

Abstract
Objectives:
The cause of hypocretin cell loss in human narcolepsy–cataplexy is unknown but has been suggested to be neurodegenerative in nature. To test this hypothesis, we evaluated the remaining hypocretin cells in human narcolepsy brains for the presence of aggregated protein inclusions, gliosis, and inflammation.
Methods:
Brains were examined by routine histologic methods for potential comorbid neurodegenerative diseases and through immunohistochemical screening for protein inclusions in the hypothalamus. Hypothalamic sections of 4 subjects with narcolepsy and 5 nonneurologic controls were examined immunohistochemically with antibodies against ubiquitin (a marker of aggregated protein), allograft inflammatory factor 1 (AIF1, a microglial activation marker), glial fibrillary acidic protein (GFAP, a reactive astrocytic marker), and hypocretin. Hypothalami of subjects with narcolepsy were additionally examined for the presence of known components of protein aggregates (tau, α-synuclein, amyloid β, and TDP-43).
Results:
Hypocretin cells were markedly decreased in all 4 subjects with narcolepsy. Ubiquitinated inclusions were not observed in the total of 96 remaining hypocretin cells in these subjects. Further, we noted that even in patients with dementia neuropathology, the lateral hypothalamic hypocretin area was spared from ubiquitinated inclusions. AIF1 and GFAP staining in the perifornical area was unremarkable.
Conclusions:
Our findings suggest that hypocretin cell loss does not involve ubiquitinated inclusions, the hallmark of most neurodegenerative diseases. The lack of increased markers of inflammation also argues against a progressive and continuous neurodegenerative process.
GLOSSARY
- (AD)
= neuropathologically consistent with Alzheimer disease;
- AIFI
= allograft inflammatory factor 1;
- CERAD
= Consortium to Establish a Registry for Alzheimer's Disease;
- DAB
= diaminobenzidine;
- Fx
= fornix;
- GFAP
= glial fibrillary acidic protein;
- IgG
= immunoglobulin G;
- LBD
= Lewy body disease;
- NA
= not available;
- NFT
= neurofibrillary tangle;
- PBS
= phosphate-buffered saline;
- PLM
= periodic leg movement;
- PMI
= postmortem interval;
- PSG
= polysomnography;
- REML
= REM latency;
- SL
= sleep latency;
- SOREMP
= sleep onset REM period;
- 3V
= third ventricle.
Studies in animals and humans have shown that narcolepsy–cataplexy is caused by impaired hypocretin (also called orexin) neurotransmission.1,2 Impaired hypocretin transmission due to single hypocretin gene mutations is rare in humans.1 Rather, in most cases of narcolepsy with cataplexy, hypothalamic hypocretin cells are lost through an unknown and highly selective process that does not alter the closely intermingled melanin-concentrating hormone cell population.1,2 Speculation on the causes of this cell loss have included an autoimmune process (based on the tight HLA-DQB1*0602 association) or degeneration of hypocretin neurons.3 Repeated attempts to identify an autoimmune process have so far failed,4 suggesting that it is necessary to explore other potential mechanisms.
In neurodegenerative disorders, specific neuronal subtypes are often affected by a process associated with the accumulation of intracellular protein aggregates, but whether these aggregates cause cell death or protect against neurodegeneration remains unresolved. These aggregates generally contain ubiquitin, a “tag” that is added to proteins that are targeted for degradation.5 Recently, analysis of ubiquitinated protein revealed the novel TDP-43 protein as a cause of amyotrophic lateral sclerosis.6,7 Hypocretin cell numbers are decreased in several neurodegenerative disorders, including Parkinson disease,8,9 multiple system atrophy,10 and Huntington disease.11 In these diseases, protein inclusions were identified in small numbers of hypocretin cells,8 indicating that these cells can be affected by neurodegenerative processes. Ubiquitin-containing deposits have also been identified in the hypothalamus of patients with Niemann–Pick disease type C,12 a disorder known to cause secondary narcolepsy.
