

Abstract
Complex modulation of voltage-gated Ca2+ currents through the interplay among Ca2+ channels and various Ca2+-binding proteins (CaBPs) is increasingly being recognized. The K+ channel interacting protein 2 (KChIP2), originally identified as an auxiliary subunit for KV4.2 and a component of the transient outward K+ channel (Ito), is a CaBP whose regulatory functions do not appear restricted to KV4.2. Consequently, we hypothesized that KChIP2 is a direct regulator of the cardiac L-type Ca2+ current (ICa,L). We found that ICa,L density from KChIP2−/− myocytes is reduced by 28% compared to ICa,L recorded from WT myocytes (P<0.05). This reduction in current density results from loss of a direct effect on the Ca2+ channel current, as shown in a transfected cell line devoid of confounding cardiac ion currents. ICa,L regulation by KChIP2 was independent of Ca2+ binding to KChIP2. Biochemical analysis suggested a direct interaction between KChIP2 and the CaV1.2 α1C subunit N-terminus. We found that KChIP2 binds to the N-terminal inhibitory (NTI) module of α1C and augments ICa,L current density without increasing CaV1.2 protein expression or trafficking to the plasma membrane. We propose a model in which KChIP2 impedes the NTI module of CaV1.2, resulting in increased ICa,L. In the context of recent reports that KChIP2 modulates multiple KV and NaV currents, these results suggest that KChIP2 is a multimodal regulator of cardiac ionic currents.
Keywords: Ion channels, mouse models, amino terminal inhibitory module, auxiliary subunit, CaV1.2
Introduction
The KV4 subfamily of voltage-gated K+ channels underlies the A-currents in neuronal tissue (IA)1 and the fast-inactivating transient-outward current in the heart (Ito,f).2 The K+ Channel Interacting Proteins (KChIPs) were discovered as a family of four alternatively spliced accessory subunits for the A-type K+ currents in neuronal tissue.3, 4 Whereas all KChIPs are expressed in brain3, 4, only KChIP2 is found in heart, where it interacts with KV4.2 and KV4.3 to form the native Ito,f.5, 6 KV4 subunits co-localize with all KChIP subtypes when expressed in cell lines, and the addition of any KChIP increases the KV4-current density.7, 8 Further, KChIP1, KChIP2 and KChIP3 slow KV-current inactivation and speed recovery from inactivation.3, 5, 9 KChIP4 has been described as an inactivation suppressor of the KV4 current, where it abolishes fast inactivation.10
The KChIPs are neuronal calcium-sensor (NCS) proteins11 and contain four Ca2+-binding EF-hand motifs, of which the last three bind Ca2+.3 EF-hand mutations that ablate Ca2+ binding disrupt K+ channel modulation, but preserve binding affinity to KV4.3 NCS proteins and KChIPs share structural homology with the superfamily of Ca2+-binding proteins (CaBP)12, 13, several of which regulate ICa,L in the heart. Most prominently, calmodulin (CaM)14-17 and Ca2+-binding protein 1 (CaBP1)18 differentially govern Ca2+-dependent inactivation of ICa,L via interactions with the amino- (N) or carboxy- (C) termini of the pore-forming α1C subunit, CaV1.2. Other members of the CaBP superfamily variously affect different L-type Ca2+ channels (e.g. CaV1.3)19, 20 or non-L-type Ca2+ channels (e.g., CaV2.1)21, suggesting an extensive interplay among various Ca2+ channels and CaBPs.
Whether KChIP2 regulates Ca2+ channels has not been explored, although emerging evidence implicates KChIP2 as a modulator of channels other than KV4, particularly in the heart. For example, in cell lines KChIP2 impairs trafficking of KV1.522, and KChIP2 silencing has been reported to eliminate the sodium current (INa) in cultured neonatal cardiomyocytes.23 Thus, in the present study, we hypothesized that KChIP2 modulates the L-type Ca2+ current. Using both ventricular myocytes and a heterologous expression system, we show that KChIP2 increases ICa,L. Furthermore, biophysical and functional experiments show that KChIP2 binds specifically to the amino-terminal inhibitory (NTI) domain of CaV1.2 in a Ca2+-independent manner. This KChIP2-NTI association causes an increase in the Ca2+-current density, independent of protein synthesis and trafficking.
Materials and Methods
Electrophysiological recordings in disaggregated ventricular myocytes
We studied adult (10-12 weeks), male C57BL6 WT and re-derived KChIP2−/− mice.24 All animal experiments were approved by the Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health). Genotyping was performed using DNA isolated from tail samples, followed by PCR amplification of KChIP2 and neomycin. ECGs obtained from anesthetized WT and KChIP2−/− mice (Online Figure I) showed ST-segment elevation specific to the KChIP2−/− mice, consistent with earlier reports from the KChIP2-/- mice originally generated in the 129SV background.24
Hearts were excised from anesthetized (0.1 mg/g ketamine, 0.01 mg/g xylazine, IP) mice and myocytes were disaggregated from the left ventricular free wall. Membrane recordings were made within 6 hours of myocyte isolation using standard whole-cell voltage-clamp protocols. Patch pipettes (1-2 MΩ) contained (in mM): 80 aspartic acid, 10 NaCl, 70 CsOH, 40 CsCl, 2 MgCl2, 10 EGTA, 10 HEPES-Na, 2 ATP-Na2, and 0.1 GTP-Na2 (pH 7.2). Cells were superfused with (in mM): 135 NaCl, 10 CsCl, 1 CaCl2, 1 MgCl2, 5 HEPES-Na, 10 dextrose, and 0.01 tetrodotoxin (pH 7.4; T 37°C). Current-voltage curves were generated as nifedipine-sensitive (5 μM) difference-currents using 400-ms depolarizing steps from a holding potential of −40 mV as described.25 Currents are expressed as peak-inward density; cell capacitances were similar for WT and KChIP2−/− myocytes.
