

Abstract
Tumour necrosis factor (TNF)-α is crucial for resistance to Trypanosoma cruzi acute infection, but there is scant information on its role during the chronic phase. To address this issue, we analysed whether a short treatment with a TNF-α blocker affected the course and characteristics of chronic disease in a rat experimental model of T. cruzi infection. An anti-TNF-α agent (infliximab) was administered during the chronic phase for a period of 4 weeks (3 mg/kg/week), while control infected rats were inoculated with saline physiological solution. Search for parasites yielded non-successful results in all infected groups, irrespective of treatment. Nevertheless, the presence of T. cruzi kDNA in heart tissue was detected in infected and infected plus treated animals. Because infliximab might induce changes in the anti-parasite cytokine response, circulating levels of interleukin (IL)-10, interferon-gamma and nitric oxide were evaluated. An increase in IL-10 levels was observed only in the infected group treated with the anti-TNF-α blocker compared to the remaining groups (P < 0·05). A clear attenuation of histological damage associated with a diminution of cardiac TNF-α mRNA expression was observed in the infected and treated animals compared to the infected and non-treated group. Blocking of TNF-α during a relatively short period in chronically infected rats did not lead to evident parasite reactivation but reduced myocarditis severity significantly, indicating a role of this cytokine in the pathogenesis of chronic myocardial damage.
Keywords: Chagas' reactivation, chronic chagasic myocarditis, infliximab treatment, Trypanosoma cruzi, tumour necrosis factor-α
Introduction
Chagas' disease, caused by the protozoan Trypanosoma cruzi, is one of the most important endemic parasitoses in Latin America [1]. Acute infection is usually oligosymptomatic and, once resolved, evolves to an indeterminate and/or chronic form of disease. The most important clinical manifestation of Chagas' disease is chronic myocarditis, which affects 30% of infected individuals [2,3]. So far, the factors underlying distinct clinical outcomes are not understood completely. However, there is a general consensus that the cytokine-mediated immune response plays an essential role both in protection and disease pathogenesis [4,5]. Tumour necrosis factor (TNF)-α has been identified as one of the cytokines playing important and opposite roles during Chagas' disease. During acute T. cruzi infection, several studies demonstrate an essential role of TNF-α in the host defence, triggering phagocytic macrophage activation and inflammation [6,7]. At the same time, elevated TNF-α levels are correlated with pathology, including excessive inflammation, cachexia and death [8–11]. Neutralization of TNF-α or abrogation of its functionality during acute phase results in increased parasite burden, ameliorated cachexia and reduced myocardial inflammatory infiltrates [11]. Because TNF-α appears to be involved in the development of immunoglobulin (Ig)G antibody response, a deficient humoral response may account partly for the impaired parasite control seen in mice devoid of TNF-α effects [12].
In the lifelong chronic phase, histological and/or molecular techniques show T. cruzi derived-antigens or parasite persistence and inflammatory response of variable severity in diverse host tissues. Human chagasic myocarditis appears to be associated with a low parasite load [13,14] and increased presence of TNF-α[15,16], suggesting that a persistent stimulus may induce TNF-α synthesis, favouring control of subclinical infection, but at the same time the development of a pathological response. The nature of the stimulus may be due to the presence of the parasite, its antigens or mimetic parasite antigens [17].
The role of TNF-α during the chronic phase has been studied much less. Human studies only show an association between TNF-α levels and severity of pathology [15,16]. Experimental studies in TNF-α or TNF-receptor (TNF-R) knock-out animals are not feasible, as they die during the acute phase. In view of the protective and pathological roles of TNF-α, the question arises of whether treatment with monoclonal antibodies against TNF-α, once the acute infection is resolved, will affect the long-term outcome of this trypanosomiasis. Besides its intrinsic value, the question is clinically relevant. Approximately 20 million people are infected with T. cruzi, for which the potential consequences of anti-TNF-α therapy in infected individuals presenting co-morbidities suitable for this intervention need to be investigated.
To address this issue, we carried out a study in a well-characterized rat model of chronic chagasic infection developed in our laboratory [18–21]. In this model, challenge with T. cruzi in inbred strain ‘l’ rats results in a self-resolving acute phase followed by a chronic infection in which most rats develop a mild to intense focal myocarditis [18–21]. The data indicate that short TNF-α blocking during the chronic phase did not produce patent T. cruzi reactivation but reduced myocarditis severity significantly; the hallmark of Chagas' disease.
Materials and methods
Rats and experimental infection
Male ‘l’ rats were bred at the animal facilities from the School of Medicine of Rosario. Animals were housed under a 12-h light/12-h dark schedule (lights on starting at 07:00 h) with free access to food and water. All animal procedures were carried out in accordance with the institutional guidelines. Young animals (3 weeks old) were injected subcutaneously with 106 viable trypomastigotes of T. cruzi (Tulahuén strain). Parasites were maintained by serial passages in BALB/c suckling mice.
