

Abstract
Buruli ulcer (BU) is the third most common mycobacterial disease in immunocompetent hosts. BU is caused by Mycobacterium ulcerans, which produces skin ulcers and necrosis at the site of infection. The principal virulence factor of M. ulcerans is a polyketide-derived macrolide named mycolactone, which has cytotoxic and immunosuppresive activities. We determined the severity of inflammation, histopathology and bacillary loads in the subcutaneous footpad tissue of BALB/c mice infected with 11 different M. ulcerans isolates from diverse geographical areas. Strains from Africa (Benin, Ghana, Ivory Coast) induced the highest inflammation, necrosis and bacillary loads, whereas the strains collected from Australia, Asia (Japan, Malaysia, New Guinea), Europe (France) and America (Mexico) induced mild inflammation. Subsequently, animals were infected with the strain that exhibited the highest (Benin) or lowest (Mexico) level of virulence in order to analyse the local immune response generated. The Mexican strain, which does not produce mycolactone, induced a predominantly T helper type 1 (Th1) cytokine profile with constant high expression of the anti-microbial peptides beta defensins 3 and 4, in co-existence with low expression of the anti-inflammatory cytokines interleukin (IL)-10, IL-4 and transforming growth factor (TGF)-β. The highly virulent strain from Benin which produces mycolactone A/B induced the opposite pattern. Thus, different local immune responses were found depending on the infecting M. ulcerans strain.
Keywords: animal model, Buruli ulcer, cytokines, immunopathology, Mycobacterium ulcerans
Introduction
Buruli ulcer (BU), which is considered the third most common mycobacterial disease in immunocompetent individuals after tuberculosis and leprosy [1,2], is a skin disease caused by Mycobacterium ulcerans characterized by the production of ulcers and massive necrosis at the site of infection [2]. Three clinical stages of BU have been described: pre-ulcerative (which can present as a nodule, plaque, papule or oedema), ulcerative and healed (scar) [3]. Histological studies have shown extensive necrosis of the skin and subcutaneous tissue with minimal inflammation during the ulcerative stage, whereas numerous granulomas are found in the healing stage [2,3]. The prevalence of BU has been increasing dramatically in West Africa during the last 10 years and no completely efficient drug treatment is yet available. Surgical excision remains the best current therapy [2].
M. ulcerans has been detected in stagnant or slowly moving water from many tropical and temperate parts of the world [1,4]. Mycobacterial infection is acquired when skin injury comes into contact with the contaminated water [2], but there is some evidence for vector-mediated transmission in laboratory mice by the bite of aquatic insects (Naucoridae) that are infected with this mycobacterium [5,6].
In contrast to other pathogenic mycobacteria, which grow as facultative intracellular organisms inside macrophages, M. ulcerans occurs in lesions primarily as extracellular microcolonies [2]. The principal virulence factor of M. ulcerans is a polyketide-derived macrolide named mycolactone, which has cytotoxic, analgesic and immunosuppressive activities [7,8], and is encoded in a 174-kb plasmid [9]. It has been reported that disease severity is related to the type and amount of mycolactone produced by each strain, i.e. African strains, which produce the greatest number and quantity of mycolactones, and are associated with more severe forms of the disease [10]. Mycolactone is capable of causing cytopathic effects and apoptosis in different cell types [11].
From the immunological perspective, suppression of tumour necrosis factor (TNF)-α, interleukin (IL)-2 and IL-10 secretion by high-density lipid fractions of M. ulcerans growth media has been reported in in vitro assays [7]. BU patients can mount a humoral immune response, as evidenced by antibody production against mycobacterial antigens, but at the same time they suffer profound systemic T cell anergy to mycobacterial antigens [12]. It has been proposed that the uptake of M. ulcerans by phagocytes may orientate the host immune response towards a T helper 1 (Th1) type, ineffective for clearance of extracellular bacteria. Then the anti-phagocytic and cytotoxic properties of mycolactone may help the organisms to evade the immune system [13]. Moreover, high levels of interferon (IFN)-γ expression but undetectable IL-10 production have been demonstrated in lesions from patients suffering the nodular stage, whereas high levels of IL-10 but low or undetectable IFN-γ expression have been found in lesions from patients with the ulcerative form [14]. Similarly, a high percentage of IFN-γ-positive cells is found in situ in granulomas from chronic ulcerative lesions, whereas high percentages of IL-10 and transforming growth factor beta (TGF)-β-positive cells exist in the early ulcerative form of the disease without granulomas [15]. Because the level of virulence among M. ulcerans strains is related to the type of produced mycolactone and the specific local immune response, we used the experimental BU model in the footpads of BALB/c mice to assess the extent of inflammation, bacillary loads and histopathological findings, comparing a wide range of M. ulcerans clinical isolates from diverse parts of the world. Then, we analysed the local immunopathological changes during infections produced by selected strains with either high or low virulence.
Material and methods
Experimental model of BU in mice
We used 11 different and well-characterized Mycobacterium ulcerans strains from the collection of the Mycobacteriology Unit at the Tropical Medicine Institute, Antwerp, Belgium. The year and geographical site of strain isolation, as well as the type of mycolactone produced by each strain, is showed in Table 1. All the strains were wild-type, low-passage clinical isolates. Bacilli were cultured in Middlebrook 7H9 liquid media supplemented with oleic acid albumin dextrose complex (OADC), and after 6 or 8 weeks of growth they were recovered in sterile endotoxin-free isotonic saline solution, aliquoted and maintained at −70°C until use. Before use, bacilli were adjusted at 5 × 105/40 µl and their viability was checked using a fluorescein diacetate stain.
Table 1.
