

Abstract
B cells play an important role in the pathogenesis of both systemic and organ-specific autoimmune diseases. Autoreactive B cells not only produce autoantibodies, but are also specialized to present specific autoantigens efficiently to T cells. Furthermore, these B cells can secrete proinflammatory cytokines and can amplify the vicious cycle of self-destruction. Thus, B cell-directed therapies are potentially an important approach for treating autoimmune diseases. On the other hand, like T cells, there are subsets of B cells that produce anti-inflammatory cytokines and are immunosuppressive. These regulatory B cell subsets can protect against and ameliorate autoimmune diseases. Thus targeting B cells therapeutically will require this balance to be considered. Here we summarize the roles of pathogenic and regulatory B cells and current applications of B cell-directed therapy in autoimmune diseases. Considerations for future development of B cell-directed therapy for autoimmune diseases have also been discussed.
Keywords: autoimmune disease, B cell, immunotherapy
B cells in health and diseases
There has been a major research focus on T cells in autoimmune disease for many years and clinical trials of T cell targeted therapy have also been carried out in a number of different autoimmune diseases [1–3]. However, the roles of B cells in autoimmune diseases that were thought previously to be T cell-mediated have been increasingly appreciated. B cells are usually divided into two lineages – B1 and B2 cells. B1 cells, the producers of natural antibodies, originate from bone marrow or fetal liver [4] and are located mainly in the peritoneal and pleural cavities in mice [5,6]. They are more like innate immune cells, and essential for immune defence against encapsulated bacteria. In contrast to B1 cells, B2 or conventional B cells comprise the adaptive arm of the immune system. Upon encountering antigen, mature B2 cells are activated, expand and generate short-lived plasma cells. Some activated B2 cells differentiate further in germinal centres to become memory B cells or long-lived plasma cells. Although there are mechanisms in place to protect against autoimmunity, given the complexity of the immune system it is likely that potentially self-reactive B cells could develop and escape to the periphery. Indeed, studies have suggested that about 50–75% of newly produced B cells are self-reactive and are subject to three distinct tolerance mechanisms, including clonal deletion, receptor editing and anergy [7–11]. However, even in healthy individuals, the autoreactive B cells are only reduced to 6–20% of mature B cells [10]. Loss of control of these autoreactive B cells may lead to autoimmune diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE).
The pathogenic roles of B cells in autoimmune diseases involve several pathways that include production of autoantibodies, immune complex formation, cytokine and chemokine secretion, as well as ectopic neolymphogenesis [12–14].
The role of autoantibodies in different autoimmune diseases varies [15]. The presence of autoantibodies may predict or precede the development of autoimmune diseases in humans [16–18]. Antibodies, such as rheumatoid factor in rheumatoid arthritis and antinuclear antibodies in SLE, can also be used as a marker of disease activity. Autoantibodies can induce neonatal lupus syndromes through maternal transfer of antibodies [19]. Furthermore, maternal transmission of autoantibodies was also shown to be involved in the development of spontaneous diabetes in the non-obese diabetic (NOD) mouse model of human type 1 diabetes [20], although the precise role of these maternally transmitted antibodies has not been elucidated.
In some autoimmune diseases the binding of the autoantibodies to the target self-antigen causes the disease, for example in Graves' disease, where binding of autoantibodies to the thyroid stimulating hormone (TSH) receptor mimics the effect of endogenous TSH but is not subject to the normal endocrine feedback mechanisms. In other instances, such as in myasthenia gravis, binding of the autoantibodies to the acetylcholine receptor can interfere with neuromuscular cell function [13]. Thus, the autoantibodies in these disease are involved directly in the pathogenesis of disease.
Autoantibodies may also form immune complexes that deposit in specific tissues and cause detrimental inflammation. This is well known for inducing renal damage in SLE, where immune complex deposition in the kidneys activates the complement cascade, damaging the basement membrane of glomeruli leading to nephritis. This is also a mechanism for the induction of arthritis and vasculitis in anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis [13].
There has been much interest in the role of B cells as antigen-presenting cells in diseases such as autoimmune diabetes and multiple sclerosis, as shown in the animal models of these diseases. B cells are also major cytokine producers in autoimmune disorders [21,22]. The production of interleukin (IL)-2 and interferon (IFN)-γ by B cells promotes T helper type 1 (Th1) differentiation, and tumour necrosis factor (TNF)-α secreted by B cells can further amplify the Th1 phenotype [23] and might contribute indirectly to the tissue damage in autoimmune diseases. In combination with transforming growth factor (TGF)-β, IL-6 produced by B cells promotes Th17 differentiation and might play a role in the pathogenesis of multiple sclerosis [21,24].
Finally, B cells have been suggested to play an important role in the formation and maintenance of new lymphoid structures [25]. Ectopic neolymphogenesis has been observed in many autoimmune diseases, such as multiple sclerosis, diabetes, rheumatoid arthritis and thyroiditis [26–29]. These areas, which have very similar appearances to lymph nodes, have an arrangement of cells within which B cells may generate inflammatory signals that serve to activate T cells and dendritic cells and thus amplify local disease.
In addition to the pathogenic roles of B cells in autoimmune diseases, B cells also have regulatory functions and this has not been well appreciated. Recent studies have indicated the existence of distinct B cell subsets that suppressed the progression of autoimmune disease or improved the recovery from inflammation [30–33]. The following B cell-mediated regulatory mechanisms have been postulated: (i) production of IL-10, which directly inhibits inflammatory cascades and restores the Th1/Th2 balance [34]; (ii) production of TGF-β1 that induces apoptosis of effector T cells [22]; (iii) recruitment of different subsets of regulatory T cells [35]; (iv) function as secondary antigen-presenting cells (APC) to dampen activated CD4+ T cells directly [36,37]; and (v) secretion of immunoglobulin (Ig)G and IgA that can neutralize harmful soluble factors and activate dendritic cells (DC)/macrophages via the stimulatory Fc receptor to enhance the clearance of apoptotic cells that are a potential source of self-antigens for activating self-reactive T cells [38]. Secreted IgG could also dampen immune responses by activation of inhibitory Fc receptors [39].
Thus, it is clear that B cell have dual roles in health and disease – immunopathogenic and immunoregulatory. The best immunotherapy will inhibit the pathogenic function but enhance the regulatory function of B cells.