We hypothesized that hypocretin cell neurodegeneration occurs in narcolepsy, a process likely marked by the presence of protein aggregates. We examined rare remaining hypocretin cells of patients with narcolepsy for the presence of ubiquitinated inclusions, and potential astrocytic or microglial reactions, processes associated with active neurodegeneration. We also examined hypothalamic sections from 4 subjects with narcolepsy for the presence of known components of protein aggregates (tau, α-synuclein, amyloid β, and TDP-43).
METHODS
This study was approved by the ethical committees of the collaborative institutes. Written informed consent was obtained from all patients or families. We examined brain tissue from 4 patients with narcolepsy (50% men, aged 78 ± 8 years, 75% Japanese, postmortem interval [PMI] 9.6 ± 7.5 hours) and 5 nonneurologic controls (2 men and 3 women, without a clinical history of unexplained excessive daytime sleepiness, aged 77 ± 8 years, 60% Japanese, PMI 6.7 ± 2.0 hours). None of these subjects were used in previous neuropathologic studies.1,2,13 Demographic data are shown in the table. In most cases (7 of 9), brains were first studied by routine neuropathologic examination. Right brain hemispheres were fixed postmortem with 10% formalin, embedded in paraffin, and 8-μm sections from cortices, hippocampus, basal ganglia, midbrain, and pons cerebellum were prepared. These sections were stained using the hematoxylin and eosin, Klüver–Barrera, methenamine silver, and Gallyas–Braak methods. We used Consortium to Establish a Registry for Alzheimer's Disease (CERAD) scoring14 and Braak staging15 when Alzheimer neuropathology was noted.
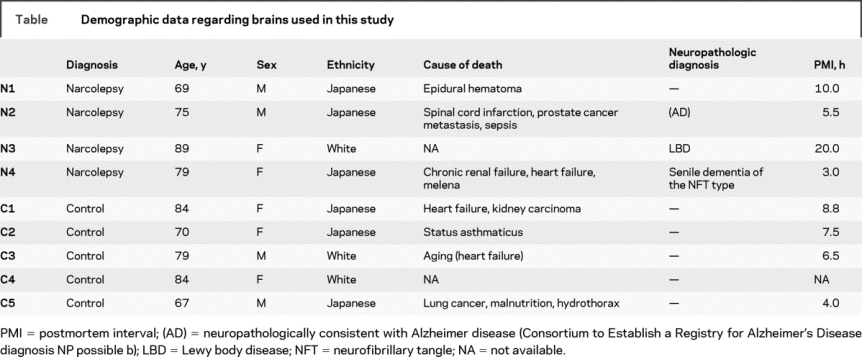
Table Demographic data regarding brains used in this study
Seven brain blocks containing hypothalamus (3 narcolepsy and 4 controls) were also obtained and fixed in 4% paraformaldehyde, cut into 40-μm cryosections and used for immunohistochemical staining. Hypothalamic structure orientation was determined using human atlas coordinates.16 Four stained sections (spaced at approximately 800 μm) covering the hypocretin cell–containing perifornical area, were selected. This area spans from the mammillary bodies (optic chiasm + 10 mm) to an area immediately proximal to where the fornix makes contact with the third ventricle (optic chiasm + 7 mm). Paraffin-embedded hypothalamic sections were used for immunostaining in the 2 subjects (1 narcolepsy and 1 control) whose hypothalamic tissue was unavailable in frozen sections. For these, representative 8-μm thin coronal sections (corresponding to atlas figure 28, optic chiasm + 8 mm) were selected for evaluation.
