

Abstract
Enzyme pro-drug suicide gene therapy has been hindered by inefficient viral delivery and gene transduction. To further explore the potential of this approach, we have developed AdIU1, a prostate-restricted replicative adenovirus (PRRA) armed with the herpes simplex virus thymidine kinase (HSV-TK). In our previous Ad-OC-TK/ACV phase I clinical trial, we demonstrated safety and proof of principle with a tissue-specific promoter-based TK/pro-drug therapy using a replication-defective adenovirus for the treatment of prostate cancer metastases. In this study, we aimed to inhibit the growth of androgen-independent (AI), PSA/PSMA-positive prostate cancer cells by AdIU1. In vitro the viability of an AI- PSA/PSMA-expressing prostate cancer cell line, CWR22rv, was significantly inhibited by treatment with AdIU1 plus GCV (10 μg ml-1), compared with AdIU1 treatment alone and also cytotoxicity was observed following treatment with AdIU1 plus GCV only in PSA/PSMA-positive CWR22rv and C4-2 cells, but not in the PSA/PSMA-negative cell line, DU-145. In vivo assessment of AdIU1 plus GCV treatment revealed a stronger therapeutic effect against CWR22rv tumors in nude mice than treatment with AdIU1 alone, AdE4PSESE1a alone or in combination with GCV. Our results demonstrate the therapeutic potential of specific-oncolysis and suicide gene therapy for AI-PSA/PSMA-positive prostate cancer gene therapy.
Keywords: suicide gene therapy, M6 prostate-specific promoter, HSV-TK, prostate cancer
Introduction
Prostate cancer is still the leading cancer diagnosed in American men. The incidence of prostate cancer is age-dependent and has steadily increased over the last several decades.1 Localized prostate cancer can be managed effectively with surgery or radiation, whereas advanced and metastatic disease eventually progresses to an androgen-independent (AI) state with limited treatment options. The aging population of men with an increase in prostate cancer incidence combined with an absence of successful therapies for advanced disease require the development of novel therapies.
Suicide gene therapy is an attractive approach to increasing drug selectivity toward cancer cells. Tumor-specific suicide gene therapy using a tissue-specific promoter is a rational treatment strategy for prostate cancer.2-6 Herpes simplex virus thymidine kinase (HSV-TK)-based suicide gene therapy has been used to target prostate cancer for over a decade.7-10 The pro-drug, ganciclovir (GCV) is phosphorylated by HSV-TK to its monophosphate form, which is rapidly converted to di- and triphosphate forms by cellular kinases, the latter of which is toxic to cells. The GCV triphosphate is incorporated into DNA during cell division, causing single-strand DNA breaks and inhibition of DNA polymerase10-12 and causes DNA chain termination, which leads to programmed cell death.
Tumor-specific oncolytic adenoviruses have been effective and safe treatment options for patients with metastatic disease. Several studies have demonstrated the importance of tissue-specific vectors, revealing systemic toxicity with the administration of high doses of nonspecific vectors. Through the use of prostate-specific promoters and enhancers, the expression of a therapeutic gene or adenoviral replication factor can be limited to cells that contain the appropriate activators and transcription factors. Currently, kallikrein 2, PSA, rat probasin and osteocalcin (OC) are each under extensive investigation as regulators of prostate-restricted replication adenoviruses.13-18 We have demonstrated that both the prostate-specific antigen (PSA) and OC promoters could transcriptionally regulate the HSV-TK gene in a prostate-specific manner both in vitro and in vivo.19 This tissue-specific HSV-TK production combined with pro-drug administration inhibited the growth of androgen-independent (AI)-PSA-producing cells in vitro, in animal models of human prostate cancer and in patients with prostate cancer enrolled in a phase I clinical trial of OC promoter-based HSV-TK gene therapy.19,20 Others have demonstrated similar in vitro and in vivo efficacy as well as safe administration to men with locally advanced and metastatic prostate cancer.21,22 More recently, Freytag et al. 23-25demonstrated the safety and efficacy of a conditionally replicating, non-tissue-specific adenovirus containing the suicide genes TK and CD when combined with external beam radiation therapy. Although Ad5-CD/TKrep/pro-drugs/radiation therapy demonstrated a promising result in locally recurrent prostate cancer, the expression of the CD/TK fusion gene under the control of strong universal CMV promoter also showed lack of tissue specificity, which severely impairs the safety of this virus. Besides, Ad5-CD/TKrep is based on the E1B 55k deleted virus, del520/ONXY-015. Recent studies indicated that the replication of del520 virus is p53-independent.26-29 In addition, del520 exhibits substantial replication in certain normal cells. These complications will potentially limit the use of Ad-CD/TKrep. Nevertheless, Ad5-CD/TKrep warrants further development as a suicide gene therapy vector.
We recently developed a prostate-specific chimeric enhancer, PSES, by combining enhancers from PSA and PSMA genes. PSA and prostate-specific membrane antigen (PSMA) are prostate-specific biomarkers expressed by the majority of prostate tumors and non-cancerous epithelium. The main prostate-specific enhancer activity of the PSA enhancer core is located in a 189 bp region called AREc3, and the main prostate-specific enhancer activity of the PSMA enhancer core is located in a 331 bp region called PSME(del2). The combination of these two regulatory elements, AREc3 and PSME(del2), called prostate-specific enhancing sequence (PSES), showed high activity specific to PSA/PSMA-positive prostate cancer cells, regardless of androgen status.30 This PSES promoter has been used to control the replication of a PRRA, which demonstrated prostate-specific replication and therapeutic efficacy both in vitro and in vivo.31 We also made a shorter form by deleting the L2 and L5 regions in AREc3 and replacing the 90 bp proximal region of PSME with a simple AP-3 binding site. This version of PSES was called M6. For this study, we developed a novel HSV-TK-armed tissue-specific replicative adenovirus, AdIU1, using the PSES promoter to drive the expression of adenoviral E1a and E4 and the M6 promoter to drive the expression of adenoviral E1b and HSV-TK. AdIU1 demonstrated selective cytotoxicity toward AI PSA/PSMA-expressing prostate cancer both in vitro and in vivo.