Molecular biology
cDNA constructs consisted of rabbit α1C (CaV1.2), and rat CaVβ2, CaVα2δ2, and GFP-KChIP2b (IRES) and GFP in pcDNA3.1. The ΔNT-CaV1.2 (amino acids 140-2171), and KChIP2-ΔEF (Asp residues at position 1 in EF-hands 2, 3 and 4 of KChIP2b mutated to Ala) were generated by standard molecular biology techniques. The GFP-α1C construct was constructed as previously described.26
Electrophysiological recordings in tsA201 cells
Transfection of 5 μg total DNA (35 mm culture dish) into tsA201 cells (ECACC, Wiltshire, UK) was performed using calcium-phosphate precipitation (Chemicon, Phillipsburg, NJ). The tsA201 cell line is an SV-40 transformed variant of the HEK293 cell line that produces large amounts of recombinant protein. Transfected cells were identified by green fluorescent protein. Patch-clamp experiments were conducted 48 hours after transfection. Pipettes (3-5 MΩ) contained (in mM): 135 Cs-MeSO3, 5 CsCl, 5 EGTA, 1 MgCl2, 10 HEPES-Na, 4 mM ATP-Na2 (pH 7.2). External solution contained (in mM): 140 TEA-Cl, 10 HEPES-Na, 5 CaCl2 (pH 7.3; T 22°C). Ba2+ containing superfusate was prepared by equimolar substitution of BaCl2 for CaCl2. Expressed currents were recorded using 400-ms depolarizing steps to −40 to +60 mV from a holding potential of −90 mV.
Protein chemistry
Immunoblotting was done using previously described methods.27 Briefly, whole-cell protein lysates were obtained from WT and KChP2−/− ventricular tissue and from transfected tsA201 cells. Myocardial protein lysates were fractionated using a protocol from Ventura et al.28 Proteins were separated on 4-20% gradient SDS-PAGE gels and transferred to PVDF membranes. Blots were probed with primary antibodies against CaV1.2 (NeuroMab, CA), GAPDH or actin (SantaCruz, CA). Fusion proteins (GST-tagged N terminus and 6xHis-tagged C terminus) were co-expressed with KChIP2 and purified as described earlier.29
Statistical analysis
Data are expressed as means±SEM. One-way ANOVA followed by Bonferroni's t test, when appropriate, was used for statistical comparison. P<0.05 was considered statistically significant.
Results
Mice Lacking KChIP2 Have Reduced ICa,L
We compared patch-clamp recordings from disaggregated left-ventricular myocytes from WT and KChIP2−/− mice to investigate whether KChIP2 regulates ICa,L. Figure 1A (left) shows representative nifedipine-sensitive current traces. The current-voltage relationship demonstrates reduced ICa,L in KChIP2−/− myocytes compared to WT myocytes, independent of test voltage (Figure 1A, right). For example, at a test voltage (Vtest) of +10 mV, ICa,L density was −16.6±1.0 pA/pF in WT myocytes (n=15) versus −12.0±0.7 pA/pF (n=18; P<0.05) in KChIP2−/−, a 28% reduction. There were no changes in ICa,L activation or steady-state inactivation (Figure 1B). The decreased current amplitude in KChIP2−/− myocytes was associated with slower Ca2+-dependent inactivation. The decay phase of the current trace was best fit with 2 exponentials. At voltage steps to +10 mV, τfast and τslow for WT myocytes were 10±1 and 61±10 ms, respectively, compared with 13±1 (P<0.05) and 81±5 ms (P=0.07), respectively, for KChIP2−/− myocytes.
Figure 1.