Cryosectioned brain samples were permeabilized with 0.3% Triton X-100 in phosphate-buffered saline (PBS) for 4 weeks. All subsequent steps were performed on free-floating sections at 4°C, interspersed with washes in PBS buffer (5 minutes 3 times). Sections were 1) treated with 0.6% H2O2 in PBS for 30 minutes; 2) blocked in 1.5% normal goat or horse serum in PBST (PBS + 0.25% Triton X-100) for 30 minutes; and 3) incubated in PBST for 24 hours with rabbit anti-ubiquitin polyclonal antibody (1:10,000, Z0458, Dako, Carpinteria, CA), rabbit anti-allograft inflammatory factor 1 (AIF1, also called ionized calcium binding adaptor molecule 1:Iba1) polyclonal antibody (1:5,000, 019-19741; Wako, Osaka, Japan), or rabbit anti-glial fibrillary acidic protein (GFAP) antibody (1:5,000, RB-087-A, Thermo, Fremont, CA) and a mouse anti-hypocretin monoclonal antibody (1:10,000 house made). Four narcolepsy hypothalamic sections were also stained with rabbit anti-phosphated tau polyclonal antibody (1:50,000, AP42217), rabbit anti–α-synuclein polyclonal antibody (1:5,000, 117518), rabbit anti–amyloid β polyclonal antibody (1:20,000, E5019), mouse anti–phosphated TDP-43 monoclonal antibody (1:5,000, pS409/41020), anti-ubiquitin antibody (polyclonal antibody Z0458 or mouse monoclonal anti-ubiquitin antibody (1:5,000, MAB1510 clone FK1, Millipore, Bellerica, MA), and anti–phosphated tau polyclonal antibody (AP422) as well as anti-hypocretin monoclonal antibody; 4) incubated with biotinylated goat anti-rabbit immunoglobulin G (IgG) or horse anti-mouse IgG antibody (1:200, Vector Laboratories, Burlingame, CA) in PBST for 1 hour; 5) incubated in ABC reagents (Vector Laboratories) in PBST for 1 hour; and 6) preincubated in 0.25 mg/mL diaminobenzidine (DAB) or DAB with 1% nickel chloride in PBST for 15 minutes followed by the addition of H2O2 as a final concentration of 0.02% for 15 minutes. Sections were then mounted and analyzed by microscopy, and an Olympus BX51 microscope (Olympus, Tokyo, Japan) equipped with charge-coupled device camera (DP70, Olympus) was used to take digital photographs of stained sections. Paraffin sections were first deparaffinized in xylene, hydrated, and stained on the slide according to the procedures described above.
RESULTS
Case reports.
Case N1 (a Japanese man) began working in a fish market at age 14 years, and recurrent daytime sleep episodes began at age 15 years. He slept frequently during work, unaware that he was affected by hypersomnia. His first cataplexy episode, at age 53 years, caused him to fall to the ground. Cataplexy occurred when he became excited or angry, and he resigned from work at age 60 years after dropping a special knife while cleaning a large fish with a fellow worker. His cataplexy continued to become aggravated, leading him to seek medical help, and he was diagnosed with narcolepsy at age 63 years. He rarely experienced hypnagogic hallucinations and sleep paralysis. His 1 nap polysomnography (PSG) at his initial visit showed shortened sleep latency (SL) and REM latency (REML) (2 minutes and 6 minutes), and indicated a sleep onset REM period (SOREMP). At age 66 years, he sustained a brain injury in a traffic accident, and MRI revealed a right frontal lesion. Subsequently, his unsteady walking led to frequent falls and injuries. He sometimes showed nocturnal occupational delirium and was hospitalized 3 times for the treatment of prolonged disoriented hallucinatory states. Nocturnal PSG at age 68 years showed lack of deep sleep but no signs of sleep apnea or periodic leg movements (PLMs). Cognitive function was not assessed, but memory disturbances were not noted when he was fully awake. He died of epidural hematoma, due to repeated head injury at age 69 years. Neuropathologic examination revealed small numbers of neurofibrillary tangles (NFTs) in the hippocampus (Braak stage I) and senile plaques in the temporal cortex (Braak stage A, CERAD age-related plaque score B). Sparse ubiquitin-positive threads were observed in the ventral part of the posterior hypothalamus (premammillary area) but not in the lateral hypothalamic hypocretin area. No ubiquitin-positive inclusions were found in the 16 remaining hypocretin cells. Additional screening of hypothalamic sections for known protein aggregates revealed 1 NFT and several neuropil threads in premammillary area. There were no amyloid β–positive senile plaques, α-synuclein–positive inclusions, Lewy neurites, TDP-43–positive inclusions, or dystrophic neurites in the hypothalamus of case N1.