Code, geographical origin, year of isolation and type of produced mycolactone of the M. ulcerans isolates used in this study.
| ITM no. | Species identification | Geographical origin | Providers | Year | Type of mycolactone |
|---|---|---|---|---|---|
| 941331 | M. ulcerans | Papua New Guinea | ITM | 1994 | C |
| 1441 | M. ulcerans | Benin | ITM | 2000 | A/B |
| 5151 | M. ulcerans | D.R. Congo (Zaire) | ITM | 1972 | A/B |
| 97483 | M. ulcerans | Ghana | ITM | 1997 | A/B |
| 941328 | M. ulcerans | Malaysia | ITM | 1994 | A/B |
| 5114 | M. ulcerans | Mexico | PL | 1953 | – |
| 5147 | M. ulcerans | Australia | JS | 1961 | C |
| 8756 | M. shinshuense | Japan | ATCC 33728 | 1980 | D |
| 940511 | M. ulcerans | Ivory Coast | ITM | 1994 | A/B |
| 970680 | M. ulcerans | Togo | ITM | 1997 | A/B |
ITM, Institute of Tropical Medicine, Antwerp, Belgium; ATCC, American Type Culture Collection; JS, J. Stanford, School of Pathology, London, UK; PL, P. Lavalle, Centro Dermatologico Pascua, Mexico City, Mexico. M. ulcerans, Mycobacterium ulcerans; M. shinshuense, Mycobacterium shinshuense.
The experimental model was set up in 6–8-week-old male BALB/c mice. Groups of 36 animals were inoculated intradermally in both footpads, with 5 × 105/40 µl of each different strain. Preliminary experiments performed in our laboratory indicated that this was the best dose. The control group received isotonic saline solution through the same route. Experiments were performed in P3 biosafety facilities in accordance to institutional guidelines for animal care and experimentation. To determine the level of inflammation, footpad swelling was recorded in each mouse at selected time-points using an engineering micrometer. The whole experiment was repeated and results were pooled.
Quantification of bacillary loads by colony-forming units (CFUs) determination
Three footpads, right or left, collected from infected and control mice at each time-point were frozen immediately in liquid nitrogen and kept at −72°C until use. Tissues were homogenized with a Polytron homogenizer (Kinematica, Luzern, Switzerland) in sterile tubes containing 3 ml phosphate-buffered saline (PBS) 1×-Tween 80 (0·05%). The concentrated suspensions and decimal dilutions were cultured in 7H9 + OADC solid medium at 30°C in a 5% CO2 atmosphere and colonies were counted after 8 weeks of culture.
Preparation of skin samples for histopathology, immunohistochemistry and automated morphometry
Groups of four mice infected with each of the 11 strains and the control were killed by exsanguination at 4, 14, 21, 28, 60 and 120 days post-infection; one footpad (right or left, alternately) was dissected immediately and fixed in absolute ethanol for 24 h, dehydrated and embedded in paraffin. Skin sections of 5 µm thickness were mounted on silane-treated slides, deparaffinized and stained with haematoxylin and eosin (H&E) and Ziehl–Neelsen stain.
Due to the striking differences in inflammation that we found between the strains from Benin and Mexico, we selected these for detailed comparison of the local immune response induced throughout the course of the disease. The same paraffin-embedded material prepared for the histopathological studies was used to determine the local cytokine production by immunohistochemistry. Skin and subcutaneous sections from mice infected with either of these two strains and from controls at each time-point were deparaffinized and maintained in HEPES buffer 1× (HEPES, NaCl, CaCl2). The endogenous peroxidase activity was blocked with 6% H2O2 dissolved in PBS 1× + 0·1% sodium azide and incubated for 1 h. After blocking with normal swine sera, tissue sections were incubated with primary specific polyclonal rabbit antibodies overnight at 4°C at optimal dilutions, against TNF-α, IFN-γ, TGF-β, IL-4, IL-10, murine beta defensin 3 (mBD-3) and mBD-4 (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA). Secondary biotinylated antibodies [anti-rabbit–biotin immunoglobulin (Ig)G or anti-goat–biotin IgG] were used to detect primary bound antibodies, followed by horseradish peroxidase (HPR)-conjugated avidin. Enzyme-linked antibodies revealed by 3,3-diaminobenzidine (DAB)/hydrogen peroxide for 5–10 min at room temperature. Tissue sections were counterstained with haematoxylin.
For determination of inflammatory cell subsets, at each time-point, right or left footpads from three mice infected with Benin or Mexico strains were dissected immediately, frozen by immersion in liquid nitrogen and kept at −72°C until use. Tissues were embedded into a cryoprotecting medium (OCT) and 5-µm thick sections were obtained and mounted on silane-covered slides, fixed in acetone (at −20°C) for 10 min and then in chloroform for 20 min at room temperature, and maintained in HEPES buffer 1× for at least 5 min. The endogenous peroxidase activity was blocked as described above. After blocking with normal swine sera, tissue sections were incubated with primary antibodies against murine CD4 T lymphocytes, CD8 T cells and macrophages CD11b (all from BD Pharmingen, San Diego, CA, USA) overnight at 4°C, using predetermined optimal dilutions. Secondary biotinylated antibodies were used to detect bound primary antibodies and revealed by incubation with 3,3-diaminobenzidine/hydrogen peroxide for 5–10 min at room temperature. Tissue sections were counterstained with Harris haematoxylin.
For morphometry, at least three random fields from each section from three different mice and for each time-point and each strain were studied. The total number of inflammatory cells, specifically around the necrotic ulcerative area, was quantified and the cytokine-immunostained cells or positive cells for each subset marker were determined using an automated image analyser (Q Win Leica, Milton Keynes, UK), then the percentage of immunostained cells was estimated.