Materials and methods
Cells and cell culture
HER 911E4 cells, derived from adenoviral E1 (bp 79-5789)-immortalized HER 911 (human embryonic retinoblastoma) cells, express adenoviral E4 proteins under control of the tetR promoter. HER 911E4 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), hygromycin B (0.1 mg ml-1; Calbiochem, San Diego, CA) and doxycycline (2 μg ml-1; Sigma, St Louis, MO). AI, androgen receptor (AR) and PSA/PSMA-positive prostate cancer cell lines C4-2 and CWR22rv, and AI, AR and PSA-negative cell lines DU-145 and PC3 were cultured in RPMI 1645 supplemented with 10% FBS and 1% P/S. Human renal cell carcinoma RCC29, human testicular cancer cell line Tera-1 and human colon cancer cell line HT-29 were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and 1% P/S. The breast cancer cell line MCF 7 was cultured in MEM with 10% FBS and 1% P/S with 1mm sodium pyruvate. HER 293 cells were cultured in MEM with 10% FBS and 1% P/S and 0.1 mm non-essential amino acids. The cells were maintained at 37 °C in a 5% CO2 incubator.
Luciferase assay
The tissue-specific activity of M6 was investigated by transient transfection for luciferase assay. A certain number of cells (2 × 105 cells per well) were plated for 24 h. Plasmid DNA, pGL3/M6/TATA and pGL3/TATA were delivered into the cells with DOTAP (Roche, Indianapolis, IN) following the manufacturer’s protocol. DNA (0.5-1 μg) was mixed with lipid at room temperature before addition to a well containing 1 ml of serum-free and phenol red-free RPMI 1640 medium. After 15 min, DNA-lipid complexes were added to the well and incubated for 5 h at 5% CO2 and 37 °C. DNA-lipidcontaining medium was then replaced with 1 ml culture medium. After 2 days, the cells were collected and lysed in 250 μl passive lysis buffer (Promega, Madison, WI). Cell lysates were vortexed for a few seconds and spun for 3 min. A supernatant (10 μl) was mixed with 50 μl of luciferase substrate (Promega, Madison, WI) and measured with a femtometer (Zylux, Germany). The luciferase activity was determined by being divided by the basal activity represented by transfection of pGL3/TATA.
Construction of the prostate-restricted replicative adenovirus AdIU1
The construction of the backbone for AdE4PSESE4 was described earlier.31 To construct AdIU1, the CMV-EGFP expression cassette in AdE4PSESE1a was replaced by an M6-HSV-TK expression cassette. HSV-TK and E1b in the left arm was under the transcriptional control of M6 and E4 and E1a in the right arm was controlled by a PSES promoter. Figure 1 illustrates the structure of each virus used in this study. The adenoviral genome was released from the cloning vector by digestion with Pac I restriction enzyme and transfected into HER 911E4 cells using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA). The plate was incubated at 37 °C under 5% CO2 for 7-10 days after transfection until a cytopathic effect was observed. AdIU1 was further amplified in HER 911E4 cells. The recombinant adenoviruses were purified by CsCl gradient centrifugation. All gradient-purified viral stocks were dialyzed in a dialysis buffer (1 mm MgCl2,10mm Tris-HCl (pH 7.5) and 10% glycerol) for 24 h at 4 °C, with three buffer changes. Aliquots of purified virus were stored at -70 °C. The viral titer was determined by Adeno-XTM Rapid Titer system (BD Biosciences, Palo Alto, CA) following the manufacturer’s protocol. Briefly, a dilution of the viral stock in question was used to infect HER 911 E4 cells. Forty-eight hours later, these cells were fixed and stained with antibody specific to the adenovirus hexon protein. A signal was detected after a secondary antibody conjugated with horseradish peroxidase amplified the signal of the anti-hexon antibody. Subsequent exposure to a metal-enhanced diaminobenzidine substrate turned the infected cells dark brown. Then the titer of the stock in question could be determined by counting the number of brown cells in a given area. Each stained cell corresponded to a signal infectious unit (IFU).
Figure 1.

(a) Sequences of the enhancer core of the PSA gene, AREc3, located in the 4.3 kb upstream of PSA promoter. Sequence analysis of AREc3 revealed six putative GATA transcriptional factor-binding sites besides the reported three androgen-response elements. (b) Sequences of PSME located in the third intron in the PSMA gene (FOLH1). PSME is characterized by the repeat sequence (marked by underline and bold). Several potential transcription factor-binding sites, such as AP-1, AP-3 and SRY/SOX, are indicated. (c) pGL3/m6/TATA (0.5 μg) was transfected into various cell lines (2 × 105 cells for each). After 2 days, cells were harvested, lysed with passive lysis buffer and analyzed for luciferase activity. This experiment was conducted in the absence of androgen. Luciferase activity was determined by being divided by the basal activity represented by transfection of pGL3/TATA. pGL3/m6/TATA is active only in PSA/PSMA-positive LNCaP and C4-2 cells.
Viral replication assay
CWR22rv, C4-2, PC-3 and DU-145 cells were seeded in 6-well plates (1 × 106 cells per well) 1 day before to viral infection and subsequently infected with AdIU1 or AdE4PSESE1a (2 IFU per cell). The media were changed 24 h after infection, and the viral supernatants were harvested 3 days after infection. The cells were examined under light microscopy daily for up to 5 days. Then the titers of the harvested viral supernatants were determined by titer assay. HER 911E4 cells were seeded in 96-well plates (5 × 103 cells per well) 1 day before infection. The cells were infected with serial volume dilutions of the harvested supernatants, ranging from 1 to 10-11 μl per well, with each row of 8 wells receiving the same dose of virus. The media were changed on day 4, and the cells were examined under the microscope on day 7. The dose of the produced viruses was represented as an LD50 value, the dilution factor that caused a cytopathic effect in at least 4 wells of cells in a single row on a 96-well plate by day 7.