B cell-directed therapy for autoimmune diseases
Given their pathogenic roles in autoimmune disease, B cells could be important target for immune intervention. A number of different approaches to target B cells and their functions are available, as discussed in the next sections. There are three main strategies to treat autoimmune disorders using B cell-directed therapy: (i) targeting B cell-specific surface molecules with depleting antibodies; (ii) induction of B cell apoptosis by targeting BCR signalling; and (iii) blocking B cell survival and activation factors (Fig. 1).
Fig. 1.
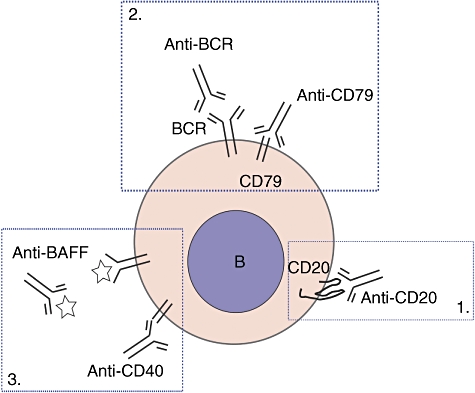
Strategies for B cell-directed therapy. 1. Targeting B cell-specific surface molecules. 2. Targeting B cell receptor (BCR) to induce B cell apoptosis. 3. Blockade of B cell survival or activation.
Targeting B cell-specific surface molecules
Antibodies against B cell surface molecules, such as CD19, CD20, CD21, CD22 and CD23 have been developed for B cell-directed therapy in animal studies [13,40,41]. However, only a few of these antibodies have been used in clinical trials [12]. Rituximab, a human/murine chimeric anti-human CD20 (hCD20) monoclonal antibody that depletes most B cells, has been studied widely in recent years. Rituximab was approved initially for treating patients with non-Hodgkin's lymphoma [42]. Interestingly, autoimmune disorders in some patients who also suffered from lymphoma showed significant clinical improvement in co-existing autoimmune disease, such as cold agglutinin haemolytic anaemia [43]. In other cases where rheumatoid disease was present in patients with lymphoma [44], improvement of rheumatoid disease was observed after rituximab treatment, although apart from the rituximab the other components of the chemotherapy probably also played a role in this improvement. However, remission of autoimmune disorders in patients with lymphoma promoted B cell depletion as a therapy for a variety of autoimmune diseases [12,45–47]. The incidence of severe side effects of this therapy is low when treating patients with autoimmune diseases, and most reactions have occurred at the time of infusion. Major theoretical severe side effects have not been encountered thus far in trials which have used this treatment [48].
One of the advantages of rituximab treatment is that rituximab can deplete peripheral human B lymphocytes successfully for a period ranging from 3 months to more than a year without significant change in the level of serum antibodies, as plasma cells are generally not affected. However, B cells located in various lymphoid organs have different sensitivities to anti-CD20 monoclonal antibody (mAb) treatment [49]. B cell depletion by anti-CD20 is mediated in vivo through FcRγ, particularly FcγRI and FcγRIII, expressed on monocytes and macrophages which are responsible for the removal of B cells to which the anti-CD20 antibodies have bound [50]. Studies have indicated that more than 90% of circulating B cells in blood can be depleted within minutes of treatment, whereas depletion of B cells in lymph nodes and spleen is less effective [49,51,52]. It is interesting that B cells in the mouse peritoneal cavity are more resistant to the treatment [53]. In MRL/lpr and NOD mice, B cells in the spleen were also found to be less sensitive to anti-CD20 mAb treatment compared with B cells in peripheral blood [51,52]. Further analysis of different subsets of splenic B cells revealed that follicular B cells were sensitive, but marginal zone (MZ) B cells were resistant to anti-CD20 mAb treatment and germinal centre B cells were most resistant to this therapy [49]. The resistance to depletion was due mainly to low accessibility of anti-CD20 to the cells of the extravascular microenvironment [49]. Given that integrins mediate long-term B cell retention in the splenic marginal zone [54], combination therapy with anti-integrin α4 and anti-αL promoted the mobility of MZ B cells and led to efficient depletion of MZ B cells by anti-CD20 mAb [49]. Further understanding of the dynamics of B cell depletion, as well as B cell circulation dynamics and retention in the tissue microenvironment, will be important for the most effective use of this strategy of targeting B cell surface molecules to treat autoimmune diseases. This is discussed in detail in the review by Leandro et al.[55]. In addition, the use of integrin blockade could improve depletion efficiency.
To date, rituximab has been used off-label in more than 18 autoimmune diseases, with a good safety record [56]. Although efficacy varies among patients with different autoimmune diseases, cumulative data have suggested a beneficial role of rituximab therapy, particularly in patients with rheumatoid arthritis who were unresponsive to tumour necrosis factor (TNF)-blocking agents. With success in other autoimmune diseases, B cell depletion therapy has received growing attention as a possible treatment for type 1 diabetes (T1D). In parallel with a TrialNet sponsored trial of rituximab treatment in patients with type 1 diabetes (http://www.diabetestrialnet.org/patientinfo/studies.htm), we generated a transgenic NOD mouse expressing human CD20 (hCD20/NOD). The hCD20/NOD mouse allowed us to test B cell depletion therapy with rituximab in NOD mice, the best-studied animal model of human T1D. Our study demonstrated that temporary depletion of B cells by anti-human CD20 monoclonal antibody prevented and reversed autoimmune diabetes in a proportion of hCD20/NOD mice [51]. This was mediated, at least in part, through the induction of regulatory T and B cells, and modulation of antigen-presenting cell function [51]. Using anti-mouse CD20 monoclonal antibody, Xiu and colleagues also showed that the B cell-directed therapy could prevent autoimmune diabetes in NOD mice, which supported our findings [57]. The clinical trial using rituximab to treat patients with type 1 diabetes has yet to be reported (http://www.diabetestrialnet.org/patient info/studies.htm).
Furthermore, a recent study using the toxin calicheamicin-conjugated anti-CD22 monoclonal antibody to deplete B cells in NOD mice demonstrated that B cell depletion could prevent and reverse spontaneous diabetes [58]. Anti-CD22 antibody (epratuzumab) depletes 30% of peripheral B cells and reduces reponsiveness of B cells from patients with SLE to B cell stimuli such as lipopolysaccharide (LPS), cytosine-phosphate-guanosine (CpG) and anti-CD40 [59]. Epratuzumab has also been shown in two phase II studies, one in SLE and the other in Sjögren's syndrome, to be well tolerated. Toxin-conjugated anti-CD22 is also in clinical trials in SLE [12]. The use of anti-CD22 antibody is another approach to target B cells in autoimmune diseases.