Kinetics of cytokine gene expression during M. ulcerans infection determined by real-time reverse transcription–polymerase chain reaction (RT–PCR)
After killing the mice, footpads from three different mice at each time-point infected with M. ulcerans from Benin and Mexico were removed, and the tissue was frozen immediately by immersion in liquid nitrogen. RNA was isolated using the reagent trizol (Gibco BRL, Gaithersburgh, MD, USA), as described previously [6,11]. cDNA was synthesized by using Moloney murine leukaemia virus reverse transcriptase (Gibco BRL) and oligo-dT priming. Quality and quantity of RNA were evaluated through spectrophotometry (260/280) and on agarose gels. Reverse transcription of the mRNA was performed using 5 µg RNA, 2 µM oligo-dT 15 primer (Promega, Madison, WI, USA), 10 units/µl ribonuclease inhibitor (Invitrogen, Carlsbad, CA, USA), 1× RT buffer, 0·5 mM of each dNTP and 4 units Omniscript Reverse Transcriptase (Qiagen, Inc., Valencia, CA, USA). Real-time PCR was performed using the 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA) and Quantitect SYBR Green Mastermix kit (Qiagen, Inc.). Standard curves of the quantified and diluted PCR product, as well as negative controls, were included in each PCR run. Specific primers were used for the following targets: glyceraldehyde-3-phosphate dehydrogenase (G3PDH): 5′-CATTGTGGAAGGGCTCATGA-3′, 5′-GGAAGGCCATGCCAGTGAGC-3′; TNF-α: 5′-TGTGGCTTCGACCTCTACCTC-3′, 5′-GCCGAGAAAGGCTGCTTG-3′; IFN-γ: 5′-GGTGACATGAAAATCCTGCAG-3′, 5′-CCTCAAACTTGGCAATACTCATGA-3′; IL-4: 5′-CGTCCTCACAGCAACGGAGA-3′, 5′-GCAGCTTATCGATGAATCCAGG-3′; IL-10: 5′-AAAGGCACTGCACGACATAGC-3′, 5′-TGCGGAGAACGTGGAAAAAC-3′; TGF-β: 5′-AGGGCTACCATGCCAACTTCT-3′, 5′-CCGGGTTGTGTTGGTTGTACA-3′; mBD-3: 5′-TCTGTTTGCATTTCTCCTGGTG-3′, 5′-TAAACTTCCAACAGCTGGAGTGG-3′; mBD-4: 5′-TCTGTTTGCATTTCTCCTGGTG-3′, 5′-TTTGCTAAAAGCTGCAGGTGG-3′; and macrophage inflammatory protein (MIP-1b/ccl 4): 5′-TCTCTCCTCTTGCTCGTGGC-3′, 5′-ATTGGTGCTGAGAACCCTGG-3′.
Cycling conditions used were: initial denaturation at 95°C for 15 min, followed by 40 cycles at 95°C for 20 s, 60°C for 20 s and 72°C for 34 s. Quantities of the specific mRNA were measured according to the corresponding gene-specific standard. The mRNA copy number of each cytokine was related to 1 million copies of mRNA encoding the G3PDH gene.
Results
Inflammation, bacillary loads and histopathology
Figure 1 shows the kinetics of the inflammatory response reported as footpad thickness. During the first month, a similar mild inflammation was seen in the footpad of animals infected with any of the strains. Then, after 2 and 4 months of infection, a huge inflammation with ulcer formation was produced by M. ulcerans isolated from Africa (Benin, Ghana, Togo), while the rest of the strains collected from Australia, Asia (Japan, Malaysia, New Guinea), Europe (France) and America (Mexico) induced mild inflammation without ulcer formation. Thus, according to these results it was possible to separate these isolates into two groups, with high and low virulence. Benin 1441 strain induced the highest inflammation with extensive necrosis and ulceration, which motivated us to kill the animals for humanitarian reasons 2 months after infection. In contrast, strain Mexico 5114 produced the lowest inflammatory response.
Fig. 1.
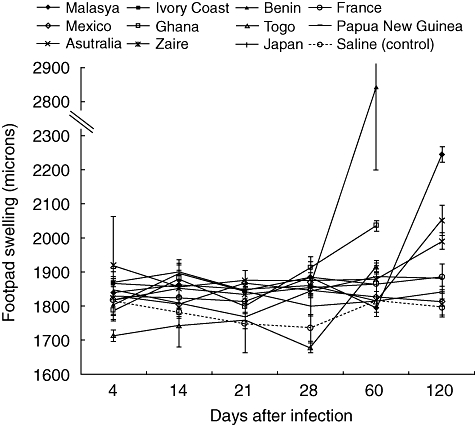
Inflammation during infection induced by different Mycobacterium ulcerans strains. Kinetics of inflammation determined by footpad thickness measured with an engineering micrometer at the indicated days after infection with 11 different M. ulcerans isolates. Animals infected with strains from Benin, Ghana and Ivory Coast showed massive necrosis and inflammation after 2 months of infection and were killed for humanitarian reasons.
Figure 2 shows the bacillary loads determined by CFU counts. All the M. ulcerans strains yielded similar bacillary loads during the first month post-infection. Then, after 2 months, animals infected with strain Benin 1441 showed a sixfold higher CFU count than mice infected with strain Mexico 5114, while the others showed intermediate bacillary loads. After 4 months of infection, almost no bacilli growth was determined in animals infected with the Mexican strain. Thus, we decided to perform histopathology and immunological studies comparing the strains from Benin and Mexico.
Fig. 2.
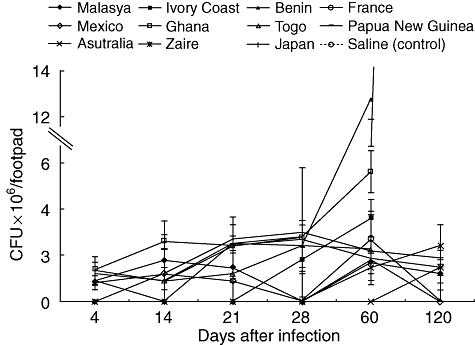
Kinetics of bacillary loads in footpads infected with different Mycobacterium ulcerans isolates. Skin and subcutaneous tissues from BALB/c footpads infected with different M. ulcerans isolates were dissected at the time-points indicated and used to determine colony forming units. Data are expressed as mean and standard deviation from three different animals per time-point and strain.