Western blot analyses for E1a and HSV-TK expression
To detect the tissue-specific expression of Ad5E1a and HSV-TK, 1 × 106 cells in 60 mm dishes were infected with AdIU1. Each cell line was infected with standardized doses of virus.31 Cells were harvested and lysed in 100 μl of cell lysis buffer (50 mm Tris-HCl (pH 7.4), 1% NP-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm PMSF, 1 μg ml-1 Aprotinin, 1 μg ml-1 leupeptin, 1 μg ml-1 pepstatin, 1 mm Na3VO4 and 1 mm NaF) 48 h post-viral infection. Lysates were centrifuged at 14 000 r.p.m. for 20 min and the supernatants were collected. Protein concentration was estimated by dye binding assay (Bio-Rad, Hercules, CA). Protein (25 μg) was loaded onto a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The proteins were transferred to polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA) and the membrane was probed with antibodies reactive to Ad5E1a protein (BD Bioscience) or TK polyclonal serum (provided by M Black, Department of Pharmaceutical Sciences, Washington State University, Pullman, WA). and visualized by an enhanced chemiluminescence kit (Amersham Life Science, Piscataway, NJ).
Dose-dependent in vitro cell-killing assay
CWR22rv and DU145 cells were seeded onto 24-well plates at a density of 1.5 × 105 or 1 × 104 cells per well respectively. After 24 h, the cells were infected with 0.0002-2 IFU per cell of AdIU1 or AdE4PSESE1a. Twenty-four hours after infection, the media were removed and replaced by fresh media with or without GCV (10 μg ml-1). Media with or without GCV were changed every 2 days. Viable cells were analyzed by crystal violet assay 7 days post-infection.
Time-dependent in vitro killing assay
CWR22rv and DU-145 cells were plated in 24-well plates. Cells were divided into four treatment groups, no treatment, AdIU1 (0.2 IFU per cell), GCV and AdIU1 (0.2 IFU per cell) plus GCV. The media were changed 24 h after infection, and GCV (10 μg ml-1) was added 24 h after media change. Cell viability was analyzed on days 1, 3, 5 and 7 by crystal violet assay.
In vivo evaluation of AdIU1 therapy
All animal methods and procedures were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee (IACUC). CWR22rv xenografts were established by injecting 2 × 106 cells subcutaneous (s.c.) in the flanks of 6-week-old male, athymic nude mice. The injected mice were castrated 3 days after cell injection. Mice with similar tumor sizes (3-5 mm) were divided into four groups receiving, AdE4PSESE1a (control PRRA), AdE4PSESE1a plus GCV, AdIU1, or AdIU1 plus GCV treatment. Virus particles (2 × 109) of either AdE4PSESE1a or AdIU1 in 100 μl PBS were injected intratumorally. Five days after virus injection, GCV (40 mg kg-1 body weight) was administered intraperitoneally two times daily for 10 days. Tumor sizes were measured every 5 days, and the following formula was applied to calculate tumor volume length × width2 × 0.5236. Mice were killed and tumors harvested for histological examination 30 days after injection.
Histology and immunohistochemistry
Tumors were harvested, immediately fixed in formalin and embedded in paraffin. The tissue sections were stained with hematoxylin and eosin according to the standard protocol. For immunohistochemistry, tumor sections were deparaffinized, rehydrated and heated in a microwave oven for 20 min in activity antigen retrieval solution (10 mm citric buffer, pH 6.0). Endogenous peroxidase was inactivated with 3% hydrogen peroxide solution. The slides were rinsed with distilled water, washed two times with PBS for 3 min and incubated with Superblock (Scytek Laboratories, Burlingame, CA) in a humidified chamber for 1 h at room temperature. After rinsing with PBS, the slides were incubated with avidin (Vector Laboratories, Inc., Burlingame, CA) for 15 min, washed with PBS and blocked with biotin in a humidified chamber for 15 min at room temperature. A monoclonal mouse antibody to adenovirus type 5 (Abcam, Cambridge, MA) was applied. The slides were incubated with primary antibodies overnight in humidified chambers at 4 °C. After PBS rinse, a biotinylated secondary antibody was applied to the slides and incubated for 1 h. After washing with PBS, slides were incubated with avidin-peroxidase complex reagent (Vector Laboratories, Inc., Burlingame, CA, USA) for 30 min, washed once with PBS, stained with freshly prepared diaminobenzidine solution for 15 min and counterstained with hematoxylin.
In situ terminal deoxynucleotide transferase-mediated nick end labeling assay
The in situ apoptosis detection kit was purchased from Roche Diagnostics. Tumor tissue sections were deparafinized using a sequential xylene protocol and rehydrated through gradients of ethanol and distilled water. Slides were treated with 10 nmol l-1 Tris solution containing 1 μg ml-1 proteinase K for 15 min. All slides were rinsed with PBS and incubated with 100 μl terminal deoxynucleotidyl transfererase-mediated nick end labeling (TUNEL) reaction mixture (or 100 μl control labeling solution for negative control) in a humid chamber at 37 °C for 30 min. The slides were washed three times with PBS and incubated with 100 μl TUNEL POD solution in a humid chamber at 37 °C for 30 min. After washing with PBS, the slides were stained with freshly prepared diaminobenzidine solution for 10 min, rinsed with PBS and counterstained with hematoxylin.
Results
Construction of a shorter form of PSES, M6
We initially constructed an HSV-TK armed PRRA by replacing the CMV-GFP expression cassette from AdE4PSESE1a31 with a PSES-HSV-TK expression cassette. However, the virus did not propagate well in 911E4 cells, limiting its clinical utility (Ahn M et al., unpublished data). We believe that the problem was the packaging size limitation of the adenovirus. We modified the PSES sequence by deleting the non-functioning sequences, thereby shortening the total insert size.30 Our linker mutation study indicated that L2 and L5 likely did not have a function (Figure 1a). In addition, the activity of a 90-bp upstream region in AREc3 of PSME del2 was mediated by AP-3 (Figure 1b).32 Utilizing this information, we decided to make a shorter form of PSES, called M6, by deleting the L2 and L5 sequence of AREc3 and replacing the 90-bp upstream sequence of PSME del2 with a simple AP-3 binding sequence. Figure 1c demonstrates that M6 retained tissue-specific activity in PSA and PSMA-positive cells. Replacing the PSES sequence with M6 sequence resulted in a 106 bp decrease and normal propagation in 911E4.