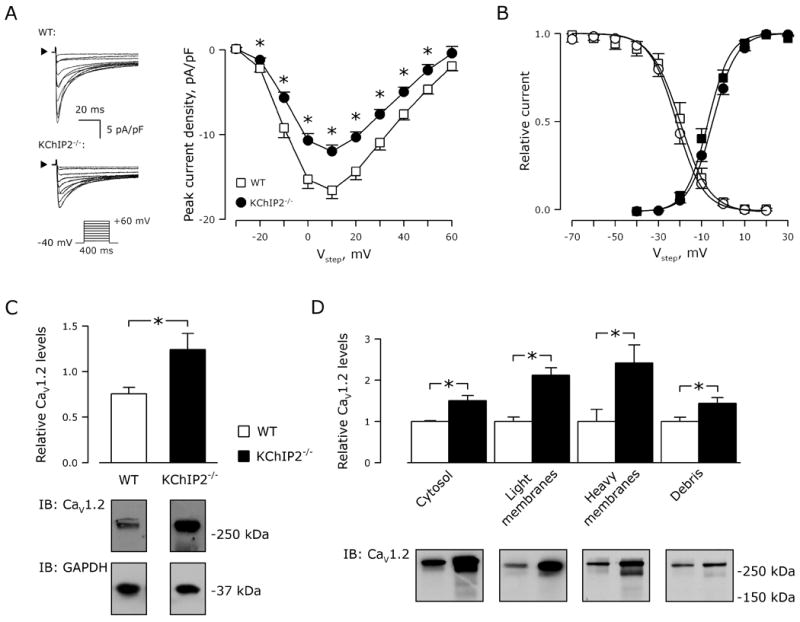
KChIP2−/− myocytes have decreased ICa,L. A, Representative nifedipine-sensitive current traces normalized to cell capacitance (left panels; arrowheads indicate 0 pA), and current-voltage relationship for whole-cell Ca2+ currents (right panel) from cardiomyocytes isolated from WT (squares, n=15) and KChIP2−/− myocytes (circles, n=18). *, P<0.05 versus WT. Inset, voltage protocol. B, Current activation (filled symbols) and steady-state inactivation (non-filled symbols) recorded from WT (squares) and KChIP2−/− (circles) myocytes. Solid lines represent sigmoidal fits to the data, where comparable V0.5-values (activation, WT: −7±1 versus KChIP2−/−: −5±1 mV; inactivation, WT: −20±2 versus KChIP2−/−: −22±1 mV; P>0.05 for both) and k-values (activation, WT: 5±1 versus KChIP2−/−: 6±2; inactivation, WT: −7±1 versus KChIP2−/−: −6±1; P>0.05 for both) were obtained. C, Representative immunoblot for CaV1.2 (below) and quantification (above) of whole-cell lysates from WT (n=4) and KChIP2−/− (n=4) ventricles. GAPDH was used as control for equal loading, and was used for normalizing CaV1.2 levels. *, P<0.05. D, Protein fractionation and immunostaining for CaV1.2. Quantification of immunoblots of CaV1.2 (above) from fractions predominantly containing cytosol, light membranes, heavy membranes, and debris, respectively. *, P<0.05. Below: Representative immunoblots.
The reduced ICa,L could not be explained by less CaV1.2 protein: rather, we found a 64% increase in CaV1.2 protein in whole-cell lysates from KChIP2−/− ventricular tissue, compared to WT (P<0.05; Figure 1C). When we partitioned protein lysates from WT and KChIP2−/− ventricles into cytosolic, light membrane (including the plasma membrane), heavy membrane (including the sarcoplasmic reticulum) and debris (predominantly nuclei) fractions, we observed significantly more CaV1.2-immunoreactive protein in all KChIP2−/− fractions (Figure 1D), suggesting that the decreased ICa,L in KChIP2−/− mice does not result from deficient protein trafficking. Thus, despite an increase in CaV1.2 protein, the absence of KChIP2 attenuates ICa,L in myocytes.
KChIP2 Augments Ca2+ Currents in a Heterologous Expression System
The reduced ICa,L in KChIP2−/− myocytes could be due to either loss of a direct modulatory effect upon CaV1.2 or secondary to unidentified electrical remodeling processes that diminish ICa,L. To distinguish between these possibilities, we tested whether KChIP2 directly modulates ICa,L in a heterologous expression system. The CaV1.2 α1C subunit and the Ca2+ channel accessory subunits β2 and α2δ were expressed with or without KChIP2 in tsA201 cells and whole-cell patch-clamp recordings were compared. Currents from cells expressing KChIP2 showed a ∼2-fold increase in peak density (Figure 2A) without any change in the activation or steady-state inactivation kinetics (Figure 2B). Scaling peak current to unity revealed no effect of KChIP2 co-expression on current decay (Figure 2C, top), and pedestal currents measured at 300 ms were comparable between KChIP2-positive and -negative cells (Figure 2C, bottom).
Figure 2.
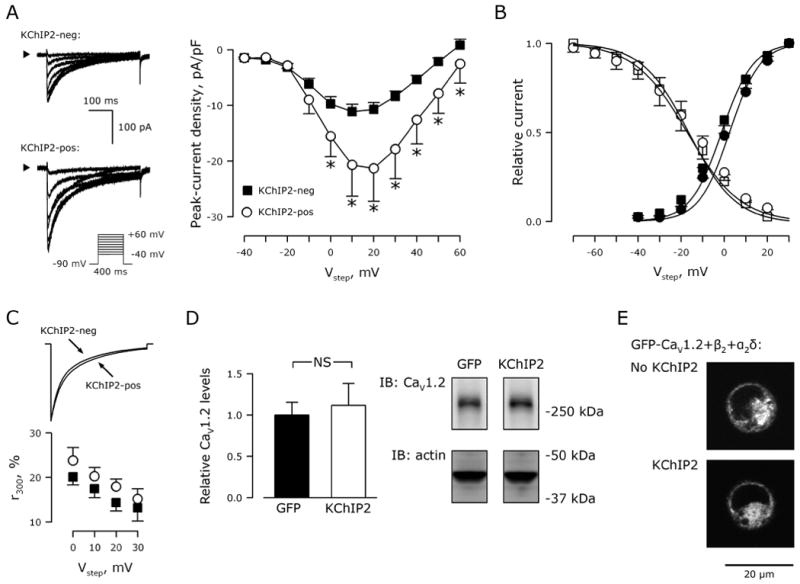
KChIP2 augments peak CaV1.2 current in transfected tsA201 cells. A, Representative current traces from tsA201 cells transfected with the CaV1.2 α1C, β2, and α2δ subunits with and without KChIP2. Arrowheads indicate 0 pA. Inset, voltage protocol. Right, Ca2+ current-voltage relationship from KChIP2-transfected cells (circles, n=14) and GFP-transfected cells (KChIP2-neg; squares, n=18). Data presented as mean±SEM; *, P<0.05 versus KChIP2-neg). B, Current activation (filled symbols) and steady-state inactivation (non-filled symbols) recorded in KChIP2-negative (squares) and -positive (circles) cells. Solid lines represent sigmoidal fits to the data, where comparable V0.5- and k-values were obtained (not shown). C, Comparable current-decay time course exemplified with scaled amplitudes. Below, residual current at 300 ms relative to peak current amplitude in KChIP2-negative (squares) and -positive (circles) cells. D, Representative immunoblot for CaV1.2 (right) and quantification (left) of whole-cell lysates from tsA201 cells transfected with CaV1.2, β2, and α2δ with GFP or KChIP2. Transfections were done in triplicates. Actin was used as control for equal loading, and was used for normalizing CaV1.2 levels. NS, P>0.05. E, Representative fluorescent confocal images of tsA201 cells transfected with GFP-labeled CaV1.2, β2, and α2δ with and without KChIP2.