After 4 days of infection, M. ulcerans strain Mexico 5114 induced oedema and an inflammatory infiltrate composed predominantly of macrophages and lymphocytes, with few neutrophils. This type of inflammation was maintained until day 14 post-infection, when some epithelioid activated macrophages were also seen. At day 21 after infection, mild oedema was observed with chronic inflammation without necrosis. One week later, a well-organized inflammatory infiltrate was seen, containing macrophages and lymphocytes with dilated blood vessels and numerous distended lymphatic vessels. Ziehl–Neelsen staining showed numerous extracellular bacilli (Fig. 3a,b). At days 60 and 120 post-infection, the inflammatory infiltrate was distributed homogeneously in the subcutaneous and muscle tissue, and numerous epithelioid macrophages and lymphocytes were seen forming granuloma-like structures in co-existence with occasional mycobacteria, without significant histological changes in the adjacent epidermis (Fig. 3c,d). In contrast, strain Benin 1441 induced oedema and inflammation dominated by neutrophils with few macrophages and lymphocytes at the site of inoculation after 4 days of infection. At day 14 post-infection, extensive and diffuse inflammation dominated by lymphocytes and macrophages was seen in the subcutaneous tissue, in co-existence with slight epidermal acanthosis. One week later, there was mild oedema and focal areas of necrosis with numerous dilated blood vessels. At day 28 post-infection, these necrotic areas were larger with extensive oedema and few inflammatory cells (Fig. 3e), but abundant chronic inflammatory infiltrate was seen in the perinecrotic areas. At day 60 after infection, there were huge areas of necrosis affecting the whole epidermis with extensive detachment (acantholysis) and ulcer formation (Fig. 3f). Dermis and subcutaneous tissue showed extensive oedema and massive necrosis with eosinophilic debris and scarce inflammatory cells, whereas the neighbouring areas showed abundant lymphocytes and macrophages near the blood vessels (Fig. 3g). Acid-fast staining showed massive numbers of extracellular bacilli in the necrotic areas (Fig. 3h).
Fig. 3.
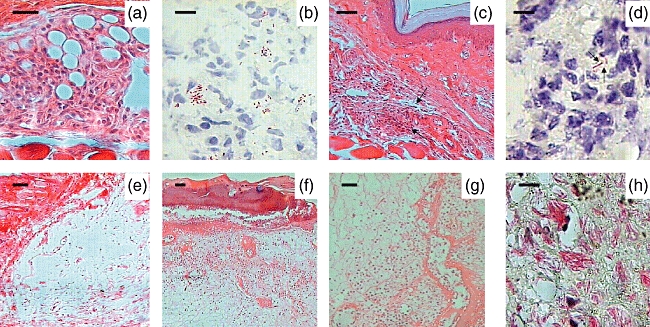
Representative histopathology of BALB/c subcutaneous footpad tissue infected with Mycobacterium ulcerans strains from Mexico and Benin. (a) A well-organized inflammatory infiltrate containing lymphocytes and macrophages is seen after 28 days of infection with strain 5114 from Mexico (400×, bar = 50 µ). (b) In the same lesion there are numerous extracellular bacilli as demonstrated by Ziehl–Neelsen staining (1000×, bar = 10 µ). (c) After 2 months of infection with M. ulcerans from Mexico, lymphocytes and macrophages are organized in small nodules forming granuloma-like structures (arrows) (40×, bar = 200 µ). (d) In these lesions, occasional bacilli were found (arrows) (1000×, bar = 10 µ). (e) Extensive oedema with slight inflammatory infiltrate is seen after 1 month of infection with strain 1441 from Benin (100×, bar = 100 µ). (f) After 2 months of infection, the Benin strain induced extensive oedema, massive necrosis with acantholysis and a slight inflammatory infiltrate (40×, bar = 200 µ). (g) In the same section, the connective tissue around the necrotic areas shows abundant chronic inflammatory infiltrate (40×, bar = 200 µ). (h) These necrotic areas show massive amount of extracellular bacilli revealed by Ziehl–Neelsen staining (1000×, bar = 10 µ).
Immunohistochemistry and automated morphometry of the local subsets of inflammatory cells and cytokine expression
The Benin strain induced extensive necrosis with paucicellular inflammation during late infection, but the tissue around these necrotic areas showed numerous inflammatory cells, which were similar in number to those counted in the subcutaneous footpad tissue from mice infected with the strain from Mexico (Figs 3g and 4). When these inflammatory cells were compared using immunohistochemistry and automated morphometry, similar percentages of CD4 immunostained cells were found in mice infected with either strain, except at day 60 post-infection when 30% more positive cells were seen in mice infected with the Mexican strain (Fig. 4). Similar percentages of CD8 immunostained cells were also seen in mice infected with either strain, except at day 28 of infection, when twofold more CD8+ cells were found in mice infected with the Benin strain. In contrast, a higher percentage of macrophages was induced by the Benin strain throughout the infection, except at day 60 when a similar percentage of macrophages was seen (Fig. 4).
Fig. 4.
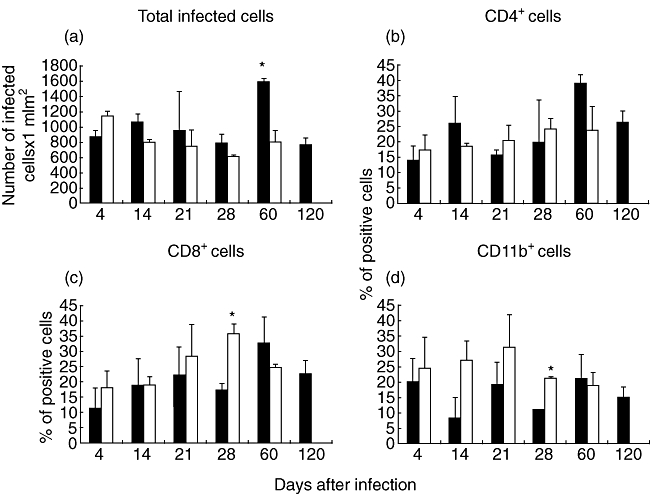
Kinetics of inflammatory cell numbers and percentage of cellular subsets during infection with Mycobacterium ulcerans strains from Mexico and Benin. The total number of inflammatory cells (a) and the percentage of CD4 (b), CD8 (c) and macrophages CD11b+ (d) was determined by immunohistochemistry and automated morphometry in frozen sections from animals infected with strain 5114 from Mexico (black bars) and isolate 1441 from Benin (white bars). Three different randomly chosen areas at 400× magnification from the subcutaneous tissue of four different mice per time-point were studied. Data are expressed as means and standard deviation; asterisks represent statistical significance (P < 0·005).