Construction of a TK-armed PRRA
AdIU1 was constructed by replacing the CMV-GFP expression cassette in AdE4PSESE1a31 with a M6-HSV-TK expression cassette to extend the therapeutic potential of the PSES-based PRRA (Figure 2). HER 911E4 cells were transfected with recombinant adenoviral cosmid linearized by Pac I restriction enzyme digestion, and AdIU1 was propagated in HER 911 E4 cells as described in Materials and methods. CsCl-purified AdIU1 was titered with the Adeno-XTM rapid titer kit and expressed as infectious units (IFU). To assess the prostate specificity and viral replication efficiency of AdIU1, an in vitro viral replication assay was performed. PSA/PSMA-positive and -negative cells were infected with a dose of AdIU1 different from adenovirus’s similar infectivity.31 AdIU1 replicated as efficiently as AdE4PSESE1a in PSA/PSMA-positive C4-2 and CWR22rv cells (Table 1). The replication was diminished three- to fourfold in PSA/PSMA-negative PC-3 and DU-145. Demonstrating the fact that AdIU1 replication was tightly controlled by PSES and restricted to PSA/PSMA-positive cells.
Figure 2.

Schematic illustration of AdIU1. AdIU1 was constructed by placing adenoviral E1a and E4 genes under the control of PSES to direct adenovirus replication, and HSV-TK gene, a pro-drug enzyme gene, under the control of m6 enhancer to maximize cell-killing activity through a bystander effect.
Table 1.
Tissue/tumor-specific replication ability of AdIU1a
| Cell lines | Input dosesb (IFU) |
Output viral dosesc (LD50)d |
|
|---|---|---|---|
| AdlU1 | AdE4PSESE1a | ||
| C 4-2 | 6.6 × 104 | 106 | 106 |
| CWR22rv | 2 × 104 | 106 | 106 |
| PC 3 | 2.3 × 105 | 102 | 102 |
| DU-145 | 1.6 × 105 | 5 × 102 | 5 × 102 |
Cells were seeded and infected with AdE4PSESE1a or AdIU-1, and the supernatants were harvested for titer assay as described in ‘Materials and methods’.
Input viral doses mean the virus doses used to infect cells (IFU).
Output viral doses mean the virus doses produced and used for LD50 assay.
The virus production was expressed as an LD50 value (thedilution factor that caused a CPE in at least 4 wells of cells in a row of 8 wells on a 96-well plate on day 7).
Western blotting analysis of adenovirus E1a and herpes simplex virus thymidine kinase proteins expression
Androgen-independent, PSA/PSMA-positive CWR22rv and C4-2, as well as AI, PSA/PSMA-negative DU-145 and PC-3 were infected with standardized doses of AdIU1. Forty-eight hours after AdIU1 infection, cell lysates were collected and western blot was performed using monoclonal antibodies against Ad5E1a or polyclonal HSV1-TK antiserum. Protein expression of both Ad5E1a and HSV1-TK was detected following AdIU1-infected, PSA/PSMA-positive CWR22rv and C4-2 cells. On the other hand, the expression of Ad5E1a and HSV1-TK proteins, in AdIU1 infected PSA/PSMA-negative PC-3 and DU-145 cells were low or undetectable (Figure 3). This result indicated that PSES and M6 promoter retained its prostate specificity to mediated E1a and HSV-TK expression in AI, PSA/PSMA-positive cells.
Figure 3.
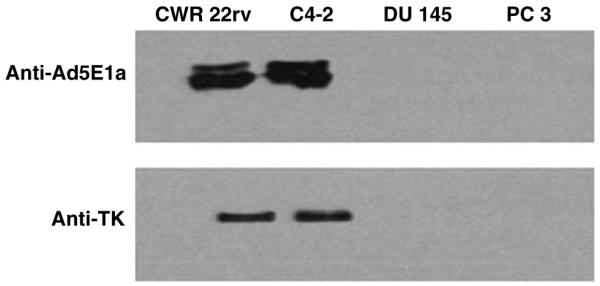
Expression of Ad5 E1a and HSV1-TK proteins by AdIU1 was evaluated in different cell lines. CWR22rv, C 4-2, PC-3 and DU-145 were infected with AdIU1. Forty-eight hours after viral infection, cell lysates were collected and western blot was performed. A large amount of E1a proteins ranging in size from approximately 35-46 kDa and HSV-TK (40-50 kDa) were detected in AdIU1-infected CWR22rv and C 4-2 cells. AdIU1-infected DU-145 and PC-3 did not express a detectable amount of E1a and HSV-TK proteins.
Selective cell killing activity of AdIU1 plus GCV against AI, PSA/PSMA-positive human prostate cancer cells in vitro
An in vitro GCV sensitivity assay was performed. Each cell line, CWR22rv, C4-2 and DU-145 were seeded in triplicate in 24-well plates at a density of 1 × 105 or 2 × 104 cells per well and were incubated with increasing concentrations of GCV (0-100 μg ml-1). Cell viability was determined after 5 days using crystal violet assay, and a corresponding IC50 dose was determined for each cell line (data not shown). An optimal non-toxic GCV treatment dose was determined to be 10 μg ml-1.
To evaluate the selective cytotoxicity of AdIU1 and AdE4PSESE1a viruses, we infected each cell line with wide dose ranges (0.0002-2 IFU per cell) of virus, and then treated infected cells with or without GCV (10 μg ml-1) (Figure 4a). The growth of AI, PSA/PSMA-positive human prostate cancer cell line, CWR22rv was significantly inhibited by 0.0002 IFU of AdIU1 in the presence of GCV. AdIU1 without GCV had similar killing activity as AdE4PSESE1a either in the presence or absence of GCV. The growth of the AI, AR and PSA/PSMA-negative cell line, DU-145 was unaffected by either virus with or without GCV. In a time course experiment, CWR22rv and DU-145 cells were seeded in -24-well plates. The 24 wells were divided into four groups, no treatment, AdIU1, GCV alone and AdIU1 plus GCV treatment. The GCV alone group demonstrated limited cytotoxicity confirming that GCV (10 μg ml-1)was not toxic to either of the prostate cancer cell lines. The CWR22rv cell line demonstrated cell growth inhibition at day 7 after AdIU1 exposure. The killing activity was significantly enhanced when GCV was administered following AdIU1 infection (Figure 4b). The DU-145 cell line demonstrated limited cytotoxicity in all four treatment regimens (Figure 4c).