Case N2 (a Japanese man) had a maternal family history of hypersomnia. Frequent napping episodes at school began at age 12 years, followed by the onset of terrifying hypnagogic hallucinations and sleep paralysis. He described the illusion of a robber entering his bedroom, sitting on his face, and preventing him from moving or screaming for help. His first episode of cataplexy was at age 18 years, and attacks occurred with laughter or excitement (such as winning at Mahjong, or when scolding children). He worked in his father's woodworking company, taking 2 naps on weekdays, and was diagnosed with narcolepsy at age 33 years. His initial 1 nap PSG showed shortened sleep and REM latencies (SL 2 minutes, REML 2 minutes), which were also consistently seen in follow-up 1 nap PSG studies later in his life. After age 70 years, he sometimes stood still or moved during the night while half asleep. His nocturnal PSG at age 72 years showed mild sleep disturbances with an apnea hypopnea index of 9.8, a PLM index of 5.8, and a 7.5-minute stage-1 REM period. At age 74 years, he underwent surgery for bile duct metastasis of testicular carcinoma followed by hormone therapy. He subsequently became anxious and entered a disoriented, agitated, and confusional state at night. Brain MRI at age 74 years revealed age-related mild brain atrophy, but cognitive function was not assessed. He died of spinal cord infarction and sepsis at age 76 years. Neuropathologic examination showed substantial NFT in the neocortex (Braak stage V) and senile plaques in precentral gyrus (Braak stage C, CERAD age-related plaque score C). Neuropathologic diagnosis for AD was CERAD NP possible b.14 In hypothalamic tissues, several ubiquitin-positive threads and abundant neuropil threads and NFT were observed, mainly in the ventral or medial parts of the posterior hypothalamus including the tuberomammillary nucleus, which is consistent with previous reports showing regional preference of NFT pathology in the hypothalamus.21 No ubiquitin-positive inclusions were found in the lateral hypothalamic area or in 53 remaining hypocretin cells. There were no amyloid β–positive senile plaques, α-synuclein–positive inclusions, Lewy neurites, TDP-43–positive inclusions, or dystrophic neurites in the hypothalamus of case N2.
Detailed clinical data of case N3 (a white woman) were not available. Her narcolepsy symptoms of excessive daytime sleepiness and typical cataplexy started in her 20s, and she was diagnosed with narcolepsy. Her nocturnal PSG and Multiple Sleep Latency Test results met International Classification of Sleep Disorders 1 narcolepsy criteria. She had comorbid dementia later in her life, and died of dementia at age 89 years. Immunohistochemical examination revealed many tau-positive NFTs and neuropil threads (Braak stage V) and moderate amounts of senile plaques in the neocortex (CERAD age-related plaque score B). Abundant α-synuclein–positive Lewy neurites and Lewy bodies were also found in the neocortex. The neuropathologic diagnosis was Lewy body disease. No ubiquitinated inclusions were found in the lateral hypothalamic area or in 7 remaining hypocretin cells. Several NFTs and neuropil threads and many Lewy neurites were observed in the ventral part of the posterior hypothalamus. There were no senile plaques, α-synuclein–positive inclusions, TDP-43–positive inclusions, or dystrophic neurites in the hypothalamus of case N3.