As in myocytes, the reduced current density in the absence of KChIP2 could not be attributed to reduced CaV1.2 protein expression or decreased trafficking to the plasma membrane: we observed no differences in protein levels with or without KChIP2 co-expression (Figure 2D). Further, analysis of GFP-labeled CaV1.2 distribution in tsA201 cells showed that membrane targeting of CaV1.2 was unaffected by KChIP2 (Figure 2E). The membrane-to-whole-cell GFP fluorescence ratio normalized to cell size was comparable in KChIP2-negative (5.9±1.6 arbitrary units; n=18) and KChIP2-positive cells (5.6±0.9 arbitrary units, P>0.05; n=19).
To test specifically for a role for KChIP2 in trafficking CaV1.2 to the plasma membrane, we compared currents from cells transfected with CaV1.2 (without β2) to cells transfected with CaV1.2 and KChIP2. As expected, in the absence of a β2 subunit, currents were barely detectable (Online Figure II). The addition of KChIP2 did not generate larger currents, suggesting that KChIP2 cannot replace the role of β subunits in trafficking the pore-forming β subunits to the plasma membrane. Since KChIP2 modulated ICa,L in a heterologous expression system essentially stripped of all other ionic currents, we conclude that KChIP2 acts directly upon the CaV1.2 channel.
KChIP2 affects CaV1.2 currents in a Ca2+-independent manner
Next, we determined whether the modulatory effects of KChIP2 are dependent upon Ca2+. When we substituted Ba2+ for Ca2+as charge carrier, the addition of KChIP2 still increased current density of CaV1.2 channels expressed in tsA201 cells (Figure 3A). This suggests that Ca2+ permeation through the channel does not alter KChIP2-mediated effects. To test for a contribution of Ca2+ binding to the KChIP2 EF hands, we constructed a Ca2+-insensitive KChIP2 with Asp -to-Ala mutations in EF hands 2-4 KChIP2-ΔEF.3 Compared to WT-KChIP2, this triple EF-hand mutant produced a similar increase in the peak-current density when expressed in tsA201 cells in the presence of CaV1.2, β2 and α2δ (Figure 3B). Thus, Ca2+ binding to KChIP2 does not influence the KChIP2-mediated increase in CaV1.2 current density. Interestingly, we observed a 7±2 mV depolarizing shift in the activation curve of the Ca2+ current with this mutant KChIP2 (P<0.05 versus WT-KChIP2). This shift persisted when Ba2+ replaced Ca2+ as the charge carrier (data not shown).
Figure 3.
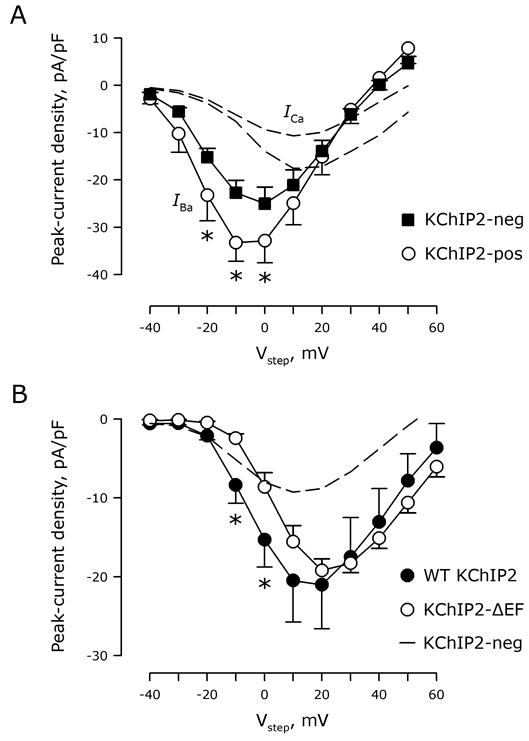
KChIP2 affects ICa,L in a Ca2+-independent manner. A, Ba2+ currents obtained in KChIP2-negative (squares) and -positive (circles) cells as a function of test voltage (Vstep). *, P<0.05 versus KChIP2-negative cells. For comparison, current-voltage relationships attained with Ca2+ as charge carrier (Figure 2A) are reproduced with dotted lines. B, Peak-current density as a function of test voltage (Vstep) in cells transfected with CaV1.2, β2, and α2δ subunits with either WT-KChIP2 (filled circles) or the calcium-insensitive KChIP2-ΔEF (non-filled circles). *, P<0.05 versus KChIP2-ΔEF (nWT-KChIP2=14; nKChIP2-ΔEF=10). Dotted line illustrates the current-voltage relations in the absence of KChIP2 (Figure 2A).