B cell depletion monotherapy using monoclonal antibodies to molecules expressed by B cells has clinical efficacy in a number of different autoimmune diseases, and the approved use of rituximab in patients with RA has been particularly successful. The factors affecting the efficiency of B cell depletion include: (i) the expression pattern of the targeted molecule on B cells, including the stage in B cell development and/or differentiation at which the molecule is expressed and level of expression; (ii) factors affecting the sensitivity of targeted B cells to antibodies, such as tissue microenvironment and mobility of B cells; and (iii) status of effector cells [such as natural killer (NK) cells, macrophages] involved in removal of antibody-targeted B cells, including the availability and activation of these effector cells. To achieve better depletion of B cells, experience from anti-CD20 studies in NHL patients suggested that modulation of the Fc portion of the antibodies can also improve B cell killing [60,61]. Thus, increasing experience with the use of these targeted monoclonal antibodies should be combined with further basic research to maximize the efficacy of B cell depletion.
Induction of B cell apoptosis by targeting BCR signalling
The early studies of therapy targeting B cells focused on BCR signalling using anti-Ig reagents. Polyclonal anti-Ig antibodies used at the neonatal stage could deplete newly generated immature B cells and/or other surface Ig-expressing B cells in mice [20,62,63]. This approach, however, was less efficient when used in adult animals that had a high level of circulating immunoglobulins. Nevertheless, monoclonal antibodies against surface IgM were successful in decreasing circulating B cells in animal models of experimental autoimmune thyroiditis, SLE and T1D [63–65]. A strategy similar to the one recently used with anti-CD3 that targets part of the T cell receptor signalling complex was also developed for B cells targeting CD79. CD79 is expressed almost exclusively on B cells and comprises CD79a and CD79b, which are B cell receptor (BCR)-associated transmembrane signalling proteins. Like CD20, CD79 is not expressed in the late plasma cell stage of B cell differentiation. Antibodies against CD79a or CD79b have been tested in B cell neoplastic disease but induced only minimal B cell depletion [66]. Thus, the development of reagents targeting the BCR for humans has been hampered by a low efficiency of B cell depletion [66]. However, Li and colleagues demonstrated in a recent study that combination treatment using anti-CD79a and anti-CD79b decreased B220+CD19+ B cells significantly in peripheral blood, bone marrow and spleen compared with other reagents, and this led to amelioration of the lupus-like disease in MRL/lpr mice [40]. Other effects, not seen with anti-CD20, were noted, such as an increase in the double-negative and putatively regulatory T cell population in the spleen. However, MRL/lpr mice have a mutant Fas receptor and regulatory effects of the double-negative T cell population are likely to occur through Fas–Fas ligand interactions. Thus, it is most likely that the effects of treatment with anti-CD79a and b occur through changes in the B cell population rather than through double-negative T cells. Overall, although targeting the BCR directly using reagents such as anti-IgM or anti-CD79 may have some limitations, they may still provide an option in certain autoimmune diseases such as SLE [40].
Blockade of B cell survival and activation
Inhibition of B cell activation and survival is an alternative strategy for targeting B cells and the following approaches have been applied to inhibit B cell survival and action.
Blockade of CD40–CD40L interaction
It is known that the CD40–CD40L co-stimulatory interaction promotes B cell proliferation and survival [67,68]. Blocking this interaction prevents B cell activation and class switching in both germinal centres and extrafollicullar sites [69,70]. This approach, however, does not eliminate B cells. An earlier clinical trial of CD40–CD40L blockade using anti-CD40L showed efficacy in idiopathic thrombocytopenia and improved the clinical score in patients with SLE [71,72]. However, due to the expression of CD40 on activated endothelial cells [73] and CD40L on human platelets [74,75], contributing to thrombotic problems arising with the use of anti-CD40L, the clinical application of CD40–CD40L blockade is in doubt.
Neutralizing BAFF/APRIL and receptors
BAFF (B cell-activating factor belonging to the TNF family) is produced by neutrophils, monocytes, macrophages, dendritic cells and T cells [76]. In addition to its expression on a small proportion of CD4+ and CD8+ T cells, the receptor for BAFF is also found on B cells and it is important for B cell proliferation and an essential survival factor for B cells [77,78]. A proliferation inducing ligand (APRIL), another member of the TNF family, has close homology to BAFF and is produced by similar cells. BAFF binds to three known receptors, including transmembrane activator and CAML interactor (TACI), B cell maturation antigen (BCMA) and BAFF receptor (BAFF-R or BR3). APRIL also binds to two of the three receptors used by BAFF, TACI and BCMA, but not BAFF-R [79].
Elevated BAFF has been found in patients with Sjögren's syndrome, SLE and autoimmune neurological diseases [80–83], which has provided a therapeutic opportunity for manipulating BAFF. Animal studies have suggested that neutralizing anti-BAFF antibody could prevent autoimmune diabetes in NOD mice through depletion of follicular and marginal zone B lymphocytes. This treatment enhanced negative selection of autoreactive B cells, abrogating the production of serum insulin autoantibodies and reduced the severity of islet inflammation [84].
Targeting the BAFF/APRIL signalling pathway using belimumab, a humanized monoclonal antibody against soluble BAFF and which neutralizes BAFF, has been shown to be beneficial in patients with moderate to severe RA in clinical trials. Patients who failed to respond to disease-modifying agents, and in some cases also had no benefit from TNF inhibitors, showed remission with belimumab compared with placebo [85]. However, it is not clear at present if this treatment will move forward for general availability in therapy for RA. In a phase II clinical trail, belimumab has also shown promising clinical efficacy in patients with SLE [86] and it is currently in phase III clinical trials. Similarly, TACI-Ig and BCMA-Ig act as decoy receptors and also have the effect of neutralizing soluble BAFF. TACI-Ig has been shown in animal studies to reduce both mature B cells and transitional T2 cells as well as circulating immunoglobulin [87]. When tested in collagen-induced arthritis, the use of TACI reduced progression of disease. TACI-Ig has been used in phase I clinical trials in SLE and RA and was found to be well tolerated [88,89].
Thus, the strategy of targeting B cell survival offers another option for B cell-directed therapy, although the depletion is not as complete as that seen with rituximab, and the outcome of the various trials that target BAFF/APRIL in RA and SLE are awaited.