Verification of infliximab bioactivity in ‘l’ rats
The anti-TNF-α chimeric monoclonal antibody, infliximab (Remicade; Shering Plough, Heist-op-den-Berg, Belgium), was employed for neutralizing TNF-α. To validate its neutralizing activity, ad-hoc experiments were carried out. First, two groups of acutely infected young rats were treated with infliximab (3 mg/kg, three times for 1 week) by the intraperitoneal (i.p.) route or physiological saline solution, respectively, beginning 1 day after infection. An additional control group was inoculated with infliximab at the same time-points. Parasite circulating load was evaluated until 10 days post-infection (p.i.) by direct microscopic blood observation. Secondly, two groups of young rats were injected with a lethal dose of lipopolysaccharide (LPS) (5 mg/kg); one group was inoculated simultaneously with a single dose of infliximab (5 mg/kg, i.p.) and the other group with physiological saline solution (0·1 ml, i.p.). As an additional control, one group of rats received only infliximab. Mortality was evaluated for 7 days.
Treatment groups and study design
Young male rats were separated randomly into four groups: controls non-infected (Co); non-infected but treated with the anti-TNF-α blocking antibody (infliximab); infected (Tc); and infected and treated with the anti-TNF-α blocking antibody (Tc + infliximab). In this model, circulating parasites disappear 30 days p.i. resulting in the end of the acute phase, with chronic myocardial lesions being well established (or starting to be noticeable) following 10 weeks of infection [18]. Infliximab administration was initiated after 12 weeks p.i. (3 mg/kg; three times a week, i.p.) for 4 consecutive weeks. Co and Tc groups were inoculated with physiological saline at the same time. Infliximab was found to have a half-life of 11 days in rats following a dose of 8 mg/kg [22], for which the 3 mg/kg administered for nearly 48 h is likely to be highly efficacious in neutralizing endogenous TNF-α.
On completion of the experiments (120 days p.i.), rats were anaesthetized with ketamine/xylazine, blood was collected by cardiac puncture and serum was stored at −20°C for further assessment of TNF-α, interferon (IFN)-γ, interleukin (IL)-10 and nitric oxide (NO)-derived metabolites and specific antibody levels. Different tissues were removed for histopathology.
Parasitological tests
Different approaches were undertaken to detect bloodstream forms of T. cruzi. First, parasitaemia was assessed by direct standardized microscopic observation of 5 µl of heparinized tail venous blood, either before (90 days p.i.) or following infliximab treatment (120 days p.i.). Data were expressed as positive (+) or negative (−) samples or parasites/ml when applicable (400× magnification). Following infliximab treatment and to corroborate the presence or not of parasites in circulation, blood from each infected rat (treated or non-treated) was inoculated into five highly susceptible newborn mice (30 µl of blood/mice). After transference, mice were inspected for mortality and searched for the presence of trypomastigotes throughout a 30-day period. Xenodiagnosis of infected rats was also assessed as described previously after only infliximab treatment [23].
Polymerase chain reaction (PCR) for identification of kDNA from T. cruzi
A standard PCR procedure was used, following strict decontamination procedures and the use of disposable material. The method was used on either blood or tissue samples. Two hundred µl of blood were obtained from the tail of each rat, before and after infliximab treatment. Blood samples were transferred to guanidine–ethylenediamine tetraacetic acid (EDTA)-containing tubes, boiled for 15 min and stored at 4°C until DNA extraction. After treatment with the TNF-α blocker, small fragments of fresh heart were collected and homogenized in a glass tissue grinder. The samples were mixed thoroughly with guanidine–EDTA, boiled and stored at 4°C. DNA was extracted by using phenol–chloroform–isoamyl alcohol followed by ethanol precipitation. Finally, the solution was suspended in free-endonuclease sterile water. DNA amplification was carried out in 50 µl of a mixture containing Taq buffer, 4 mM MgCl2, 0·2 mM of each deoxynucleoside triphosphate, 1·25 U Taq polymerase (Fermentas, Hanover, MD, USA) and 1 mM of each primer (Operon, Köln, Germany). Amplifications comprised a sequence of 330 base pairs (bp), a T. cruzi-specific repetitive minicircle sequence (forward primer #121: 5′-AAATAATGTACGGG(g/T)GAGATGCATGA-3′; reverse primer #122: 5′-GGTTCGATTGGGGTTGGTGTAATATA-3′) [24]. The PCR reaction was initiated with 3 min of denaturalization at 94°C, followed by five cycles of amplification, each consisting of 45 s at 94°C, 45 s at 68°C and 45 s at 72°C, and 36 cycles consisting of 45 s at 94°C, 45 s at 64°C and 45 s at 72°C in an Eppendorff cycler and a final cycle of 10 min at 72°C. We analysed the PCR product in a 1% agarose gel stained with ethidium bromide. Positive and negative T. cruzi blood samples were used as internal controls of PCR. This PCR protocol has a 0·1% sensitivity of kDNA after hybridization with a specific probe which corresponds to one intact parasite, or 0·01% of the T. cruzi DNA fragment circulating in the blood or tissue from an infected host.
