS16–SS21. [PubMed] [Google Scholar]
- 20.Sako Y, Yanagida T. Nat Rev Mol Cell Biol. 2003;4:SS1–SS5. [PubMed] [Google Scholar]
- 21.Kim S, Lim YT, Soltesz EG, De Grand AM, Lee J, Nakayama A, Parker JA, Mihaljevic T, Laurence RG, Dor DM, Cohn LH, Bawendi MG, Frangioni JV. Nat Biotechnol. 2004;22:93–97. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao X, Cui Y, Levenson R, Chung L, Nie S. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 23.Chen FQ, Gerion D. Nano Lett. 2004;4:1827–1832. [Google Scholar]
- 24.Derfus AM, Chan WCW, Bhatia SN. Nano Lett. 2004;4:11–18. doi: 10.1021/nl0347334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoshino A, Fujioka K, Oku T, Suga M, Sasaki YF, Ohta T, Yasuhara M, Suzuki K, Yamamoto K. Nano Lett. 2004;4:2163–2169. [Google Scholar]
- 26.Kirchner C, Liedl T, Kudera S, Pellegrino T, Muñoz Javier A, Gaub HE, Stölzle S, Fertig N, Parak WJ. Nano Lett. 2005;5:331–338. doi: 10.1021/nl047996m. [DOI] [PubMed] [Google Scholar]
- 27.Ding L, Stilwell J, Zhang T, Elboudwarej O, Jiang H, Selegue JP, Cooke PA, Gray JW, Chen FF. Nano Lett. 2005;5:2448–2464. doi: 10.1021/nl051748o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parak WJ, Gerion D, Zanchet D, Woerz AS, Pellegrino T, Micheel C, Williams SC, Seitz M, Bruehl RE, Bryant Z, Bustamante C, Bertozzi CR, Alivisatos AP. Chem Mater. 2002;14:2113–2119. [Google Scholar]
- 29.Gerion D, Pinaud F, Williams SC, Parak WJ, Zanchet D, Weiss S, Alivisatos AP. J Phys Chem B. 2001;105:8861–8871. [Google Scholar]
- 30.Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Nat Biotechnol. 2003;21:47–51. doi: 10.1038/nbt767. [DOI] [PubMed] [Google Scholar]
- 31.Jaiswal JK, Simon SM. Trend Cell Biol. 2004;14:497–504. doi: 10.1016/j.tcb.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 32.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN. GenomeBiology. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engelman JA, Berg AH, Lewis RY, Lin A, Lisanti MP, Scherer PE. J Biol Chem. 1999;274:35630–35638. doi: 10.1074/jbc.274.50.35630. [DOI] [PubMed] [Google Scholar]
- 34.Luschen S, Scherer G, Ussat S, Ungefroren H, Adam-Klages S. Exp Cell Res. 2004;293:196–206. doi: 10.1016/j.yexcr.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, Sun P. Mol Cell Biol. 2002;22:3389–3403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang CY. Nat Cell Biol. 2005;7:909–915. doi: 10.1038/ncb1291. [DOI] [PubMed] [Google Scholar]
- 37.Haq R, Brenton JD, Takahashi M, Finan D, Finkielsztein A, Damaraju S, Rottapel R, Zanke B. Cancer Res. 2002;62:5076–5082. [PubMed] [Google Scholar]
- 38.Petropoulou C, Trougakos IP, Kolettas E, Toussaint O, Gonos ES. FEBS Lett. 2001;509:287–297. doi: 10.1016/s0014-5793(01)03150-7. [DOI] [PubMed] [Google Scholar]
- 39.Munakata H, Yosizawa Z. Biochim Biophys Acta. 1980;623:412–417. doi: 10.1016/0005-2795(80)90270-6. [DOI] [PubMed] [Google Scholar]
- 40.Cowley DO, Muse GW, Van Dyke T. Mol Cell Biol. 2005;25:7796–7802. doi: 10.1128/MCB.25.17.7796-7802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meraldi P, Sorger PK. EMBO J. 2005;24:1621–1633. doi: 10.1038/sj.emboj.7600641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanoosthuyse V, Hardwick KG. Trends Cell Biol. 2005;15:231–233. doi: 10.1016/j.tcb.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Tang Z, Shu H, Oncel D, Chen S, Yu H. Mol Cell. 2004;16:387–397. doi: 10.1016/j.molcel.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 44.Margolis RL. Dev Cell. 2004;7:634–635. doi: 10.1016/j.devcel.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 45.Ouyang B, Lan Z, Meadows J, Pan H, Fukasawa K, Li W, Dai W. Cell Growth Differ. 1998;9:877–885. [PubMed] [Google Scholar]
- 46.Hansen DV, Loktev AV, Ban KH, Jackson PK. Mol Biol Cell. 2004;15:5623–5634. doi: 10.1091/mbc.E04-07-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Letwin K, Mizzen L, Motro B, Ben-David Y, Bernstein A, Pawson T. EMBO J. 1992;11:3521–3531. doi: 10.1002/j.1460-2075.1992.tb05435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delaval B, Ferrand A, Conte N, Larroque C, Hernandez-Verdun D, Prigent C, Birnbaum D. Oncogene. 2004;23:4516–4522. doi: 10.1038/sj.onc.1207593. [DOI] [PubMed] [Google Scholar]
- 49.Lauffart B, Howell SJ, Tasch JE, Cowell JK, Still IH. Biochem J. 2002;363:195–200. doi: 10.1042/0264-6021:3630195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abaza A, Soleilhac JM, Westendorf J, Piel M, Crevel I, Roux A, Pirollet F. J Biol Chem. 2003;278:27844–27852. doi: 10.1074/jbc.M304522200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edsall LC, Cuvillier O, Twitty S, Spiegel S, Milstien S. J Neurochem. 2001;76:1573–1584. doi: 10.1046/j.1471-4159.2001.00164.x. [DOI] [PubMed] [Google Scholar]
- 52.Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. J Biol Chem. 1998;273:23722–23728. doi: 10.1074/jbc.273.37.23722. [DOI] [PubMed] [Google Scholar]
- 53.Matsusaki M, Larsson K, Akagi T, Lindstedt M, Akashi M, Borrebaeck CAK. Nano Lett. 2005;5:2168–2173. doi: 10.1021/nl050541s. [DOI] [PubMed] [Google Scholar]
- 54.Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. Nat Cell Biol. 2005;7:126–136. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 55.Hughes M, Dobric N, Scott IC, Su L, Starovic M, St-Pierre B, Egan SE, Kingdom JC, Cross JC. Dev Biol. 2004;271:26–37. doi: 10.1016/j.ydbio.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 56.St-Pierre B, Flock G, Zacksenhaus E, Egan SE. J Biol Chem. 2002;277:46544–46551. doi: 10.1074/jbc.M111652200. [DOI] [PubMed] [Google Scholar]
- 57.Turley H, Wykoff CC, Troup S, Watson PH, Gatter KC, Harris AL. J Pathol. 2004;203:808–813. doi: 10.1002/path.1585. [DOI] [PubMed] [Google Scholar]
- 58.Boudjelal M, Taneja R, Matsubara S, Bouillet P, Dolle P, Chambon P. Genes Dev. 1997;11:2052–2065. doi: 10.1101/gad.11.16.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ding LH, Shingyoji M, Chen F, Hwang JJ, Burma S, Lee C, Cheng JF, Chen DJ. Radiat Res. 2005;164:17–26. doi: 10.1667/rr3354. [DOI] [PubMed] [Google Scholar]