Important considerations for the use of B cell-directed therapy and future development of B cell-directed therapy
Immune regulatory role of B cells
Like T cells, B cells also have a dual role in immunity, including autoimmunity, and the immune regulatory roles of B cells are increasingly appreciated. Thus, a complete depletion of B cells may not be the best approach for optimal efficacy in some autoimmune diseases [90]. Furthermore, B cell depletion at different disease stages could lead to opposite effects, as shown in experimental allergic encephalomyelitis (EAE), an animal model of human multiple sclerosis [30]. In the early stages of EAE, anti-CD20 treatment exacerbated disease symptoms substantially because the treatment also depleted an IL-10-producing CD1dhiCD5+ regulatory B cell subset, whereas at a later stage in the disease progression, B cell depletion suppressed dramatically the clinical signs of paralysis [30]. These results demonstrate dual roles of B cells during EAE immunopathogenesis. However, B cell depletion in both early and later stages of T1D development in NOD mice had a beneficial outcome [51,58 (#133)]. The roles of B cells vary at different stages in different autoimmune diseases. The therapeutic effect of B cell depletion for the treatment of autoimmune diseases may thus depend on the balance of the pathogenic and regulatory roles of B cells during the course of disease.
Thus, a major challenge of B cell-directed therapy lies in fully understanding the roles of B cells in disease development, such as (i) which subsets of B cells are pathogenic or regulatory; (ii) at what stage B cells are pathogenic or regulatory; and (iii) where the pathogenic B cells are located. The answers to these questions will be important to target the pathogenic B cells more specifically.
Is long-term depletion of B cells necessary for continued clinical effect?
The aetiology of most autoimmune diseases is multi-factorial, and many studies have suggested that environmental factors could play an important role in addition to genetic factors that include susceptibility or protection encoded in major histocompatibility complex (MHC) molecules. It is reasonable to believe that modulation or resetting of the immune system, such as the approach of B cell depletion, could improve disease. However, whether this will have long-term benefit has yet to be shown.
The best-studied mouse model of human type 1 diabetes, NOD mice, develops spontaneous autoimmune diabetes starting around 12 weeks of age. However, when B cells were removed in embryonic development by genetic alteration, such as in µMT–/– NOD mice, diabetes was inhibited [91–93], although there are instances where a small number of mice are able to develop disease [94]. In these mice, where B cells are deficient from birth, this induced mutation most probably has effects by altering the interactions with other developing cells of the immune system. Therefore, it is important to confirm the results of those studies by removing B cells after maturation rather at ontogeny. Recent studies showed that when B cells were removed transiently at different time-points in the course of diabetes development, the disease onset was significantly delayed or prevented in the NOD mice [51,57,58,84]. Most importantly, temporary depletion of B cells can cure type 1 diabetes in a proportion of diabetic NOD mice. These studies confirmed the early reports using µMT–/– NOD mice and, more importantly, extended our understanding that B cells play an important role in T1D development through antigen presentation and/or pro- or anti-inflammatory cytokine production [95]. Considering the data from µMT–/– NOD mice, that B cells are important in antigen presentation and the progression of the autoimmune process, it is conceivable that repeated B cell depletion therapy might have long-term benefit. However, regulatory B cells have been reported among the newly generated B cells [51,58] and repeated B cell depletion will also target the B regulatory cells. Repeated treatment may therefore not be desirable. It is clear that more studies are required to determine whether repeated treatment would be beneficial in diabetes.
It is known that rheumatoid factor (RF)-producing B cells are pathogenic in RA. The efficacy of destruction of RF-producing B cell clones by anti-CD20 antibodies and/or other agents has been tested to determine whether repeated treatment might be necessary. In a follow-up clinical trial the investigators demonstrated clinical improvement in all the RA patients, which lasted up to 43 months from a single 2-week cycle of rituximab therapy, but all patients eventually relapsed [96]. In addition, approximately half the patients relapsed at the time of repopulation of circulating B cells and rise of autoantibodies [97]. This trial demonstrated that transient B cell depletion by rituximab could induce long-term improvement in RA, although this was not permanent. The need for repeated treatment was supported by results from a recent clinical trial which suggested that repeated B lymphocyte depletion over a 5-year period is an acceptable and relatively well-tolerated therapy in RA [48]. In this study each cycle of treatment showed benefit, on average for 15 months with a range of 6–43 months, and the time to retreatment ranged from 5 to 60 months. At the time of reporting at 7 years, 19 of the original 37 patients recruited remained in the programme. The reasons for not continuing with the treatment included lack of efficacy of a relatively short time-period of response, and a small number of patients had problems with lower respiratory tract infection [48]. Cessation of treatment due to hypersensitivity was relatively uncommon. Thus, at least in RA, there is some benefit to repeated treatment, although at this time it appears that this treatment does not induce permanent remission. The benefit of repeated B cell depletion is likely to be different in different diseases. However, the possibility that the immune system of patients may be compromised due to humoral immunodeficiency remains a serious concern for repeated treatment. Although Popa et al. showed that total immunoglobulin levels remained in the normal range even after five cycles of treatment in most RA patients, IgA, IgG and IgM were below normal in a group of patients. This suggested that the repeated use of rituximab could potentially jeopardize host protective immunity, as lower respiratory tract problems were observed in a few patients in spite of the fact that a reduction in Ig was not associated with infections or adverse clinical events following repeated treatment [48]. Thus far, however, evidence suggests that repeated B cell depletion, on balance, can have long-term benefits, at least in patients with RA.
In summary, transient depletion of B cells can ameliorate autoimmune diseases and repeated treatment can prevent disease relapse (Fig. 1). However, the optimal timing for repeated B cell depletion remains a difficult decision due to the heterogeneity of patient populations. Thus, patient-tailored treatment regimens may be required for the best therapeutic outcome. From the safety viewpoint, continued close monitoring is required for repeated treatment. Most importantly, the identification and analysis of biomarkers, such as unique feature(s) of repopulated B cell subsets combined with autoantibody profile and Ig level, could improve this therapy significantly.
Could combination treatment improve efficacy?
Although rituximab, as monotherapy, has been utilized in a variety of autoimmune diseases, with increasing understanding of the safety of this and other similar reagents it is possible that rituximab could be used in combined therapy, i.e. together with other Food and Drug Administration (FDA)-approved drug(s) such as anti-inflammatory agents. It is conceivable that rituximab could be used as part of a monoclonal antibody cocktail simultaneously or in a sequential treatment protocol.