Figure 4.

Dose- or time-dependent in vitro killing assay. 1.5 × 105 CWR22rv and DU-145 cells were seeded in 24-well plates, infected by serial dilutions of AdIU1 from 0.0002 to 2 IFU per cell, with replicative-deficient adenovirus AdE1aPSESE4 as a control, and then treated with or without GCV (10 μg ml-1). Seven days after infection, cells were stained with crystal violet (a). CWR 22rv and DU-145 were treated with 0.2 IFU per cell of AdIU1 (■), 10 μg ml-1 of GCV (▲) or AdIU1 plus GCV (●). A group of cells without treatment (◆) were used as a control. At days 1, 3, 5 and 7, crystal violet staining was performed to detect attached cells. Then 1% SDS was added to lyse the cells and for OD590 reading. Cell survival rate curves were drawn to evaluate the killing activity of AdIU1. The growth of AdIU1-infected cells was significantly inhibited by the addition of ganciclovir (GCV) (10 μg ml-1), especially in CWR22rv (b). On the other hand, the growth of DU-145 (c) was not inhibited by AdIU1 plus GCV treatment.
In vivo growth inhibition of CWR22rv xenograft by AdIU1/GCV
Human prostate CWR22rv4 xenograft tumors were induced by s.c. injection of CWR22rv cells into the flanks of athymic nude mice. The mice were castrated 3 days after CWR22rv inoculation to test whether AdIU1 or AdE4PSESE1a was able to eliminate AI tumors in a castrated host. After tumor formation, the mice were randomized into four treatment groups (AdIU1, AdE4PSESE1a, AdIU1 plus GCV and AdE4PSESE1a plus GCV). After randomization to groups the mice were injected intratumorally with AdIU1 or AdE4PSE-SE1a on day 0. On day 5, groups receiving GCV treatments were injected with GCV (40 mg kg-1 body weight) two times daily for 10 days. Tumor volumes were measured every 5 days (Figure 5). AdIU1/GCV effectively inhibited the growth of CWR22rv xenografts. Light microscopic observation of hematoxylin and eosinstained tissue sections from tumors injected with AdIU1/GCV showed substantial treatment effect and a large amount of fibrosis (Figure 6) in contrastto other groups. In addition, we observed that all necrotic tumors stained positive for apoptosis by TUNEL assay (Figures 7A-D). We observed no significant difference in apoptosis between both groups at day 30. Anti-adenovirus type 5 E1a immunohistochemical staining revealed that extensive viral infection existed throughout the AdIU1, AdE4PSESE1a and AdE4PSESE1a plus GCV treatment group tumors (Figures 7e-h); however, adenovirus staining was absent in the AdIU1 plus GCV treatment group.
Figure 5.
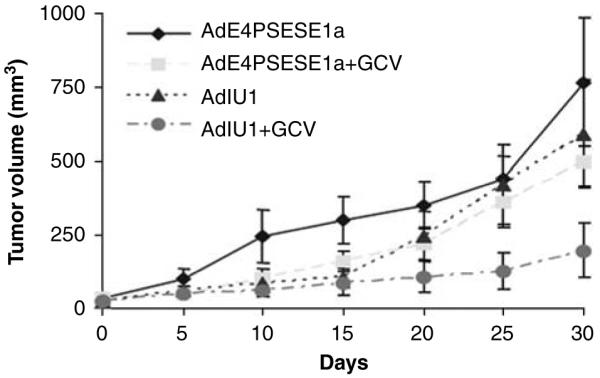
Tumor growth inhibition by AdIU1 plus GCV in a xenograft prostate model. CWR22rv prostate tumor xenografts were established subcutaneous in athymic nude mice. Tumors were treated with AdE4PSE-SE1a (◆, n=7), AdE4PSESE1a plus/GCV (■, n=8), AdIU1 (▲, n=8) or AdIU1 plus GCV (●, n=8). Viruses were delivered by intratumoral injection at day 0, and GCV (80 mg kg-1 of body weight day-1) was administered two times a day for 10 days. Tumor volumes were measured every 5 days. Treatment with AdIU1 plus GCV significantly inhibited the growth of CWR22rv tumors compared with treatment of the AdIU1group.
Figure 6.

Histologic representations of virus-treated tumors. Tumor sections of AdIU1 plus GCV (a), AdIU1 (b), AdE4PSESE1a plus GCV (c) and AdE4PSESE1a (d) treatment groups were stained with hematoxylin and eosin. The lower left panel of each picture was taken at low power ( × 4 magnification), and the upper right panel is magnified to × 40 (focused on white box). Tumors treated with AdIU1 plus GCV (a) demonstrated marked fibrosis and infiltration of fibroblasts (arrows). AdE4PSESE1a alone and plus GCV and AdIU1 alone treated tumors revealed healthy foci of tumor cells embedded within large areas of necrosis.
Figure 7.

Apotosis signal and Ad5E1a expression in subcutaneous tumors. All necrotic areas showed apoptotic signals by TUNEL Assay (A, AdIU1 plus GCV, B, AdIU1, C, AdE4PSESE1a plus GCV, D, AdE4PSESE1a). In the AdIU1 with GCV group, E1a expression was rare (e, AdIU1 Puls GCV, f, AdIU1, g, AdE4PSESE1a plus GCV, h, AdE4PSESE1a).