KChIP2 modulates ICa,L through binding to the N-terminal domain of CaV1.2
Other Ca2+-binding proteins affecting CaV1.2 function, like CaM and CaBP1, associate with CaV1.2 at the C- and/or the N-termini14, 15, 17, 30, 31, suggesting likely targets for KChIP2 interaction. Hence, we expressed the C- and N-termini of CaV1.2 as fusion proteins tagged with 6xHis and GST, respectively, and tested whether they were able to bind KChIP2 in a recombinant bacterial co-expression system. Figure 4A shows a representative Coomassie-stained SDS-PAGE of the proteins purified after affinity chromatography: KChIP2 co-purifies with the region of the N terminus previously shown to support CaM binding (amino acids 60-120; 17), but not with GST alone. A construct with amino acids 1-70 of CaV1.2 showed no KChIP2 co-purification (data not shown). In contrast, KChIP2 did not interact with the CaV1.2 C terminus. The C-terminal domain of CaV1.2 cannot be purified as a soluble protein in the absence of a true binding partner32 and we did not recover any soluble CaV1.2 C terminus when KChIP2 was co-expressed (Figure 4A). In contrast, the CaV1.2 C-terminal domain was purified when CaM was co-expressed as a binding partner. Thus, KChIP2 likely mediates its effect on ICa,L through an interaction with the CaV1.2 N terminus. To address the importance of the calcium-binding properties of KChIP2, we repeated the co-purification experiments when Ca2+ was chelated by EGTA. Under this condition, KChIP2 still efficiently associated with the N-terminal domain of CaV1.2 (data not shown), consistent with the absence of a Ca2+-dependent effect upon ICa,L amplitude (Figure 3).
Figure 4.
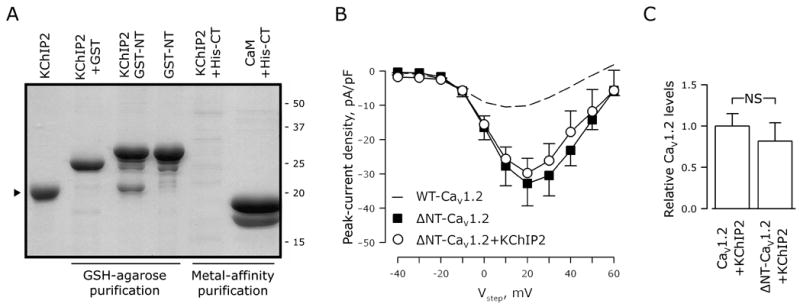
The inhibiting amino terminus of CaV1.2 interacts with KChIP2. A, Coomassie-stained SDS-PAGE of GSH-agarose or metal-affinity purified protein samples. The carboxy- and amino-termini of CaV1.2 tagged with 6xHis or GST, respectively, were expressed in a recombinant bacterial system. KChIP2 or calmodulin (CaM) was co-expressed as indicated above each lane. KChIP2 co-purifies with GST-tagged CaV1.2 amino-terminus (GST-NT), but not with GST alone or with the His-tagged carboxy-terminus (His-CT). B, Peak Ca2+-current density as a function of test voltage (Vstep) in tsA201 cells transfected with ΔNT-CaV1.2, β2, and α2δ with KChIP2 (circles) or without KChIP2 (squares). No significant differences were found (nKChIP2-pos=8; nKChIP2-neg=9; mean±SEM). Dotted line indicates WT-CaV1.2 in the absence of KChIP2 (from Figure 2A), to illustrate the augmented current density secondary to deletion of the N-terminal domain. C, Ablation of the amino terminus does not alter protein expression of CaV1.2 in transfected tsA201 cells. NS, P>0.05; n=3 transfections per group.
To confirm functionally that KChIP2 modulates CaV1.2 through its interaction with the α1C N-terminus, we deleted the first 139 amino acids of α1C (ΔNT-CaV1.2) and tested this truncated channel in patch-clamp experiments. The current recorded from the ΔNT-CaV1.2 construct in the absence of KChIP2 was increased about 3 fold compared to WT-CaV1.2 (Figure 4B), as previously reported.33 The increased current density seen with the ΔNT-CaV1.2 construct relative to WT-CaV1.2 was not secondary to changes in protein levels of CaV1.2 (Figure 4C), consistent with pervious results.33, 34 With its binding site in the CaV1.2 N-terminus now absent, KChIP2 no longer produced an increase in ICa,L current density (Figure 4B). These data suggest that KChIP2 affects ICa,L through a direct association with the N-terminal domain of CaV1.2. Since these heterologous expression systems contain β and α2δ subunits, we cannot formally exclude the possibility that KChIP2 interactions with these auxiliary subunits contribute to the observed modulation.
Discussion
The novel findings in this study are that KChIP2 modulates ICa,L by interacting with the N-terminal domain of CaV1.2 and does so in a Ca2+-independent manner. Moreover, this modulation increases ICa,L amplitude without requiring additional CaV1.2 channel protein.
Generally, amplitudes of ionic currents can be augmented by adding functional ion channels to the sarcolemma (N), by increasing the likelihood of the channel to be in the open state (PO), or by improving conductance of the individual channel (i). Although the present study did not determine which (combination) of these 3 mechanisms was underlying the KChIP2-mediated increase in whole-cell Ca2+-current amplitude, the unaltered protein expression (Figure 2D) and localization (Figure 2E) make it unlikely that the KChIP2-mediated increase in amplitude derives from improved trafficking of functional channels to the membrane. Consistent with this logic, sub-cellular fractions from WT myocytes actually had lower levels of CaV1.2 protein than KChIP2−/− myocytes (Figure 1D) even though current amplitude was higher in the WT myocytes.