The first possible approach is the combination of rituximab with cytokines that can promote B cell depletion. In treatment of lymphoma, therapies using cytokines such as granulocyte colony-stimulating factor (G-CSF) and IL-2 in combination with rituximab have shown an increased therapeutic effect [98–100]. It is possible that rituximab treatment in combination with cytokines might enhance the efficiency of B cell depletion and therefore have better therapeutic efficacy for treating autoimmune diseases.
Other studies have shown that B cell depletion was less effective in autoimmune-prone mice compared with mice on a non-autoimmune-prone genetic background [49,52]. There are a number of possible reasons for this, including refractoriness to depletion that is inherited with the autoimmune susceptibility or due to the autoimmune state [52]. One possible reason for this resistance is impaired clearance by macrophages, which may contribute to resistance to depletion [52]. It is interesting that, in humans, it has been noted that efficiency of B cell depletion when rituximab treatment has been used in lymphoma [60] and SLE [101] relates to expression of the FcγRIIIa genotype. In addition, repeated rituximab treatment could lead to the loss of CD20 expression, as seen in lymphoma patients [102]. This might also be a problem in repeated rituximab treatment for autoimmune diseases. Therefore, combination therapy could provide a solution by inducing up-regulation of CD20 expression, such as with IFN-α, which could then enhance the effect of therapy as shown in treating lymphoma [103].
In addition to cytokines, rituximab could also be used with other well-accepted therapeutic reagents, such as intravenous immunoglobulins (IVIg) and plasma exchange. IVIg has been used widely to treat some autoimmune diseases [104,105]. A recent clinical study suggested that the combination of IVIg and rituximab was effective in patients with refractory pemphigus vulgaris, decreasing the levels of pathogenic anti-keratinocyte IgG4 antibodies but replacing immunoglobulins to prevent problems with humoral immunodeficiency [106]. The use of intravenous immunoglobulin in itself had been reported to be of benefit, although not in all cases, and the efficacy of the treatment in the study by Ahmed and colleagues was attributed to the rituximab [106].
Thus, there are possible combination therapies with rituximab that may either be considered to improve the efficacy of anti B cell-directed therapy or to reduce potential side effects that may be associated with this type of treatment.
Potential problems with the use of B cell-directed treatment
Therapy targeting B cells is now in clinical use in RA and is in clinical trials for a number of other autoimmune diseases. While this provides an important possibility for treatment in different diseases for which there are currently no cure, it is important to consider potential problems associated with this treatment. Firstly, will hypersensitivity reactions or development of anti-idiotype responses that block the effects of the drugs prevent repeated dosage? This is important as the indications, certainly for RA, are that repeated doses may be necessary [48]. Although this was not a major problem in this particular study, more trials will be needed to assess whether this will be a limiting feature. Secondly, and of major importance, considering the central role of B cells and antibodies in the adaptive immune response, is there a long-term risk of infection and progressive immunodeficiency? Thus far, the available studies suggest that, certainly for a single course of treatment, immunodeficiency has not been a major problem in most recipients. Probably this is more likely to be a potential concern if multiple treatments are required. At present, experience is limited, although again the study of Popa and colleagues indicate that overall immunodeficiency over 7 years was not a major feature. However, there was an increase in respiratory infections in a small number of patients in their study and this observation should be followed further. As more experience is gained from various clinical trials in other autoimmune diseases with B cell-targeted treatment, it is important to continue to weigh the benefits with the risks associated with what is undoubtedly potentially very powerful treatment.
Conclusion
Growing evidence from both animal studies and clinical trials has shown the therapeutic efficacy of B cell-directed therapy in autoimmune disorders. It is important to understand more clearly the pathogenesis of these diseases and the mechanisms of B cell depletion therapy, including immune regulation induced by the therapy. With increased knowledge, B cell-directed therapy may have an important place in treating a variety of autoimmune diseases.
Acknowledgments
The authors acknowledge funding from the Juvenile Diabetes Research Foundation (1-2007-586, 1-2007-184) and Diabetes UK (BDA-RD05/0003101) that support the research in their laboratories. C. Y. H. is a recipient of a Post-doctoral Research Fellowship from the Juvenile Diabetes Research Foundation (3-2008-426).
Disclosure
The authors declare no conflict of interest.
References
- 1.Burmester GR, Horneff G, Emmrich F. Management of early inflammatory arthritis. Intervention with immunomodulatory agents: monoclonal antibody therapy. Baillières Clin Rheumatol. 1992;6:415–34. doi: 10.1016/s0950-3579(05)80183-9. [DOI] [PubMed] [Google Scholar]
- 2.Lindberg C, Trysberg E, Tarkowski A, Oldfors A. Anti-T-lymphocyte globulin treatment in inclusion body myositis: a randomized pilot study. Neurology. 2003;61:260–2. doi: 10.1212/01.wnl.0000071852.27182.c7. [DOI] [PubMed] [Google Scholar]
- 3.Isaacs JD. T cell immunomodulation – the Holy Grail of therapeutic tolerance. Curr Opin Pharmacol. 2007;7:418–25. doi: 10.1016/j.coph.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Bofill M, Janossy G, Janossa M, et al. Subpopulations in the human fetus. J Immunol. 1985;134:1531–8. [PubMed] [Google Scholar]
- 5.Than S, Inaba M, Inaba K, Fukuba Y, Adachi Y, Ikehara S. Origin of thymic and peritoneal Ly-1 B cells. Eur J Immunol. 1992;22:1299–303. doi: 10.1002/eji.1830220527. [DOI] [PubMed] [Google Scholar]
- 6.Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 7.Cyster JG, Hartley SB, Goodnow CC. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. 1994;371:389–95. doi: 10.1038/371389a0. [DOI] [PubMed] [Google Scholar]