Case N4 (a Japanese woman) began to experience excessive daytime sleepiness at age 10 years. She fell asleep during walking and eating, awakening refreshed after 5-minute naps. Her first episode of cataplexy was at age 13 years, when she experienced sudden loss of muscle tone in her shoulders and knees after a sensation of swelling pride. Hypnagogic hallucinations and sleep paralysis began around the same time, and she described feeling as if she was being crushed by a dead body. She did not realize she was affected by hypersomnia, but was diagnosed with narcolepsy at age 55 years upon her first visit to a hospital. Her initial 1 nap PSG test showed shortened sleep and REM latencies with SOREMP (SL 3 minutes and REML 4 minutes). A resting finger tremor and memory disturbance started at age 72 years, preventing her from taking medicines appropriately. Occasional delirium at night began at age 73 years, and memory disturbances, delusions, and hallucinations in a disoriented state became progressively worse. Brain MRI at age 76 years showed marked brain atrophy in frontal and temporal lobes. Incontinence occurred at age 78 years, and she was restricted to her bed. She died of acute aggravation of chronic renal failure, heart failure, and disseminated intravascular coagulation at age 79 years. Upon neuropathologic examination, massive NFTs and neuropil threads were observed in the subiculum and entorhinal cortex, with some also in neocortex. A few senile plaques were observed selectively in the hippocampus but not in other cortical regions. Multiple small, old infarctions were found in basal ganglia. The neuropathologic diagnosis was senile dementia of the NFT type.22 No ubiquitin-positive inclusions were found in the lateral hypothalamic area or in 20 remaining hypocretin cells. A few ubiquitin-positive threads and several tau-positive neuropil threads were observed, mainly in the ventral part of the posterior hypothalamus at the premammillary level, and 1 NFT was found in the tuberomammillary nucleus, which is consistent with a previous report.21 There were no senile plaques, Lewy neuritis, α-synuclein–positive inclusions, TDP-43–positive inclusions, or dystrophic neurites in the hypothalamus of case N4.
Summary of clinical and neuropathologic findings.
All 4 patients had typical narcolepsy, showing cataplexy and SOREMPs, and all carried the HLA-DQB1*0602 allele (HLA typed at clinic or postmortem). Similar to previous reports, a dramatic (≥90%) hypocretin cell loss was observed in all 4 narcolepsy brains examined, indicating that our subjects all had typical narcolepsy with hypocretin deficiency (figure 1).

Figure 1 Ubiquitin and hypocretin immunoreactivity in hypothalamic tissue of narcolepsy and control subjects
Hypocretin cells (purple) were double stained with ubiquitin (brown) in control (A, C; case C1) and narcolepsy (B, D; case N2) hypothalamic tissues. Note that hypocretin cell were reduced in narcolepsy (B) vs control (A), and no ubiquitinated inclusions were observed in hypocretin cells in narcolepsy or control (C, D). Scale bar = 1 mm in A and B, 50 μm in C and D. Fx = fornix; 3V = third ventricle. Dashed rectangle indicates approximate area used for higher magnification.
Three of the 4 patients had comorbid dementia processes on neuropathologic examination. Many ubiquitin-positive inclusions were observed in the cortex of these subjects (figure 2), but no ubiquitinated inclusions were found in cytoplasm or nucleus of hypocretin cells in narcolepsy (regardless of comorbid dementia) (figure 1D) or in control subjects (figure 1C).

Figure 2 Ubiquitin-positive inclusions in the cortex of narcolepsy subjects with comorbid dementia
Ubiquitinated inclusions (arrows) were frequently observed in the cortex of narcolepsy patients with comorbid dementia (A; case N2; B: case N3). Scale bar = 50 μm.
Examination of hypothalamic sections for possible protein aggregates (tau, α-synuclein, amyloid β, and TDP-43) revealed that all narcolepsy cases had tau-positive NFTs and neuropil threads, showing regional preference, many in the ventral part of the posterior hypothalamus (figure 3C) but sparse in the perifornical hypocretin area (figure 3B). α-Synuclein–positive Lewy neurites were also observed (case N3). Double immunostaining for tau and ubiquitin, or for α-synuclein and ubiquitin revealed that part of these tau-positive or α-synuclein–positive structures were also positive for ubiquitin. No inclusions consisting of these components were observed in hypocretin cells of subjects with narcolepsy (figure 3B).

Figure 3 Preferential localization of tau-positive NFTs in hypothalamus of narcolepsy subject with comorbid dementia
Tau-positive threads and neurofibrillary tangles (NFTs) (brown) in hypothalamus were preferentially localized in ventral part (C) and sparse in perifornical hypocretin area (B) of narcolepsy with comorbid dementia (case N2). Only 1 NFT (arrow) was found near remaining hypocretin cell (purple; arrowhead). Note that the hypocretin cell had no tau-positive inclusions (B). Scale bar = 1 mm in A, 100 μm in B and C. Fx = fornix; 3V = third ventricle.