Discussion
Replication-defective recombinant adenoviruses have been widely studied in vitro and in vivo as a vector to deliver cancer therapeutic genes. Adenoviral-based cancer gene therapy still remains an unrealized potential for its ability to infect and transduce a variety of mammalian cells, including prostate cells,20 in a cell cycle replication-independent manner without genotoxicity. However, there are several limitations to the use of these vectors for cancer gene therapy. To overcome deficiencies of replication-deficient recombinant adenoviruses, we have developed a pair of compact regulatory elements PSES and M6 that allow for prostate-specific regulation of several adenoviral protein and HSV-TK. The main strategy is to control the expression of adenovirus E1a genes through a tissue-specific promoter. We have enhanced the earlier strategy used to restrict adenovirus replication in hepatocellular and prostate carcinomas through á-fetoprotein and PSA promoters.21,33
Gene therapy with HSV-TK as a suicide gene has been performed in a variety of tumor types in vitro, in vivo as well as in several clinical trials. Recently, Freytag et al. 23 described a replication competent oncolytic adenovirus delivering a suicide gene and therapy used in combination with radiotherapy.34-38 They demonstrated that the suicide genes CD and HSV-TK could augment the antitumor effects of oncolytic replication competent adenoviruses and also acts as a radiation sensitizer. However, Freytag’s approach lacks prostate-specific control and excludes its use in metastatic disease. We previously showed that both PSA and OC promoters can transcriptionally regulate HSV-TK gene-based therapy to inhibit the growth of AI PSA-producing cells. Earlier, a PSA-selective replication-competent adenovirus, CG787 (or CV787) was administrated intravenously to patients with hormone-refractory metastatic prostate cancer,39-41 demonstrating the potential safety of systemic administration of oncolytic vectors.
The current investigation builds on the ability of the adenovirus to infect prostate cancer cells and provide both an expanded infection and a longer exogenous gene expression with a prostate-restricted replication-competent oncolytic virus, AdIU1. In previous investigations, the PSES was developed by locating the minimal sequence, AREc3 and PSME (del2) in AREc and PSME, respectively and placing AREc3 upstream from PSME (del2).30 PSES showed high activity specifically in PSA/PSMA-positive and AI prostate cancer cells.31 L2 and L5 in AREc3 did not affect transcriptional activity and was deleted and 90-bp of proximal region of PSME was replaced by a simple AP-3 binding site. These manipulations reduced the size of PSES from 513 to 407 bp. The shorten PSES is called M6. The tissue-specific activity of M6 has been tested in several cell lines by luciferase assay showing that M6 retains strong prostate specificity being active only in PSA/PSMA-positive prostate cancer LNCaP and C4-2 cells.
In this study, we investigated the gene-directed enzyme/prodrug therapeutic effect of AdIU1, a novel PRRA expressing the M6 promoter-driven HSV-TK suicide gene. AdIU1 replicates within infected cells and kills by direct viral lysis, resulting in in vivo amplification of input viral dose. Additionally, AdIU1-infected cells produce HSV-TK to enhance killing by pro-drug administration.42 Using HSV-TK/GCV the ‘bystander effect’ is maximized by spreading to adjacent cancer cells after lysis of initially infected cells. Morris et al. 44 and Lambright et al. 43 reported that there was no advantage of adding HSV-TK/GCV therapy to a replication competent adenovirus. The result of our study suggested that AdIU1 with GCV had a better therapeutic effect than AdIU1 alone. In our animal experiment, GCV was injected 5 days after AdIU1 administration, allowing time for viral replication in the tumor. In both Morris and Lambright’s papers, GCV was given right after virus administration. Further, Freytag’s group demonstrated that HSV-TK/GCV therapy inhibited viral replication, so the therapeutic effect of combining replicating adenoviral vector and HSV-TK/GCV therapy is balanced between killing by viral replication and by HSV-TK/GCV. We believe that the major reason why Morris et al. and Lambright et al. did not see an advantage in adding HSV-TK/GCV therapy in a replicating adenovirus is because of their therapy regimen. GCV given to the animal immediately after virus injection inhibited viral replication, so the advantage of oncolysis was traded in for HSV-TK/GCV effect, resulting in a similar therapeutic effect between HSV-TK expressing replicating virus therapy with and without GCV. The in vitro tissue-specific cytotoxicity of AdIU1/GCV in CWR22rv, C4-2 and DU-145 cells was assessed. Although the growth of AI, PSA/PSMA-positive prostate cancer cell lines CWR22rv and C4-2 were significantly inhibited by a small number of AdIU1 virus particles and GCV, the growth of the AI, PSA/PSMA-negative prostate cancer cell line, DU-145 could only be inhibited by a much greater exposure to AdIU1/GCV. More importantly, the in vivo ability of intratumoral injection AdIU1/GCV to effectively inhibit growth of CWR22rv tumors in nude mice was confirmed. Histological analysis revealed a large number of fibroblasts infiltrating only AdIU1/GCV-treated tumors. Virus persistency was detected in AdIU1, AdE4PSESE1a and AdE4PSESE1a/GCV-treated tumors, but not in AdIU1/GCV-treated tumors. This observation is consistent with Freytag et al. that TK/GCV treatment inhibits adenoviral replication and suggests effective HSV-TK-mediated killing of infected and unlysed cells. In addition, it is likely that AdIU1/GCV combined with radiation therapy could have even a greater growth inhibition.
In conclusion, we have developed a prostate-restricted replicative, HSV-TK-armed adenovirus, AdIU1. AdIU1 demonstrated prostate cancer-specific-killing activity, which could be enhanced by GCV administration. Gene therapy as monotherapy against prostate cancer currently remains in its infancy. Although preventive strategies are being entertained, the ultimate clinical use of gene therapy for improving prostate cancer treatment would most likely be in combination with surgery, radiation or chemotherapy. The ability of providing prostate-specific cell killing and gene expression provides an opportunity to attempt systemic therapy with AdIU1. AdIU1 will be the focus of a phase I clinical trial for locally advanced and metastatic prostate cancer.