- 8.Nemazee D. Antigen receptor ‘capacity’ and the sensitivity of self-tolerance. Immunol Today. 1996;17:25–9. doi: 10.1016/0167-5699(96)80565-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cyster JG, Healy JI, Kishihara K, Mak TW, Thomas ML, Goodnow CC. Regulation of B-lymphocyte negative and positive selection by tyrosine phosphatase CD45. Nature. 1996;381:325–8. doi: 10.1038/381325a0. [DOI] [PubMed] [Google Scholar]
- 10.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 11.Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28:18–28. doi: 10.1016/j.immuni.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Browning JL. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nat Rev Drug Discov. 2006;5:564–76. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- 13.Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Annu Rev Immunol. 2006;24:467–96. doi: 10.1146/annurev.immunol.24.021605.090517. [DOI] [PubMed] [Google Scholar]
- 14.Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;6:394–403. doi: 10.1038/nri1838. [DOI] [PubMed] [Google Scholar]
- 15.Fritzler MJ. Challenges to the use of autoantibodies as predictors of disease onset, diagnosis and outcomes. Autoimmun Rev. 2008;7:616–20. doi: 10.1016/j.autrev.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Leslie D, Lipsky P, Notkins AL. Autoantibodies as predictors of disease. J Clin Invest. 2001;108:1417–22. doi: 10.1172/JCI14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bizzaro N. The predictive significance of autoantibodies in organ-specific autoimmune diseases. Clin Rev Allergy Immunol. 2008;34:326–31. doi: 10.1007/s12016-007-8059-5. [DOI] [PubMed] [Google Scholar]
- 18.Rose NR. Predictors of autoimmune disease: autoantibodies and beyond. Autoimmunity. 2008;41:419–28. doi: 10.1080/08916930802031686. [DOI] [PubMed] [Google Scholar]
- 19.Lee LA. Transient autoimmunity related to maternal autoantibodies: neonatal lupus. Autoimmun Rev. 2005;4:207–13. doi: 10.1016/j.autrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Greeley SA, Katsumata M, Yu L, et al. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat Med. 2002;8:399–402. doi: 10.1038/nm0402-399. [DOI] [PubMed] [Google Scholar]
- 21.Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol. 2000;1:475–82. doi: 10.1038/82717. [DOI] [PubMed] [Google Scholar]
- 22.Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–9. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 23.Menard LC, Minns LA, Darche S, et al. B cells amplify IFN-gamma production by T cells via a TNF-alpha-mediated mechanism. J Immunol. 2007;179:4857–66. doi: 10.4049/jimmunol.179.7.4857. [DOI] [PubMed] [Google Scholar]
- 24.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. 2008;358:676–88. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 25.Cyster JG, Ngo VN, Ekland EH, Gunn MD, Sedgwick JD, Ansel KM. Chemokines and B-cell homing to follicles. Curr Top Microbiol Immunol. 1999;246:87–92. doi: 10.1007/978-3-642-60162-0_11. discussion 3. [DOI] [PubMed] [Google Scholar]
- 26.Takemura S, Braun A, Crowson C, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–80. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 27.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6:205–17. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 28.Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7:344–53. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 29.Wengner AM, Hopken UE, Petrow PK, et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56:3271–83. doi: 10.1002/art.22939. [DOI] [PubMed] [Google Scholar]
- 30.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–30. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mauri C, Ehrenstein MR. The ‘short’ history of regulatory B cells. Trends Immunol. 2008;29:34–40. doi: 10.1016/j.it.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Shimomura Y, Mizoguchi E, Sugimoto K, et al. Regulatory role of B-1 B cells in chronic colitis. Int Immunol. 2008;20:729–37. doi: 10.1093/intimm/dxn031. [DOI] [PubMed] [Google Scholar]
- 33.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–50. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003;197:489–501. doi: 10.1084/jem.20021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei B, Velazquez P, Turovskaya O, et al. Mesenteric B cells centrally inhibit CD4+ T cell colitis through interaction with regulatory T cell subsets. Proc Natl Acad Sci USA. 2005;102:2010–15. doi: 10.1073/pnas.0409449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsitoura DC, Yeung VP, DeKruyff RH, Umetsu DT. Critical role of B cells in the development of T cell tolerance to aeroallergens. Int Immunol. 2002;14:659–67. doi: 10.1093/intimm/dxf032. [DOI] [PubMed] [Google Scholar]
- 37.Burke F, Stagg AJ, Bedford PA, English N, Knight SC. IL-10-producing B220+CD11c- APC in mouse spleen. J Immunol. 2004;173:2362–72. doi: 10.4049/jimmunol.173.4.2362. [DOI] [PubMed] [Google Scholar]
- 38.Jankovic D, Cheever AW, Kullberg MC, et al. CD4+ T cell-mediated granulomatous pathology in schistosomiasis is downregulated by a B cell-dependent mechanism requiring Fc receptor signaling. J Exp Med. 1998;187:619–29. doi: 10.1084/jem.187.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siragam V, Brinc D, Crow AR, Song S, Freedman J, Lazarus AH. Can antibodies with specificity for soluble antigens mimic the therapeutic effects of intravenous IgG in the treatment of autoimmune disease? J Clin Invest. 2005;115:155–60. doi: 10.1172/JCI22753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Chen F, Putt M, et al. B cell depletion with anti-CD79 mAbs ameliorates autoimmune disease in MRL/lpr mice. J Immunol. 2008;181:2961–72. doi: 10.4049/jimmunol.181.5.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dorner T, Burmester GR. New approaches of B-cell-directed therapy: beyond rituximab. Curr Opin Rheumatol. 2008;20:263–8. doi: 10.1097/BOR.0b013e3282f5e08d. [DOI] [PubMed] [Google Scholar]
- 42.Eisenberg R. Update on rituximab. Ann Rheum Dis. 2005;64(Suppl. 4):iv55–7. doi: 10.1136/ard.2005.042648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen Y, Polliack A, Zelig O, Goldfarb A. Monotherapy with rituximab induces rapid remission of recurrent cold agglutinin-mediated hemolytic anemia in a patient with indolent lympho-plasmacytic lymphoma. Leuk Lymph. 2001;42:1405–8. doi: 10.3109/10428190109097770. [DOI] [PubMed] [Google Scholar]