No differences between narcolepsy and control subjects were observed for glial response markers AIF1 (figure 4, A–D) and GFAP (figure 4, E–H) in the perifornical hypothalamic area.
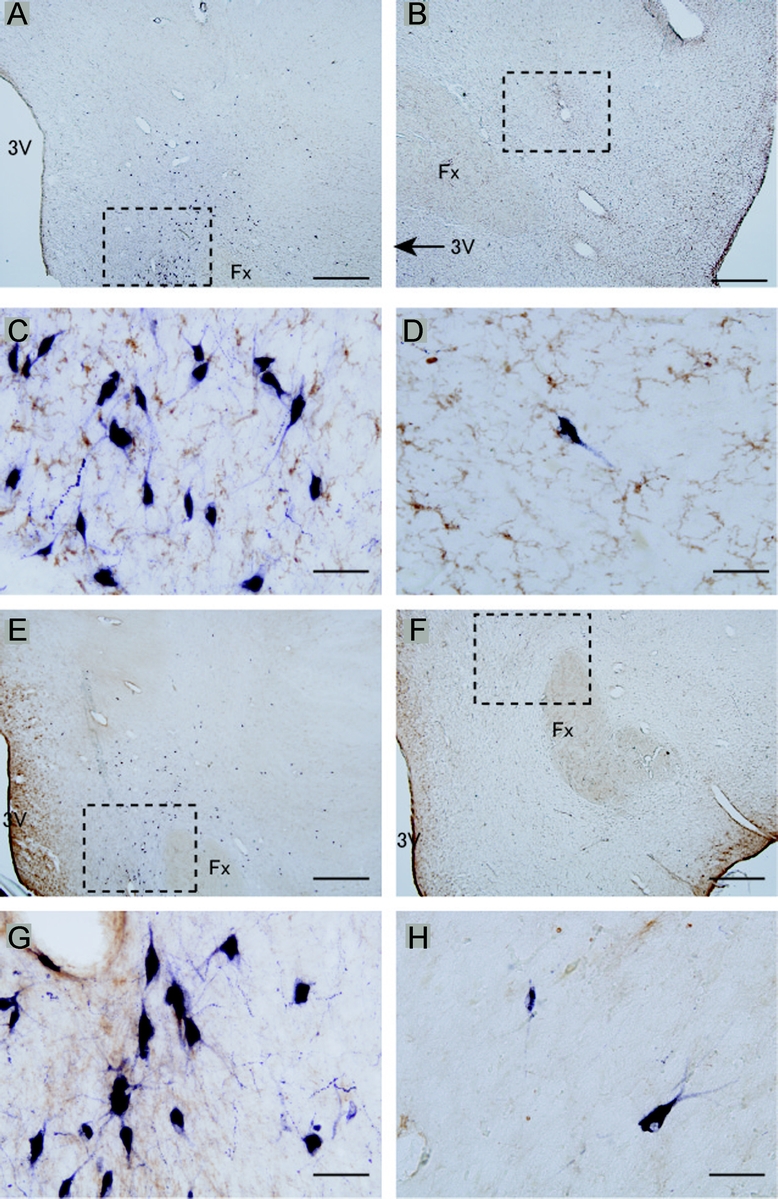
Figure 4 No increased inflammation markers in hypothalamic hypocretin area of patients with narcolepsy
A microglial activation marker allograft inflammatory factor 1 staining (brown; A–D) and a reactive astrocytic marker glial fibrillary acidic protein staining (brown; E–H) in lateral hypothalamic area were stained with hypocretin (purple). No significant differences between control (A, C, E, G; case C1) and narcolepsy (B, D, F, H; case N2) were observed. Scale bar = 1 mm in A, B, E, and F; 50 μm in C, D, G, and H. Fx = fornix; 3V = third ventricle.