Acknowledgements
We thank M Black, Department of pharmaceutical Sciences, Washington State University for providing the TK-polyclonal antibody. These studies were supported by NIH K08 CA079544-01A2 and DOD DAMD 17-03-1-0077.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Cheon J, Kim HK, Moon DG, Yoon DK, Cho JH, Koh SK. Adenovirus-mediated suicide-gene therapy using the herpes simplex virus thymidine kinase gene in cell and animal models of human prostate cancer: changes in tumour cell proliferative activity. BJU Int. 2000;85:759–766. doi: 10.1046/j.1464-410x.2000.00516.x. [DOI] [PubMed] [Google Scholar]
- 3.van der Poel HG, McCadden J, Verhaegh GW, Kruszewski M, Ferrer F, Schalken JA, et al. A novel method for the determination of basal gene expression of tissue-specific promoters: an analysis of prostate-specific promoters. Cancer Gene Ther. 2001;8:927–935. doi: 10.1038/sj.cgt.7700385. [DOI] [PubMed] [Google Scholar]
- 4.Latham JP, Searle PF, Mautner V, James ND. Prostate-specific antigen promoter/enhancer driven gene therapy for prostate cancer: construction and testing of a tissue-specific adenovirus vector. Cancer Res. 2000;60:334–341. [PubMed] [Google Scholar]
- 5.Steiner MS, Gingrich JR. Gene therapy for prostate cancer: where are we now? J Urol. 2000;164:1121–1136. [PubMed] [Google Scholar]
- 6.O’Keefe DS, Uchida A, Bacich DJ, Watt FB, Martorana A, Molloy PL, et al. Prostate-specific suicide gene therapy using the prostate-specific membrane antigen promoter and enhancer. Prostate. 2000;45:149–157. doi: 10.1002/1097-0045(20001001)45:2<149::aid-pros9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 7.Xing Y, Lu G, Xiao Y, Zeng F, Zhang Q, Xiong P, et al. Bystander effect mediated by herpes simplex virus-thymidine kinase/ganciclovir approach on prostatic cancer cells and its regulation. Zhonghua Yi Xue Za Zhi. 2002;82:1484–1487. [PubMed] [Google Scholar]
- 8.Park HS, Cheon J, Cho HY, Ko YH, Bae JH, Moon DG, et al. In vivo characterization of a prostate-specific antigen promoter-based suicide gene therapy for the treatment of benign prostatic hyperplasia. Gene Therapy. 2003;10:1129–1134. doi: 10.1038/sj.gt.3301972. [DOI] [PubMed] [Google Scholar]
- 9.Ko SC, Cheon J, Kao C, Gotoh A, Shirakawa T, Sikes RA, et al. Osteocalcin promoter-based toxic gene therapy for the treatment of osteosarcoma in experimental models. Cancer Res. 1996;56:4614–4619. [PubMed] [Google Scholar]
- 10.Springer CJ, Niculescu-Duvaz I. Prodrug-activating systems in suicide gene therapy. J Clin Invest. 2000;105:1161–1167. doi: 10.1172/JCI10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denny WA. Prodrugs for Gene-Directed Enzyme-Prodrug Therapy (Suicide Gene Therapy) J Biomed Biotechnol. 2003;2003:48–70. doi: 10.1155/S1110724303209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schepelmann S, Springer CJ. Viral vectors for gene-directed enzyme prodrug therapy. Curr Gene Ther. 2006;6:647–670. doi: 10.2174/156652306779010679. [DOI] [PubMed] [Google Scholar]
- 13.Yu DC, Sakamoto GT, Henderson DR. Identification of the transcriptional regulatory sequences of human kallikrein 2 and their use in the construction of calydon virus 764, an attenuated replication competent adenovirus for prostate cancer therapy. Cancer Res. 1999;59:1498–1504. [PubMed] [Google Scholar]
- 14.Matsubara S, Wada Y, Gardner TA, Egawa M, Park MS, Hsieh CL, et al. A conditional replication-competent adenoviral vector, Ad-OC-E1a, to cotarget prostate cancer and bone stroma in an experimental model of androgen-independent prostate cancer bone metastasis. Cancer Res. 2001;61:6012–6019. [PubMed] [Google Scholar]
- 15.DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, Goemann M, et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001;61:7464–7472. [PubMed] [Google Scholar]
- 16.Sadeghi H, Hitt MM. Transcriptionally targeted adenovirus vectors. Curr Gene Ther. 2005;5:411–427. doi: 10.2174/1566523054546189. [DOI] [PubMed] [Google Scholar]
- 17.Wu L, Matherly J, Smallwood A, Adams JY, Billick E, Belldegrun A, et al. Chimeric PSA enhancers exhibit augmented activity in prostate cancer gene therapy vectors. Gene Therapy. 2001;8:1416–1426. doi: 10.1038/sj.gt.3301549. [DOI] [PubMed] [Google Scholar]
- 18.Diamandis EP, Yousef GM. Human tissue kallikreins: a family of new cancer biomarkers. Clin Chem. 2002;48:1198–1205. [PubMed] [Google Scholar]
- 19.Hsieh CL, Gardner TA, Miao L, Balian G, Chung LW. Cotargeting tumor and stroma in a novel chimeric tumor model involving the growth of both human prostate cancer and bone stromal cells. Cancer Gene Ther. 2004;11:148–155. doi: 10.1038/sj.cgt.7700665. [DOI] [PubMed] [Google Scholar]
- 20.Kubo H, Gardner TA, Wada Y, Koeneman KS, Gotoh A, Yang L, et al. Phase I dose escalation clinical trial of adenovirus vector carrying osteocalcin promoter-driven herpes simplex virus thymidine kinase in localized and metastatic hormone-refractory prostate cancer. Hum Gene Ther. 2003;14:227–241. doi: 10.1089/10430340360535788. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR. Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997;57:2559–2563. [PubMed] [Google Scholar]
- 22.Galanis E, Vile R, Russell SJ. Delivery systems intended for in vivo gene therapy of cancer: targeting and replication competent viral vectors. Crit Rev Oncol Hematol. 2001;38:177–192. doi: 10.1016/s1040-8428(01)00103-2. [DOI] [PubMed] [Google Scholar]