- 44.Wohrer S, Troch M, Zwerina J, et al. Influence of rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone on serologic parameters and clinical course in lymphoma patients with autoimmune diseases. Ann Oncol. 2007;18:647–51. doi: 10.1093/annonc/mdl467. [DOI] [PubMed] [Google Scholar]
- 45.Edwards JC, Leandro MJ, Cambridge G. B-lymphocyte depletion therapy in rheumatoid arthritis and other autoimmune disorders. Biochem Soc Trans. 2002;30:824–8. doi: 10.1042/bst0300824. [DOI] [PubMed] [Google Scholar]
- 46.Cambridge G, Leandro MJ, Teodorescu M, et al. B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006;54:3612–22. doi: 10.1002/art.22211. [DOI] [PubMed] [Google Scholar]
- 47.Ng KP, Cambridge G, Leandro MJ, Edwards JC, Ehrenstein M, Isenberg DA. B cell depletion therapy in systemic lupus erythematosus: long-term follow-up and predictors of response. Ann Rheum Dis. 2007;66:1259–62. doi: 10.1136/ard.2006.067124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popa C, Leandro MJ, Cambridge G, Edwards JC. Repeated B lymphocyte depletion with rituximab in rheumatoid arthritis over 7 yrs. Rheumatology. 2007;46:626–30. doi: 10.1093/rheumatology/kel393. [DOI] [PubMed] [Google Scholar]
- 49.Gong Q, Ou Q, Ye S, et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol. 2005;174:817–26. doi: 10.4049/jimmunol.174.2.817. [DOI] [PubMed] [Google Scholar]
- 50.Uchida J, Hamaguchi Y, Oliver JA, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–69. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu CY, Rodriguez-Pinto D, Du W, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–67. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. 2007;179:3351–61. doi: 10.4049/jimmunol.179.5.3351. [DOI] [PubMed] [Google Scholar]
- 53.Hamaguchi Y, Uchida J, Cain DW, et al. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol. 2005;174:4389–99. doi: 10.4049/jimmunol.174.7.4389. [DOI] [PubMed] [Google Scholar]
- 54.Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science. 2002;297:409–12. doi: 10.1126/science.1071632. [DOI] [PubMed] [Google Scholar]
- 55.Leandro MJ, de la Torre I. The pathogenic role of B cells in autoantibody-associated autoimmune diseases – lessons from B cell-depletion therapy. Clin Exp Immunol. 2009;157:191–7. doi: 10.1111/j.1365-2249.2009.03978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gurcan HM, Keskin DB, Stern JN, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. 2008 doi: 10.1016/j.intimp.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Xiu Y, Wong CP, Bouaziz JD, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol. 2008;180:2863–75. doi: 10.4049/jimmunol.180.5.2863. [DOI] [PubMed] [Google Scholar]
- 58.Fiorina P, Vergani A, Dada S, et al. Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes. 2008;57:3013–24. doi: 10.2337/db08-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacobi AM, Goldenberg DM, Hiepe F, Radbruch A, Burmester GR, Dorner T. Differential effects of epratuzumab on peripheral blood B cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis. 2008;67:450–7. doi: 10.1136/ard.2007.075762. [DOI] [PubMed] [Google Scholar]
- 60.Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 61.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–7. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 62.Cooper MD, Kearney JF, Gathings WE, Lawton AR. Effects of anti-Ig antibodies on the development and differentiation of B cells. Immunol Rev. 1980;52:29–53. doi: 10.1111/j.1600-065x.1980.tb00329.x. [DOI] [PubMed] [Google Scholar]
- 63.Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes. 1997;46:941–6. doi: 10.2337/diab.46.6.941. [DOI] [PubMed] [Google Scholar]
- 64.Cerny A, Kimoto M, Hugin AW, Merino R, Izui S. Anti-IgM treatment of C57BL/6-1pr/1pr mice: depletion of B cells reduces 1pr gene-induced lymphoproliferation and mononuclear cell vasculitis. Clin Exp Immunol. 1989;77:124–9. [PMC free article] [PubMed] [Google Scholar]
- 65.Vladutiu AO. Experimental autoimmune thyroiditis in mice chronically treated from birth with anti-IgM antibodies. Cell Immunol. 1989;121:49–59. doi: 10.1016/0008-8749(89)90004-x. [DOI] [PubMed] [Google Scholar]
- 66.Zhang L, French RR, Chan HT, et al. The development of anti-CD79 monoclonal antibodies for treatment of B-cell neoplastic disease. Ther Immunol. 1995;2:191–202. [PubMed] [Google Scholar]
- 67.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–35. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 68.Miga A, Masters S, Gonzalez M, Noelle RJ. The role of CD40–CD154 interactions in the regulation of cell mediated immunity. Immunol Invest. 2000;29:111–14. doi: 10.3109/08820130009062292. [DOI] [PubMed] [Google Scholar]
- 69.Splawski JB, Fu SM, Lipsky PE. Immunoregulatory role of CD40 in human B cell differentiation. J Immunol. 1993;150:1276–85. [PubMed] [Google Scholar]
- 70.Durandy A, Honjo T. Human genetic defects in class-switch recombination (hyper-IgM syndromes) Curr Opin Immunol. 2001;13:543–8. doi: 10.1016/s0952-7915(00)00256-9. [DOI] [PubMed] [Google Scholar]
- 71.Grammer AC, Slota R, Fischer R, et al. Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154–CD40 interactions. J Clin Invest. 2003;112:1506–20. doi: 10.1172/JCI19301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patel VL, Schwartz J, Bussel JB. The effect of anti-CD40 ligand in immune thrombocytopenic purpura. Br J Haematol. 2008;141:545–8. doi: 10.1111/j.1365-2141.2008.07039.x. [DOI] [PubMed] [Google Scholar]
- 73.Phipps RP. CD40: lord of the endothelial cell. Blood. 2008;112:3531–2. doi: 10.1182/blood-2008-08-175034. [DOI] [PubMed] [Google Scholar]
- 74.Sprague DL, Sowa JM, Elzey BD, Ratliff TL. The role of platelet CD154 in the modulation in adaptive immunity. Immunol Res. 2007;39:185–93. doi: 10.1007/s12026-007-0074-3. [DOI] [PubMed] [Google Scholar]
- 75.Li N. Platelet–lymphocyte cross-talk. J Leukoc Biol. 2008;83:1069–78. doi: 10.1189/jlb.0907615. [DOI] [PubMed] [Google Scholar]
- 76.Schneider E, Moreau G, Arnould A, et al. Increased fetal and extramedullary hematopoiesis in Fas-deficient C57BL/6-lpr/lpr mice. Blood. 1999;94:2613–21. [PubMed] [Google Scholar]
- 77.Schneider P, Tschopp J. BAFF and the regulation of B cell survival. Immunol Lett. 2003;88:57–62. doi: 10.1016/s0165-2478(03)00050-6. [DOI] [PubMed] [Google Scholar]