DISCUSSION
The presence of ubiquitinated protein aggregates is a hallmark of neurodegenerative diseases.23 In this study, we used ubiquitin immunoreactivity as a marker to identify putative narcolepsy-specific protein aggregates. Because narcolepsy is associated with nearly complete disappearance of hypocretin cells, 4 narcolepsy brains were needed to study 96 hypocretin cells in detail. Ubiquitinated inclusions were not found in any of these cells (figure 1). Interestingly, ubiquitinated inclusions were not found in hypocretin cells in 3 of 4 narcolepsy patients with comorbid dementia. The few ubiquitin-positive granular or dot-like structures observed (figure 1) may be associated with aging, as previously reported.5 Additional screening of known protein aggregates showed that tau-positive NFTs were observed mainly in ventral part of posterior hypothalamus and scarcely in the perifornical hypocretin area (figure 3), confirming a previous report describing NFT localization.21 Again, no inclusions in hypocretin neurons were observed.
Our findings do not support a role for known neurodegenerative processes accompanied by ubiquitinated inclusions in the pathogenesis of narcolepsy, but it is possible, if unlikely, that the remaining cells have somehow escaped such a process. Indeed, protein aggregation is suggested to be a protective cellular response23 rather than a cause of degeneration. In this context, the failure to form ubiquitinated inclusions could reflect an increased vulnerability of hypocretin cells to yet unknown pathologic process. Nevertheless, the majority of hypocretin cells lost in narcolepsy could not involve ubiquitinated inclusions demonstrated by few ubiquitinated inclusions in the perifornical hypocretin area.
Our study of AIF1 and GFAP immunoreactivity in the perifornical area also found no active inflammation (microglial response) or residual gliosis (astrocytic fibrous network), extending our preliminary findings in a small number of subjects using HLA-DR and GFAP.1 These findings are compatible with the lack of ubiquitinated protein aggregation in remaining hypocretin cells, which would have caused inflammation along with progressive neurodegeneration. These findings are also compatible with the nonprogressive clinical course of most narcolepsy cases, i.e., rapid onset and stable symptomatology. Controversy exists regarding the increase of GFAP staining in the hypothalamus of subjects with narcolepsy.1,2 In this study, we observed no increase in GFAP staining even in narcolepsy patients with comorbid dementia. Astrocytic activation with reactive GFAP increase is a nonspecific finding associated with multiple forms of neuronal injury, including subclinical cases. Our results suggest that previous reports of increased GFAP may reflect interindividual variance rather than narcolepsy neuropathology.
We cannot completely exclude the possibility of apoptotic hypocretin cell death triggered by a neurodegenerative process at narcolepsy onset, with no residual abnormal protein deposits or traces of inflammation at the time of death. Further studies are required to explore the possibility of apoptotic hypocretin cell loss in narcolepsy.
ACKNOWLEDGMENT
The authors thank Ms. Mali Einen at Stanford University for clinical coordination; the Stanford Brain Bank for providing brain samples; Drs. Shuji Iritani, Kazuhiro Niizato, and Ken-ichi Oshima at the Tokyo Metropolitan Matsuzawa Hospital for conducting autopsies; and Dr. Juliette Faraco for critical reading and editing.
DISCLOSURE
Dr. M. Honda receives research support from the Ministry of Education, Science and Culture of Japan [Grants-in-Aid for Scientific Research: 17390324 (PI) and 19390310 (PI); Priority Areas “Comprehensive Genomics” (Collaborative researcher)]. Dr. Arai reports no disclosures. Dr. Fukazawa reports no disclosures. Dr. Y. Honda receives research support from the Ministry of Education, Science and Culture of Japan [Grant-in-Aid for Scientific Research: 18603013 (PI)]. Dr. Tsuchiya reports no disclosures. Dr. Salehi has applied for a patent at Stanford's Office of Technology Transfer related to the treatment of Down syndrome and receives research support from the Alzheimer Association Thrasher Foundation. Dr. Akiyama reports no disclosures. Dr. Mignot receives research support from the NIH [23724 (PI), MH080957 (PI), and NS062798 (PI)]. He is also funded by the Howard Hughes Medical Institute and has received research gifts from Respironics, Cephalon, Resmed, Elli Lilly, and Jazz Pharmaceuticals.