Instead of affecting the parameter N, we propose a model in which KChIP2 association with CaV1.2 increases PO by relieving the inhibitory effects of the α1C N terminus that have previously been characterized by Dascal and colleagues.30, 34 As described, α1C contains a N-terminal inhibitory (NTI) module35 that decreases the channel's maximal open probability (PO,max), in a manner that is partially relieved by the β subunit.34 We propose that KChIP2 interacts with the α1C N terminus, reducing the NTI module's effect on channel gating beyond that provided by the β subunit (Figure 5). Consistent with this hypothesis, KChIP2 binding to the N-terminal domain was established both biochemically (Figure 4A) and functionally (Figure 4B). Further, KChIP2 caused an increased current density in the absence of a shift in the activation curve (Figure 2B), consistent with modulation of the NTI module.34 Finally, KChIP2 did not increase CaV1.2 current density in the absence of the β subunit (Online Figure II), also consistent with an action on PO,max rather than an effect upon channel trafficking; however, this hypothesis has to be verified by single-channel recordings.
Figure 5.
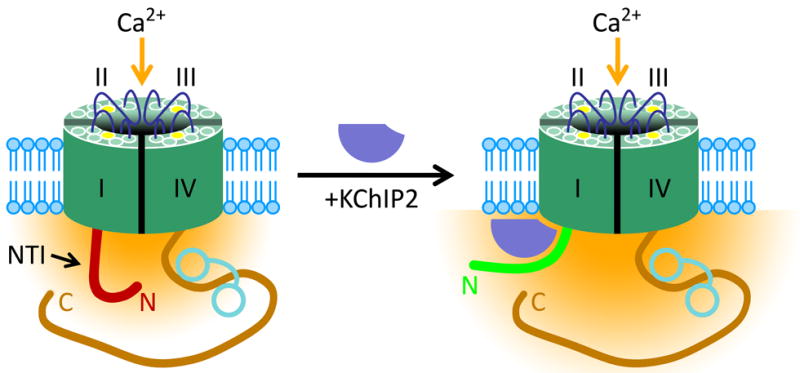
Proposed model for KChIP2 modulation of ICa,L. The N-terminal inhibitory (NTI) module impairs the Ca2+-current density (yellow) in the absence of KChIP2 (left). The N terminus translocates upon binding of KChIP2, thereby reducing the inhibitory effects on ICa,L (right). Calmodulin is illustrated as a dumbbell shaped structure on the α1C CT.
As a member of the CaBP family, KChIP2 is the newest example of a Ca2+ binding protein that regulates voltage-gated Ca2+ channels in general and CaV1.2 channels in particular. KChIP2 is unique among these CaBPs, however, in that Ca2+ binding does not appear to be required for dynamic regulation of Ca2+ channel function: we observed comparable KChIP2 influence of IBa (Figure 3A) and no change in ICa amplitude when the Ca2+-binding EF hands of KChIP2 were mutated (Figure 3B). Furthermore, KChIP2 co-purified with the N terminus both in the presence and in the absence of free Ca2+. The Ca2+-insensitive KChIP2-ΔEF mutant also binds to Kv4.2, but does not modulate the K+ current3, suggesting distinct Ca2+-dependent and –independent regulatory functions of KChIP2. Nonetheless, the depolarizing shift in the activation curve induced by the ΔEF mutant hints at an even more complex regulation of ICa,L by Ca2+ and KChIP2.
The increased CaV1.2 protein level in KChIP2−/− mice is intriguing in that KChIP family members, including KChIP2, can also act as Ca2+-dependent transcriptional repressors.36 Whether loss of KChIP2-mediated repression of CaV1.2 transcription explains the observed increase in CaV1.2 protein in the KChIP2−/− myocytes is not known but, if correct, would indicate that KChIP2 modulates CaV1.2 currents via multiple and independent mechanisms and would place more emphasis on the additional roles attributed to KChIP2, such as the modulation and regulation of cardiac NaV and KV currents.3, 6, 23 Thus, KChIP2 appears to be a multimodal regulator of several cardiac ionic currents rather than a contributor solely to Ito.
The CaV1.2 gene gives rise to multiple, tissue-specific isoforms as a consequence of alternative splicing. The NTI module is absent in short N-terminus isoforms, in which the initial 46 amino acids encoded by exon 1a are replaced by a partially homologous sequence of 16 amino acids encoded by exon 1b.34 Whereas the long isoforms containing the NTI module are predominant in the heart, vascular smooth muscle cells express only the short isoforms lacking the NTI module.37 Both short and long N-terminal isoforms are found in the brain. At present it is unknown whether KChIP2 interacts with CaV1.2 isoforms lacking the NTI module, however such association would have implications for Ca2+ entry in vascular tissue and in neuronal tissue expressing KChIP2.