- 78.Mecklenbrauker I, Kalled SL, Leitges M, Mackay F, Tarakhovsky A. Regulation of B-cell survival by BAFF-dependent PKCdelta-mediated nuclear signalling. Nature. 2004;431:456–61. doi: 10.1038/nature02955. [DOI] [PubMed] [Google Scholar]
- 79.Ng LG, Mackay CR, Mackay F. The BAFF/APRIL system: life beyond B lymphocytes. Mol Immunol. 2005;42:763–72. doi: 10.1016/j.molimm.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 80.Groom J, Kalled SL, Cutler AH, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren's syndrome. J Clin Invest. 2002;109:59–68. doi: 10.1172/JCI14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Daridon C, Devauchelle V, Hutin P, et al. Aberrant expression of BAFF by B lymphocytes infiltrating the salivary glands of patients with primary Sjogren's syndrome. Arthritis Rheum. 2007;56:1134–44. doi: 10.1002/art.22458. [DOI] [PubMed] [Google Scholar]
- 82.George-Chandy A, Trysberg E, Eriksson K. Raised intrathecal levels of APRIL and BAFF in patients with systemic lupus erythematosus: relationship to neuropsychiatric symptoms. Arthritis Res Ther. 2008;10:R97. doi: 10.1186/ar2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neurol. 2008;4:557–67. doi: 10.1038/ncpneuro0901. [DOI] [PubMed] [Google Scholar]
- 84.Zekavat G, Rostami SY, Badkerhanian A, et al. In vivo BLyS/BAFF neutralization ameliorates islet-directed autoimmunity in nonobese diabetic mice. J Immunol. 2008;181:8133–44. doi: 10.4049/jimmunol.181.11.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cohen SB. Updates from B cell trials: efficacy. J Rheumatol Suppl. 2006;77:12–17. [PubMed] [Google Scholar]
- 86.Ding C, Foote S, Jones G. B-cell-targeted therapy for systemic lupus erythematosus: an update. BioDrugs. 2008;22:239–49. doi: 10.2165/00063030-200822040-00003. [DOI] [PubMed] [Google Scholar]
- 87.Gross JA, Dillon SR, Mudri S, et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease. impaired B cell maturation in mice lacking BLyS. Immunity. 2001;15:289–302. doi: 10.1016/s1074-7613(01)00183-2. [DOI] [PubMed] [Google Scholar]
- 88.Dall'Era M, Chakravarty E, Wallace D, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007;56:4142–50. doi: 10.1002/art.23047. [DOI] [PubMed] [Google Scholar]
- 89.Tak PP, Thurlings RM, Rossier C, et al. Atacicept in patients with rheumatoid arthritis: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating, single- and repeated-dose study. Arthritis Rheum. 2008;58:61–72. doi: 10.1002/art.23178. [DOI] [PubMed] [Google Scholar]
- 90.Evans JG, Chavez-Rueda KA, Eddaoudi A, et al. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol. 2007;178:7868–78. doi: 10.4049/jimmunol.178.12.7868. [DOI] [PubMed] [Google Scholar]
- 91.Serreze DV, Chapman HD, Varnum DS, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new ‘speed congenic’ stock of NOD.Ig mu null mice. J Exp Med. 1996;184:2049–53. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Akashi T, Nagafuchi S, Anzai K, et al. Direct evidence for the contribution of B cells to the progression of insulitis and the development of diabetes in non-obese diabetic mice. Int Immunol. 1997;9:1159–64. doi: 10.1093/intimm/9.8.1159. [DOI] [PubMed] [Google Scholar]
- 93.Wong FS, Wen L, Tang M, et al. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes. 2004;53:2581–7. doi: 10.2337/diabetes.53.10.2581. [DOI] [PubMed] [Google Scholar]
- 94.Silveira PA, Johnson E, Chapman HD, Bui T, Tisch RM, Serreze DV. The preferential ability of B lymphocytes to act as diabetogenic APC in NOD mice depends on expression of self-antigen-specific immunoglobulin receptors. Eur J Immunol. 2002;32:3657–66. doi: 10.1002/1521-4141(200212)32:12<3657::AID-IMMU3657>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 95.Wong FS, Wen L. B cells in autoimmune diabetes. Rev Diabet Stud. 2005;2:121–35. doi: 10.1900/RDS.2005.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Edwards JC, Leandro MJ, Cambridge G. B lymphocyte depletion therapy with rituximab in rheumatoid arthritis. Rheum Dis Clin North Am. 2004;30:393–403. viii. doi: 10.1016/j.rdc.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 97.Cambridge G, Leandro MJ, Edwards JC, et al. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 2003;48:2146–54. doi: 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- 98.Gluck WL, Hurst D, Yuen A, et al. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-Hodgkin's lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin Cancer Res. 2004;10:2253–64. doi: 10.1158/1078-0432.ccr-1087-3. [DOI] [PubMed] [Google Scholar]
- 99.Sashida G, Takaku TI, Honda S, et al. Granulocyte colony-stimulating factor (G-CSF) could enhance Fcgamma receptor expression in neutrophils of patients with B-cell lymphoma treated with rituximab. Leuk Lymph. 2005;46:789–91. doi: 10.1080/10428190500052347. [DOI] [PubMed] [Google Scholar]
- 100.Copelan E, Pohlman B, Rybicki L, et al. A randomized trial of etoposide and G-CSF with or without rituximab for PBSC mobilization in B-cell non-Hodgkin's lymphoma. Bone Marrow Transplant. 2008;43:101–5. doi: 10.1038/bmt.2008.306. [DOI] [PubMed] [Google Scholar]
- 101.Anolik JH, Campbell D, Felgar RE, et al. The relationship of FcgammaRIIIa genotype to degree of B cell depletion by rituximab in the treatment of systemic lupus erythematosus. Arthritis Rheum. 2003;48:455–9. doi: 10.1002/art.10764. [DOI] [PubMed] [Google Scholar]
- 102.Davis TA, Czerwinski DK, Levy R. Therapy of B-cell lymphoma with anti-CD20 antibodies can result in the loss of CD20 antigen expression. Clin Cancer Res. 1999;5:611–15. [PubMed] [Google Scholar]
- 103.Sivaraman S, Venugopal P, Ranganathan R, et al. Effect of interferon-alpha on CD20 antigen expression of B-cell chronic lymphocytic leukemia. Cytokines Cell Mol Ther. 2000;6:81–7. doi: 10.1080/13684730050515804. [DOI] [PubMed] [Google Scholar]
- 104.Dalakas MC. Role of IVIg in autoimmune, neuroinflammatory and neurodegenerative disorders of the central nervous system: present and future prospects. J Neurol. 2006;253(Suppl. 5):V25–32. doi: 10.1007/s00415-006-5004-0. [DOI] [PubMed] [Google Scholar]
- 105.Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol. 2008;26:513–33. doi: 10.1146/annurev.immunol.26.021607.090232. [DOI] [PubMed] [Google Scholar]
- 106.Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006;355:1772–9. doi: 10.1056/NEJMoa062930. [DOI] [PubMed] [Google Scholar]