Mice infected with M. ulcerans strain from Mexico showed a higher percentage of IFN-γ immunostained cells than animals infected with the Benin strain, except at day 28 when similar percentages were induced by both strains (Figs 5 and 6a). Interestingly, in animals infected with the Mexican strain some macrophages showed IFN-γ immunoreactivity (Fig. 6a). A higher percentage of TNF-α immunostained macrophages was observed in mice infected with the Benin strain at days 4 and 14; during late infection, similar percentages were seen in animals infected with either strain (Figs 5 and 6). During early infection, from days 4 to 21, similar percentages of TGF-β-positive cells, preferentially macrophages, were recorded, but during late infection a higher percentage of TGF-β immunostained cells was observed in animals infected with the Benin isolate (Figs 5 and 6), Some lymphocytes and mast cells showed immunoreactivity to IL-4, and similar percentages of positive cells were detected during the whole course of infection, whereas after 21 days of infection higher percentages of IL-10-immunostained cells were induced by the Benin strain (Figs 5 and 6).
Fig. 5.
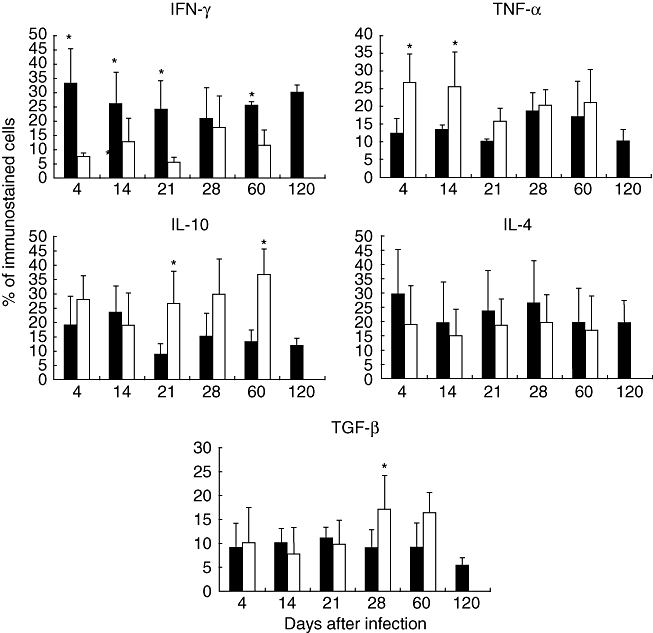
Determination of percentages of cells immunostained for cytokine production during infection with Mycobacterium ulcerans from Mexico (black bars) and Benin (white bars). Three different randomly chosen areas were used to determine, by immunohistochemistry and automated morphometry, the number of positive and negative cells in the inflammatory infiltrate of three different mice per time-point. Then, the percentage of positive cells was determined for each cytokine indicated. Data are expressed as mean and standard deviation; asterisks represent statistical significance (P < 0·005).
Fig. 6.
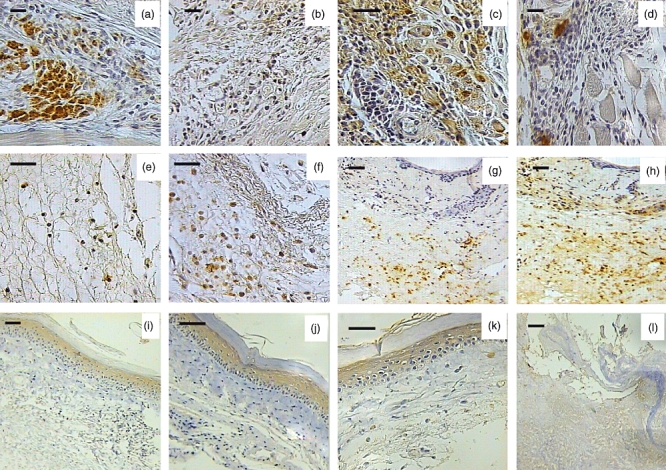
Representative photographs of immunohistochemical cytokine detection in the inflammatory infiltrate located in the mouse footpads after 60 days of infection with Mycobacterium ulcerans strains from Mexico and Benin. (a) Numerous interferon (IFN)-γ immunostained cells are seen in granulomas induced by strain 5114 from Mexico (400×, bar = 50 µ). (b) In contrast, scarce interleukin (IL)-10 immunoreactive cells are seen in the same lesion (100×, bar = 100 µ). (c) These granulomas also show a moderate number of macrophages immunoreactive for tumour necrosis factor (TNF)-α (400×, bar = 50 µ). (d) Occasional transforming growth factor (TGF)-β immunostained cells are seen in the lesions induced by the Mexican strain (400×, bar = 50 µ). (e) Scarce cells immunoreactive to IFN-γ are seen in lesions induced by strain 1441 from Benin (400×, bar = 50 µ). (f) In contrast, numerous IL-10 immunoreactive cells are seen in these lesions produced by the African strain (400×, bar = 50 µ). (g) A modest number of TNF-α immunostained cells is seen in late lesions produced by the Benin strain (100×, bar = 100 µ). (h) In comparison, more cells with immunoreactivity to TGF-β are present in late lesions induced by the African strain (100×, bar = 100 µ). (i) There is mBD-3 immunostaining in the epidermis after 28 days post-infection and 60 days (100×, bar = 100 µ). (j) Post-infection with the strain from Mexico (400×, bar = 50 µ). (k) Strong mBD-3 immunostaining is seen in the epidermis after 28 days of infection with the Benin strain (400×, bar = 50 µ). (l) In contrast, there is no mBD-3 immunostaining in the ulcerative lesions produced by the African strain after 60 days of infection (100×, bar = 100 µ).
Immunohistochemistry to detect beta-defensins showed strong mBD-3 immunostaining in the epidermis of mice infected with the Benin strain, from day 4 until day 28 post-infection. Some macrophages located in the dermal inflammatory infiltrate were also positive. Afterwards, mBD-3 expression vanished (Fig. 6). The strain from Mexico also induced rapid and strong epidermal mBD-3 immunostaining that was maintained during the whole experiment (Fig. 6).