- 23.Freytag SO, Rogulski KR, Paielli DL, Gilbert JD, Kim JH. A novel three-pronged approach to kill cancer cells selectively: concomitant viral, double suicide gene, and radiotherapy. Hum Gene Ther. 1998;9:1323–1333. doi: 10.1089/hum.1998.9.9-1323. [DOI] [PubMed] [Google Scholar]
- 24.Freytag SO, Khil M, Stricker H, Peabody J, Menon M, DePeralta-Venturina M, et al. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res. 2002;62:4968–4976. [PubMed] [Google Scholar]
- 25.Freytag SO, Stricker H, Pegg J, Paielli D, Pradhan DG, Peabody J, et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003;63:7497–7506. [PubMed] [Google Scholar]
- 26.Edwards SJ, Dix BR, Myers CJ, Dobson-Le D, Huschtscha L, Hibma M, et al. Evidence that replication of the antitumor adenovirus ONYX-015 is not controlled by the p53 and p14(ARF) tumor suppressor genes. J Virol. 2002;76:12483–12490. doi: 10.1128/JVI.76.24.12483-12490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zur Hausen H. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J Virol. 1998;72:9470–9478. doi: 10.1128/jvi.72.12.9470-9478.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 29.O’Shea CC, Soria C, Bagus B, McCormick F. Heat shock phenocopies E1B-55 K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell. 2005;8:61–74. doi: 10.1016/j.ccr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Lee SJ, Kim HS, Yu R, Lee K, Gardner TA, Jung C, et al. Novel prostate-specific promoter derived from PSA and PSMA enhancers. Mol Ther. 2002;6:415–421. doi: 10.1006/mthe.2002.0682. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Zhang YP, Kim HS, Bae KH, Stantz KM, Lee SJ, et al. Gene therapy for prostate cancer by controlling adenovirus E1a and E4 gene expression with PSES enhancer. Cancer Res. 2005;65:1941–1951. doi: 10.1158/0008-5472.CAN-04-3666. [DOI] [PubMed] [Google Scholar]
- 32.Lee SJ, Lee K, Yang X, Jung C, Gardner T, Kim HS, et al. NFATc1 with AP-3 site binding specificity mediates gene expression of prostate-specific-membrane-antigen. J Mol Biol. 2003;330:749–760. doi: 10.1016/s0022-2836(03)00640-5. [DOI] [PubMed] [Google Scholar]
- 33.Hallenbeck PL, Chang YN, Hay C, Golightly D, Stewart D, Lin J, et al. A novel tumor-specific replication-restricted adenoviral vector for gene therapy of hepatocellular carcinoma. Hum Gene Ther. 1999;10:1721–1733. doi: 10.1089/10430349950017725. [DOI] [PubMed] [Google Scholar]
- 34.Kim JH, Kim SH, Brown SL, Freytag SO. Selective enhancement by an antiviral agent of the radiation-induced cell killing of human glioma cells transduced with HSV-tk gene. Cancer Res. 1994;54:6053–6056. [PubMed] [Google Scholar]
- 35.Kim JH, Kim SH, Kolozsvary A, Brown SL, Kim OB, Freytag SO. Selective enhancement of radiation response of herpes simplex virus thymidine kinase transduced 9L gliosarcoma cells in vitro and in vivo by antiviral agents. Int J Radiat Oncol Biol Phys. 1995;33:861–868. doi: 10.1016/0360-3016(95)00134-9. [DOI] [PubMed] [Google Scholar]
- 36.Rogulski KR, Wing MS, Paielli DL, Gilbert JD, Kim JH, Freytag SO. Double suicide gene therapy augments the antitumor activity of a replication-competent lytic adenovirus through enhanced cytotoxicity and radiosensitization. Hum Gene Ther. 2000;11:67–76. doi: 10.1089/10430340050016166. [DOI] [PubMed] [Google Scholar]
- 37.Rogulski KR, Freytag SO, Zhang K, Gilbert JD, Paielli DL, Kim JH, et al. In vivo antitumor activity of ONYX-015 is influenced by p53 status and is augmented by radiotherapy. Cancer Res. 2000;60:1193–1196. [PubMed] [Google Scholar]
- 38.Freytag SO, Paielli D, Wing M, Rogulski K, Brown S, Kolozsvary A, et al. Efficacy and toxicity of replication-competent adenovirus-mediated double suicide gene therapy in combination with radiation therapy in an orthotopic mouse prostate cancer model. Int J Radiat Oncol Biol Phys. 2002;54:873–885. doi: 10.1016/s0360-3016(02)03005-5. [DOI] [PubMed] [Google Scholar]
- 39.Small EJ, Carducci MA, Burke JM, Rodriguez R, Fong L, van Ummersen L, et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol Ther. 2006;14:107–117. doi: 10.1016/j.ymthe.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 40.Dilley J, Reddy S, Ko D, Nguyen N, Rojas G, Working P, et al. Oncolytic adenovirus CG7870 in combination with radiation demonstrates synergistic enhancements of anti-tumor efficacy without loss of specificity. Cancer Gene Ther. 2005;12:715–722. doi: 10.1038/sj.cgt.7700835. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, DeWeese T, Dilley J, Zhang Y, Li Y, Ramesh N, et al. CV706, a prostate cancer-specific adenovirus variant, in combination with radiotherapy produces synergistic antitumor efficacy without increasing toxicity. Cancer Res. 2001;61:5453–5460. [PubMed] [Google Scholar]
- 42.Wang JQ, Zheng QH, Fei X, Mock BH, Hutchins GD. Novel radiosynthesis of PET HSV-tk gene reporter probes [18F]FHPG and [18F]FHBG employing dual Sep-Pak SPE techniques. Bioorg Med Chem Lett. 2003;13:3933–3938. doi: 10.1016/j.bmcl.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 43.Lambright ES, Amin K, Wiewrodt R, Force SD, Lanuti M, Propert KJ, et al. Inclusion of the herpes simplex thymidine kinase gene in a replicating adenovirus does not augment antitumor efficacy. Gene Therapy. 2001;8:946–953. doi: 10.1038/sj.gt.3301489. [DOI] [PubMed] [Google Scholar]
- 44.Morris JC, Wildner O. Therapy of head and neck squamous cell carcinoma with an oncolytic adenovirus expressing HSV-tk. Mol Ther. 2000;1:56–62. doi: 10.1006/mthe.1999.0014. [DOI] [PubMed] [Google Scholar]