Address correspondence and reprint requests to Dr. Makoto Honda, Sleep Disorders Research Project, Tokyo Institute of Psychiatry, 2-1-8 Kamikitazawa, Setagaya-ku, Tokyo 156-8585, Japan honda@prit.go.jp.
Disclosure: Author disclosures are provided at the end of the article.
Received January 27, 2009. Accepted in final form May 1, 2009.
REFERENCES
- 1.Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 2000;6:991–997. [DOI] [PubMed] [Google Scholar]
- 2.Thannickal TC, Moore RY, Nienhuis R, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000;27:469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van den Pol AN. Narcolepsy: a neurodegenerative disease of the hypocretin system? Neuron 2000;27:415–418. [DOI] [PubMed] [Google Scholar]
- 4.Scammell TE. The frustrating and mostly fruitless search for an autoimmune cause of narcolepsy. Sleep 2006;29:601–602. [DOI] [PubMed] [Google Scholar]
- 5.Gray DA, Tsirigotis M, Woulfe J. Ubiquitin, proteasomes, and the aging brain. Sci Aging Knowledge Environ 2003;2003:RE6. [DOI] [PubMed] [Google Scholar]
- 6.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 7.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006;351:602–611. [DOI] [PubMed] [Google Scholar]
- 8.Fronczek R, Overeem S, Lee SY, et al. Hypocretin (orexin) loss in Parkinson's disease. Brain 2007;130:1577–1585. [DOI] [PubMed] [Google Scholar]
- 9.Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) cell loss in Parkinson's disease. Brain 2007;130:1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benarroch EE, Schmeichel AM, Sandroni P, Low PA, Parisi JE. Involvement of hypocretin neurons in multiple system atrophy. Acta Neuropathol 2007;113:75–80. [DOI] [PubMed] [Google Scholar]
- 11.Petersen A, Gil J, Maat-Schieman ML, et al. Orexin loss in Huntington's disease. Hum Mol Genet 2005;14:39–47. [DOI] [PubMed] [Google Scholar]
- 12.Love S, Bridges LR, Case CP. Neurofibrillary tangles in Niemann-Pick disease type C. Brain 1995;118(pt 1):119–129. [DOI] [PubMed] [Google Scholar]
- 13.Crocker A, Espana RA, Papadopoulou M, et al. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology 2005;65:1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD), part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 15.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 16.Mai JK, Assheuer J, Paxinos G. Atlas of the Human Brain. San Diego: Academic Press; 1997. [Google Scholar]
- 17.Hasegawa M, Jakes R, Crowther RA, Lee VM, Ihara Y, Goedert M. Characterization of mAb AP422, a novel phosphorylation-dependent monoclonal antibody against tau protein. FEBS Lett 1996;384:25–30. [DOI] [PubMed] [Google Scholar]
- 18.Obi K, Akiyama H, Kondo H, et al. Relationship of phosphorylated alpha-synuclein and tau accumulation to Abeta deposition in the cerebral cortex of dementia with Lewy bodies. Exp Neurol 2008;210:409–420. [DOI] [PubMed] [Google Scholar]
- 19.Kametani F, Tanaka K, Ishii T, Ikeda S, Kennedy HE, Allsop D. Secretory form of Alzheimer amyloid precursor protein 695 in human brain lacks beta/A4 amyloid immunoreactivity. Biochem Biophys Res Commun 1993;191:392–398. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa M, Arai T, Nonaka T, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 2008;64:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saper CB, German DC. Hypothalamic pathology in Alzheimer's disease. Neurosci Lett 1987;74:364–370. [DOI] [PubMed] [Google Scholar]
- 22.Yamada M, Itoh Y, Otomo E, Suematsu N, Masushita M. Dementia of the Alzheimer type and related dementias in the aged: DAT subgroups and senile dementia of the neurofibrillary tangle type. Neuropathology 1996;16:89–98. [Google Scholar]
- 23.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med 2004;10(suppl):S10–S17. [DOI] [PubMed] [Google Scholar]