Kinetics of cytokine gene expression determined by real-time PCR
Figure 7 shows the comparative gene expression kinetics of cytokines and β-defensins during the course of infection induced by M. ulcerans strains from Mexico and Benin. High and progressive expression of IFN-γ, peaking at day 120, was observed in lesions induced by the strain from Mexico, whereas animals infected with the Benin strain showed significantly lower and stable expression of this cytokine. Mice infected with the Benin strain showed high TNF-α expression during the first month of infection, followed by a sharp decrease once necrosis and ulcers had appeared. In contrast, during the later phases mice infected with the strain from Mexico showed stable TNF-α expression during the whole course of infection that did not fall significantly.
Fig. 7.
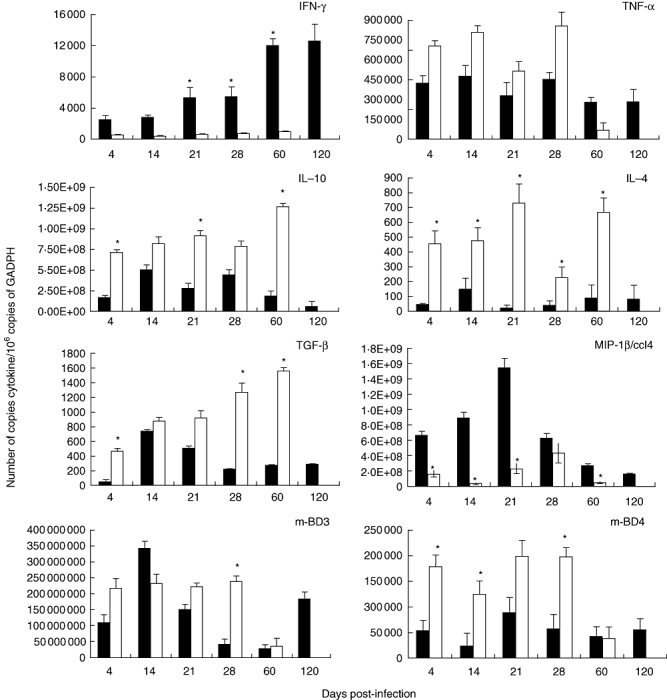
Kinetics of expression of cytokines and β-defensins during Mycobacterium ulcerans infection. Subcutaneous tissue from BALB/c mice obtained at different time-points post-infection with M. ulcerans isolate 5114 from Mexico (black bars) or strain 1441 from Benin (white bars) was used to determine the gene expression of the indicated cytokines, chemokine or β-defensins by real-time reverse transcription–polymerase chain reaction. Results are expressed as mean with standard deviation from four mice groups. Asterisks represent statistical significance between both groups (*P < 0·005).
Animals infected with the isolate from Benin showed progressively increasing expression of the anti-inflammatory immunomodulator cytokines IL-10, IL-4 and TGF-β, peaking at day 60 post-infection, when extensive necrosis and ulceration were produced. In contrast, the non-ulcerative lesions produced by strain Mexico showed lower, stable expression of these cytokines. We also determined macrophage inflammatory protein 1β (MIP-1β), a significant chemokine that attracts macrophages and lymphocytes; lesions induced by the isolate from Mexico showed high expression during early infection followed by a pronounced decrease after 1 month of infection, while lesions produced by the strain from Benin induced lower expression throughout infection.
Because β-defensins can be induced by mycobacterial infection, and the skin is a significant source of these natural anti-microbial peptides [16,17], we also determined the kinetics of expression of the genes encoding β-defensins 3 and 4 throughout the infection. Animals infected with the Benin strain showed high expression of both β-defensins during early infection, followed by a sharp decrease during the late ulcerative stage. Mice infected with the Mexican strain showed high early expression of mBD-3 for the first 3 weeks of infection, followed by a temporary decrease and high expression again at day 120 post-infection, whereas mBD-4 was expressed throughout (Fig. 7).
Discussion
There is a wide variation of the clinical presentation in BU patients, and some clinical evidence suggests that there are geographical differences in the form and severity of the disease [1]. In this regard, it has been demonstrated that a heterogeneous mixture of mycolactone variants, with identical biological activities but different potencies, can be extracted from M. ulcerans strains isolated from different geographic areas [10]. Mycolactone A/B is the most active and it has been identified in many isolates, particularly from Africa, whereas strain 5114 from Mexico did not produce this type of toxin. Mycolactone C and D are the dominant forms in strains from Australia and Asia, respectively [10]. Thus mycolactone variants are conserved within specific geographic areas, suggesting a correlation between the mycolactone profile and virulence. This correlation was confirmed by a previous study using the footpad model in BALB/c mice, demonstrating that African strains are highly cytotoxic and produce a persistent acute inflammatory infiltrate throughout the infection, whereas the low-virulence strain 5114 from Mexico induced chronic inflammation with granuloma-like structures without necrosis and ulceration [18]. We have now used the same experimental model confirming these results and extended the observations by showing, for the first time, that the highly virulent African strain Benin 1441 and the low-virulence strain 5114 from Mexico induce very different local cytokine patterns. Although this comparative study between a single mycolactone AB producer strain with a non-mycolactone-secreting strain cannot be extrapolated to define geographically linked patterns of virulence, it permitted analysis of the influence of mycolactone on the immunopathology along the infection.
The initial or pre-ulcerative phase in BU patients is characterized by a hard subcutaneous nodule or plaque. This phase could correspond to the early phase of infection (first month) in our experimental model. During this phase, both strains induced mild mixed acute and chronic inflammation, but the low-virulence strain from Mexico did not progress to the ulcerative phase and induced higher expression of IFN-γ than the Benin strain. The latter induced extensive necrosis and ulceration after 4 weeks of infection. These results are in agreement with several studies in BU patients during the pre-ulcerative phase, which reported high production of IFN-γ in peripheral blood and intralesional cells, whereas during the ulcerative phase a profound systemic anergy to mycobacterial antigens was seen, manifested by lower lymphocyte proliferation and IFN-γ production in response to stimulation with live M. ulcerans or M. bovis bacille Calmette–Guérin (BCG) [12,14,15].