Our findings may have even more relevance to arrhythmogenesis. Initially described as an auxiliary subunit for the neuronal A-type current3, KChIP2 was later shown to be important for the expression of Ito in the human heart.5 Recently, it was reported that post-transcriptional silencing of KChIP2 decreases Ito and INa, suggesting that the two channels form a structural and functional channel complex.23 The present data show that KChIP2, in addition to its other regulatory functions, modulates the cardiac Ca2+ current, implicating a broad physiological role of KChIP2 in cardiac electrophysiology. Although the arrhythmia susceptibility of KChIP2-/- knockout mice23 already pointed to a key physiological role for KChIP2, the data reported herein offer new hypotheses about the observed arrhythmogenesis. This concept is particularly relevant since mice lacking the subunit for the Ito current (KV4.2) showed no arrhythmia susceptibility38; likely one of the other modulatory roles played by KChIP2 is paramount.
Our findings also suggest a role for KChIP2 in arrhythmogenesis associated with heart failure. Since KChIP2 is downregulated in patients with heart failure39, the present findings in combination with the previous reports of KChIP2-mediated actions of KV and NaV currents23, 40, suggest that KChIP2-downregulation will have widespread effects on electrophysiology and contractile performance. Our results specifically predict that KChIP2 downregulation would decrease ICa,L, thereby highlighting a recent report in which loss-of-function mutations in the CaV1.2 α1C or the β2 subunits are associated with sudden cardiac death.41 Of particular interest, one of the mutations reported in that study, A39V in α1C, resides in the NT of α1C. It is reasonable to speculate that the reduced current observed in cells expressing the A39V mutant results from an effect upon the α1C NTI module. It is also intriguing that a common ECG phenotype reported for these patients with ICa,L loss of function was J-point elevation: as previously observed24 and as confirmed by us (Online Figure I and Online Table I), ECGs from KChIP2-/- mice also display J-point elevation. That this may be secondary to effects upon ICa,L rather than upon Ito as suggested by a report in which ECGs from Kv4.2-/- mice are indistinguishable from WT.38
Conclusion
These data provide new insights into the function of the cardiac CaV1.2 calcium channel. The effects of KChIP2 on ICa,L in both native myocytes and in heterologous expression systems strongly suggest that this protein directly augments current amplitude in the absence of altered channel density, activation or inactivation kinetics. Our data, taken together with previous studies, suggest that KChIP2 is a multimodal regulator of a range of voltage-gated cardiac K+, Na+ and Ca2+ channels.
Acknowledgments
The authors wish to thank Dr. Masahiko Hoshijima (University of California, San Diego, CA) for providing the KChIP2−/− mice.
Sources of Funding: This work was supported by USPHS-NHLBI grants HL-67101, HL-28958 and HL-76116 to MRR, and HL-088089 and HL-071165 to GSP. GSP is an Established Investigator of the American Heart Association. MBT is a Research Fellow of the Heart Rhythm Society.
Footnotes
Disclosures: None
References
- 1.Sheng M, Tsaur ML, Jan YN, Jan LY. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- 2.Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS, McKinnon D. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- 3.An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 4.Morohashi Y, Hatano N, Ohya S, Takikawa R, Watabiki T, Takasugi N, Imaizumi Y, Tomita T, Iwatsubo T. Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. The Journal of biological chemistry. 2002;277:14965–14975. doi: 10.1074/jbc.M200897200. [DOI] [PubMed] [Google Scholar]
- 5.Deschenes I, DiSilvestre D, Juang GJ, Wu RC, An WF, Tomaselli GF. Regulation of Kv4.3 current by KChIP2 splice variants: a component of native cardiac I(to)? Circulation. 2002;106:423–429. doi: 10.1161/01.cir.0000025417.65658.b6. [DOI] [PubMed] [Google Scholar]
- 6.Thomsen MB, Sosunov EA, Anyukhovsky EP, Özgen N, Boyden PA, Rosen MR. Deleting the accessory subunit KChIP2 results in loss of Ito,f and increased IK,slow that maintains normal action potential configuration. Heart Rhythm. 2009;6:370–377. doi: 10.1016/j.hrthm.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shibata R, Misonou H, Campomanes CR, Anderson AE, Schrader LA, Doliveira LC, Carroll KI, Sweatt JD, Rhodes KJ, Trimmer JS. A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4.2 potassium channels. The Journal of biological chemistry. 2003;278:36445–36454. doi: 10.1074/jbc.M306142200. [DOI] [PubMed] [Google Scholar]
- 8.Venn N, Haynes LP, Burgoyne RD. Specific effects of KChIP3/calsenilin/DREAM, but not KChIPs 1, 2 and 4, on calcium signalling and regulated secretion in PC12 cells. The Biochemical journal. 2008;413:71–80. doi: 10.1042/BJ20080441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bahring R, Dannenberg J, Peters HC, Leicher T, Pongs O, Isbrandt D. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. The Journal of biological chemistry. 2001;276:23888–23894. doi: 10.1074/jbc.M101320200. [DOI] [PubMed] [Google Scholar]
- 10.Holmqvist MH, Cao J, Hernandez-Pineda R, Jacobson MD, Carroll KI, Sung MA, Betty M, Ge P, Gilbride KJ, Brown ME, Jurman ME, Lawson D, Silos-Santiago I, Xie Y, Covarrubias M, Rhodes KJ, Distefano PS, An WF. Elimination of fast inactivation in Kv4 A-type potassium channels by an auxiliary subunit domain. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1035–1040. doi: 10.1073/pnas.022509299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat Rev Neurosci. 2007;8:182–193. doi: 10.1038/nrn2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. The Biochemical journal. 2001;353:1–12. [PMC free article] [PubMed] [Google Scholar]