Another cytokine that is crucial in the anti-mycobacterial immune response is TNF-α. Interestingly, TNF-α expression was also very high during the pre-ulcerative phase in animals infected with the Benin strain, followed by a sharp decrease during the ulcerative stage, whereas the strain from Mexico induced a constant high expression of TNF-α. These results are consistent with the observation that nodular lesions from BU patients exhibited more TNF-α expression than ulcerative lesions [14]. Thus, it seems that, as happens in tuberculosis and leprosy, M. ulcerans infection is also controlled by a Th1 response and TNF-α.
Low production of the immunomodulatory cytokine IL-10 during the whole course of infection was induced by M. ulcerans from Mexico, while the strain from Benin induced significantly higher and stable production of IL-10 during the first month of infection, followed by a twofold increase during the ulcerative stage. These results are in agreement with previous studies in BU patients that have reported low to undetectable levels of IL-10 in pre-ulcerative nodular lesions, and higher levels of this cytokine in ulcerative lesions [14]. However, our results show that even during early infection, when both strains are inducing similar inflammation and the bacillary loads are similar, the African isolate induced higher expression of IL-10, suggesting that some constitutive factors of this strain are efficient at deviating the cytokine profile towards an anti-inflammatory response. This could be a direct consequence of a very potent mycolactone accumulation [10,19,20]. In fact, all our African strains produced mycolactone A/B; however, we found differences in the severity of the inflammation and ulceration. Perhaps Benin strain 1441 produced higher amounts of mycolactone than the other tested African strains.
There are reports of a Th2 response in BU patients. Some IL-4 and IL-13 gene expression was detected in lesions from patients in French Guyana with ulcerative lesions, whereas neither cytokine was found in patients with the nodular form [14]. Similarly, following stimulation of peripheral blood cells with M. ulcerans or M. bovis BCG, Australian patients with ulcerative lesions mounted a Th2 response, whereas unaffected contacts responded mainly with a Th1 response [21]. Although the number of mRNA copies measured by real-time PCR was low, we observed low expression of IL-4 during the whole infection period in mice infected with the Mexican strain, and a significantly higher and progressive expression of this Th2 cytokine in animals infected with the African strain. In fact, Th2 cytokines exert profound IFN-γ down-regulating activity; thus, besides the direct suppression by mycolactone, production of these cytokines may be involved in the suppression of cell-mediated immunity. Moreover, in animals infected with the strain from Benin, we also observed a significantly higher and progressive expression of TGF-β, another cytokine that suppresses cell-mediated immune response efficiently. There are strong links between IL-4 and TGF-β; both IL-4 and IL-13 enhance release and activation of TGF-β related to fibrosis [22,23]. Thus, TGF-β could also participate in the development of fibrosis during the healing ulcerative stage, where numerous TGF-β immunostained macrophages have been reported in human lesions [15], as we observed in the subcutaneous tissue from mice infected with the strain from Benin. TGF-β also increases apoptosis of T cells activated by mycobacterial antigens [24,25]. Therefore, it is possible that besides mycolactone, TGF-β could participate in producing the extensive apoptosis exhibited by the inflammatory cells in ulcerative lesions, attributed previously exclusively to mycolactone. Both IL-10 and TGF-β are key mediators of regulatory T cells [26]. The high expression of both cytokines during the ulcerative stage in the lesions induced by the African strain suggests that regulatory T cells could also be involved in the suppression of the Th1 response. Some chemokines should be also important in the recruitment of inflammatory cells such as MIP-1β, whose expression was significantly higher during early lesions produced by the Mexican isolate.
Another interesting observation from our study was the high expression of mBD-3 and -4 during the pre-ulcerative phase in mice infected with the Benin strain, followed by a sharp decrease during the late ulcerative phase. This contrasted with animals infected with the low virulence strain from Mexico, which showed high and stable expression of both β-defensins. It has been demonstrated that mycobacterial infection induces production of mBD [27]. These natural anti-microbial peptides produced by epithelial cells can kill microbes and some of them have chemotactic effects on immune cells [28]. In a BALB/c model of pulmonary tuberculosis following intratracheal infection, we have demonstrated previously the rapid and high expression of mBD in the lungs during the early phase, while there was efficient control of bacillary replication. Then, during the late phase, there was a pronounced decrease in expression of mBD, while proliferation of the bacilli increased [29]. Thus, the low expression of both β-defensins during the ulcerative phase in mice infected with the virulent M. ulcerans strain could contribute to high bacillary proliferation and extensive tissue damage. It has been hypothesized that a significant proportion of the population living in endemic BU areas may be exposed to M. ulcerans but not develop the disease [1], and results from household contacts suggest that the Th1 response may prevent BU development in exposed subjects [21]. Our results suggest that another factor that can prevent M. ulcerans infection could be the production of β-defensins by the epidermis.
Acknowledgments
R. H. O. was supported by a PhD scholarship from the National Council for Science and Technology (CONACyT) México. This study was supported by the European Community (STREP contract no. 37919).
Disclosures
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in, or financial conflict with, the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1.van der Werf TS, van der Graaf WT, Tappero JW, Asiedu K. Mycobacterium ulcerans infection. Lancet. 1991;354:1013–8. doi: 10.1016/S0140-6736(99)01156-3. [DOI] [PubMed] [Google Scholar]
- 2.Asiedu K, Sherpbier R, Raviglione MC. Buruli ulcer Mycobacterium ulcerans infection. WHO Global Buruli Ulcer Initiative Report. Geneva: World Health Organization; 2000. [Google Scholar]
- 3.Guarner J, Bartlett J, Whitney EA, et al. Histopathologic features of Mycobacterium ulcerans infection. Emerg Infect Dis. 2003;9:651–6. doi: 10.3201/eid0906.020485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts B, Hirst R. Immunomagnetic separation and PCR for detection of Mycobacterium ulcerans. J Clin Microbiol. 1997;35:2709–11. doi: 10.1128/jcm.35.10.2709-2711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marsollier L, Robert R, Aubry J, et al. Aquatic insects as a vector for Mycobacterium ulcerans. Appl Environ Microbiol. 2002;68:4623–8. doi: 10.1128/AEM.68.9.4623-4628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marsollier L, André JPS, Frigui W, et al. Early trafficking events of Mycobacterium ulcerans within Naucoris cimicoides. Cell Microbiol. 2007;9:347–55. doi: 10.1111/j.1462-5822.2006.00790.x. [DOI] [PubMed] [Google Scholar]
- 7.Pahlevan AA, Wright DJM, Andrews C, George KM, Small PLC, Foxwell BM. The inhibitory action of Mycobacterium ulcerans soluble factor on monocyte/T cell cytokine production and NF-κB function. J Immunol. 1999;163:3928–35. [PubMed] [Google Scholar]
- 8.George KM, Chatterjee D, Gunawardana G, et al. Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science. 1999;283:854–7. doi: 10.1126/science.283.5403.854. [DOI] [PubMed] [Google Scholar]
- 9.Stinear TP, Mve-Obiang A, Small PLC, et al. Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proc Natl Acad Sci USA. 2004;101:1345–9. doi: 10.1073/pnas.0305877101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mve-Obiang A, Lee RE, Portaels F, Small PLC. Heterogeneity of mycolactones produced by clinical isolates of Mycobacterium ulcerans: implications for virulence. Infect Immun. 2003;71:774–83. doi: 10.1128/IAI.71.2.774-783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.George KM, Pascopella L, Welty DM, Small PLC. A Mycobacterium ulcerans toxin, mycolactone, causes apoptosis in guinea pig ulcers and tissue culture cells. Infect Immun. 2000;68:877–83. doi: 10.1128/iai.68.2.877-883.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gooding TM, Johnson PDR, Campbell DE, et al. Immune response to infection with Mycobacterium ulcerans. Infect Immun. 2001;69:1704–7. doi: 10.1128/IAI.69.3.1704-1707.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coutanceau E, Marsollier L, Brosch R, et al. Modulation of the host immune response by a transient intracellular stage of Mycobacterium ulcerans: the contribution of endogenous mycolactone toxin. Cell Microbiol. 2005;7:1187–96. doi: 10.1111/j.1462-5822.2005.00546.x. [DOI] [PubMed] [Google Scholar]
- 14.Prévot G, Bourreau E, Pascalis H, et al. Differential production of systemic and intralesional gamma interferon and interleukin-10 in nodular and ulcerative forms of Buruli ulcer. Infect Immun. 2004;72:958–65. doi: 10.1128/IAI.72.2.958-965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiszewski AE, Becerril E, Aguilar LD, et al. The local immune response in ulcerative lesions of Buruli ulcer disease. Clin Exp Immunol. 2006;143:445–51. doi: 10.1111/j.1365-2249.2006.03020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–20. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 17.Bals R. Epithelial antimicrobial peptides in host defense against infection. Respir Res. 2000;1:141–50. doi: 10.1186/rr25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliveira MS, Fraga AG, Torrado E, et al. Infection with Mycobacterium ulcerans induces persistent inflammatory responses in mice. Infect Immun. 2005;73:6299–310. doi: 10.1128/IAI.73.10.6299-6310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daniel AK, Lee RE, Portaels F, Small PLC. A analysis of Mycobacterium species for the presence of a macrolide toxin, mycolactone. Infect Immun. 2004;72:123–32. doi: 10.1128/IAI.72.1.123-132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Werf TS, Stinear T, Stienstra Y, van der Graaf WT, Small PL. Mycolactones and Mycobacterium ulcerans disease. Lancet. 2003;362:1062–64. doi: 10.1016/S0140-6736(03)14417-0. [DOI] [PubMed] [Google Scholar]
- 21.Gooding MT, Johnson PD, Smith M, Kemp AS, Robins-Browne RM. Cytokine profiles of patients infected with Mycobacterium ulcerans and unaffected household contacts. Infect Immun. 2002;70:5562–7. doi: 10.1128/IAI.70.10.5562-5567.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee CG, Homer RJ, Zhu Z, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta. J Exp Med. 2001;194:809–21. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dlugovitzky D, Bay ML, Rateni L, et al. In vitro synthesis of interferon-gamma, interleukin-4, transforming growth factor-beta and interleukin-1 beta by peripheral blood mononuclear cells from tuberculosis patients: relationship with the severity of pulmonary involvement. Scand J Immunol. 1999;49:210–7. doi: 10.1046/j.1365-3083.1999.00492.x. [DOI] [PubMed] [Google Scholar]
- 24.Mendez-Samperio P, Hernandez-Garay M, Garcia-Martinez E. Induction of apoptosis in bacillus Calmette–Guerin-activated T cells by transforming growth factor-beta. Cell Immunol. 2002;202:103–12. doi: 10.1006/cimm.2000.1662. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch CS, Johnson JL, Okwera A, et al. Mechanisms of apoptosis of T-cells in human tuberculosis. J Clin Immunol. 2005;25:353–64. doi: 10.1007/s10875-005-4841-4. [DOI] [PubMed] [Google Scholar]
- 26.Bellkaide Y, Rouse B. Nature regulatory T cells in infectious disease. Nat Immunol. 2005;6:353–60. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 27.Rivas-Santiago B, Schwander SK, Sarabia C, et al. Human β-defensin 2 is expressed and associated with Mycobacterium tuberculosis during infection of human alveolar epithelial cells. Infect Immun. 2005;73:4505–11. doi: 10.1128/IAI.73.8.4505-4511.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durr M, Peschel A. Chemokines meet defensins: the merging concepts of chemoattractants and antimicrobial peptides in host defense. Infect Immun. 2002;70:6515–7. doi: 10.1128/IAI.70.12.6515-6517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivas-Santiago B, Sada E, Tsutsumi V, Aguilar-Leon D, Contreras JL, Hernandez-Pando R. Beta-defensin gene expression during the course of experimental tuberculosis infection. J Infect Dis. 2006;194:697–701. doi: 10.1086/506454. [DOI] [PubMed] [Google Scholar]