- 13.Haeseleer F, Imanishi Y, Sokal I, Filipek S, Palczewski K. Calcium-binding proteins: intracellular sensors from the calmodulin superfamily. Biochemical and biophysical research communications. 2002;290:615–623. doi: 10.1006/bbrc.2001.6228. [DOI] [PubMed] [Google Scholar]
- 14.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 15.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ Sensor for Ca2+ -Dependent Inactivation of L-type Calcium Channels [published erratum appears in Neuron 1999 Apr;22:following 893] Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 16.Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ Channels by Gbg and Calmodulin via Interactions with N and C Termini of a1C. J Biol Chem. 2000;275:39846–39854. doi: 10.1074/jbc.M005881200. [DOI] [PubMed] [Google Scholar]
- 17.Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H, Yu K, McCoy KL, Lee A. Molecular Mechanism for Divergent Regulation of Cav1.2 Ca2+ Channels by Calmodulin and Ca2+-binding Protein-1. J Biol Chem. 2005;280:29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 19.Cui G, Meyer AC, Calin-Jageman I, Neef J, Haeseleer F, Moser T, Lee A. Ca2+-binding proteins tune Ca2+-feedback to Cav1.3 channels in mouse auditory hair cells. J Physiol. 2007;585:791–803. doi: 10.1113/jphysiol.2007.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, Fuchs PA, Yue DT. Switching of Ca2+-Dependent Inactivation of CaV1.3 Channels by Calcium Binding Proteins of Auditory Hair Cells. J Neurosci. 2006;26:10677–10689. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA. Differential modulation of Ca(v)2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–217. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Guo W, Mellor RL, Nerbonne JM. KChIP2 modulates the cell surface expression of Kv 1.5-encoded K(+) channels. J Mol Cell Cardiol. 2005;39:121–132. doi: 10.1016/j.yjmcc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 23.Deschenes I, Armoundas AA, Jones SP, Tomaselli GF. Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J Mol Cell Cardiol. 2008;45:336–346. doi: 10.1016/j.yjmcc.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyen-Tran VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR, Chien KR. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 25.Protas L, Barbuti A, Qu J, Rybin VO, Palmiter RD, Steinberg SF, Robinson RB. Neuropeptide Y is an essential in vivo developmental regulator of cardiac ICa,L. Circ Res. 2003;93:972–979. doi: 10.1161/01.RES.0000099244.01926.56. [DOI] [PubMed] [Google Scholar]
- 26.Grabner M, Dirksen RT, Beam KG. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proc Natl Acad Sci U S A. 1998;95:1903–1908. doi: 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozgen N, Dun W, Sosunov EA, Anyukhovsky EP, Hirose M, Duffy HS, Boyden PA, Rosen MR. Early electrical remodeling in rabbit pulmonary vein results from trafficking of intracellular SK2 channels to membrane sites. Cardiovasc Res. 2007;75:758–769. doi: 10.1016/j.cardiores.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ventura A, Maccarana M, Raker VA, Pelicci PG. A cryptic targeting signal induces isoform-specific localization of p46Shc to mitochondria. The Journal of biological chemistry. 2004;279:2299–2306. doi: 10.1074/jbc.M307655200. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Wang HG, Xie H, Pitt GS. Ca2+/CaM controls Ca2+-dependent inactivation of NMDA receptors by dimerizing the NR1 C termini. J Neurosci. 2008;28:1865–1870. doi: 10.1523/JNEUROSCI.5417-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ channels by gbeta gamma and calmodulin via interactions with N and C termini of alpha 1C. The Journal of biological chemistry. 2000;275:39846–39854. doi: 10.1074/jbc.M005881200. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, Kim SA, Kirk EA, Tippens AL, Sun H, Haeseleer F, Lee A. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J Neurosci. 2004;24:4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 33.Shistik E, Ivanina T, Blumenstein Y, Dascal N. Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. The Journal of biological chemistry. 1998;273:17901–17909. doi: 10.1074/jbc.273.28.17901. [DOI] [PubMed] [Google Scholar]
- 34.Kanevsky N, Dascal N. Regulation of maximal open probability is a separable function of Ca(v)beta subunit in L-type Ca2+ channel, dependent on NH2 terminus of alpha1C (Ca(v)1.2alpha) J Gen Physiol. 2006;128:15–36. doi: 10.1085/jgp.200609485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei X, Neely A, Olcese R, Lang W, Stefani E, Birnbaumer L. Increase in Ca2+ channel expression by deletions at the amino terminus of the cardiac alpha 1C subunit. Receptors Channels. 1996;4:205–215. [PubMed] [Google Scholar]
- 36.Savignac M, Pintado B, Gutierrez-Adan A, Palczewska M, Mellstrom B, Naranjo JR. Transcriptional repressor DREAM regulates T-lymphocyte proliferation and cytokine gene expression. EMBO J. 2005;24:3555–3564. doi: 10.1038/sj.emboj.7600810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saada N, Dai B, Echetebu C, Sarna SK, Palade P. Smooth muscle uses another promoter to express primarily a form of human Cav1.2 L-type calcium channel different from the principal heart form. Biochemical and biophysical research communications. 2003;302:23–28. doi: 10.1016/s0006-291x(03)00097-4. [DOI] [PubMed] [Google Scholar]
- 38.Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, Schwarz TL, Demolombe S, Nerbonne JM. Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res. 2005;97:1342–1350. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- 39.Radicke S, Cotella D, Graf EM, Banse U, Jost N, Varro A, Tseng GN, Ravens U, Wettwer E. Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovascular research. 2006;71:695–703. doi: 10.1016/j.cardiores.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 40.Frank An W, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 41.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]