TNF-α mRNA expression
Hearts, removed after infliximab treatment, were stored in liquid nitrogen until use. RNA was extracted from homogenized hearts with TRizol (Gibco, New York, USA) following instructions given by the suppliers. Two micrograms of total RNA were reverse-transcribed to first-strand cDNA by standard procedures using 50 µg/ml oligodeoxythymidylic acid (oligo-dT) anchor primers, 5 mM deoxyribonucleoside triphosphate (dNTP) mixture, 1 mM dithiothreitol (DTT), 50 U RNAsaOut (Gibco) and 200 U reverse transcriptase (SuperScript II–RNAsa H; Gibco) in the buffer supplied. The room temperature mixture was amplified in a conventional PCR procedure using 4 mM MgCl2, 5 mM dNTPs, 10 pmol each oligonucleotide and 1 U Taq DNA polymerase (Gibco) in the buffer supplied. The following primers were used for amplification: TNF-α forward primer 5′-ATGAGCACAGAAAGCATGATC-3′ and reverse primer 5′-CAGAGCAATGACTCCAAAGTA-3′; β-actin forward primer 5′-CGTGACATCAAAGAGAAGCTGGTGC-3′ and reverse primer 5′-GCTCAGGAG-GAGCAATGATCTTGAT-3′ (Operon). Reactions were performed in an Eppendorf thermal cycler, with the cycling parameters consisting of 94°C for 1 min, 60°C for 1 min and 72°C for 2 min with a final extension step of 10 min at 72°C for 35 cycles. The size of amplicons was 700 bp for TNF-α and 600 bp for β-actin. PCR products were separated on a 1·5% agarose gel and visualized with ethidium bromide staining. Estimation of TNF-α transcripts expression levels was performed by standardization with internal control of β-actin, using a densitometric scanning video camera and gelpro 32 analysis program.
Antibody responses
Blood samples were collected before and after treatment with the TNF-α blocker and allowed to clot, with the sera being stored at −20°C. Individual serum was analysed in duplicate for specific IgM and total IgG using a commercial antibody capture enzyme-linked immunosorbent assay (ELISA), coated with recombinant T. cruzi antigens (Chagatest recombinant 3·0; Wiener Laboratories, Rosario, Argentina) adapted for the detection of rat antibodies, as performed previously [25].
Histopathology
Hearts, skeletal muscles, subcutaneous lymph nodes, spleens and thymuses were removed after infliximab or control treatment and fixed in buffered formalin for histopathology. For hearts, five paraffin-embedded 5 µm sections of each animal were stained with haematoxylin and eosin for evaluation of inflammatory foci, tissue parasitism, myocarditis and fibrosis. Foci of myocarditis were classified as shown previously [26]. Briefly, normal tissue (score 0); mild foci: slight infiltration with damage of one or two myocardial fibres (score 1); moderate-sized foci: aggregated infiltrates compromising three to five muscle fibres (score 2); and intense foci: heavy accumulation of mononuclear cells with destruction of more than five muscle fibres (score 3). In this manner a rat showing three foci of chronic myocarditis (two mild, one moderate) had a score of 4. Sections from each organ were examined by an experienced pathologist blinded to the study groups.
Immunohistochemistry
Heart tissues were analysed for the surface phenotype of inflammatory cells. Serial 5–7 µm-thick sections of the heart were prepared, fixed in cold acetone and subjected to indirect immunoperoxidase staining. To identify the inflammatory cells, mouse monoclonal anti-rat antibodies (anti-CD4, CD8 and ED1 – a marker for inflammatory macrophages – from Serotec, Oxford, UK) and peroxidase labelled secondary antibodies (Sigma) were used. In addition, the immune reaction was controlled by omitting primary antibodies (negative control) and by using the full procedure on spleen sections (positive control). The material was counterstained with Mayer's haematoxylin. Slides were examined using a light microscope (Zeiss, Germany). The stained cells were identified by a partial or complete dark-coloured ring outlining their cell membrane. Cell counts were acquired by counting the total number of positively stained cells in five non-contiguous sections for each animal and analysing 50 microscopic fields in each section.
Cytokine assays and nitrite evaluation
Serum cytokines were measured by specific two-site ELISA according to the manufacturer's specifications. ELISA kits for TNF-α (limit of detection 12·5 pg/ml) were purchased from R&D (Minneapolis, MN, USA) or Pharmingen (San Diego, CA, USA) in the case of IL-10 (limit of detection 15 pg/ml) and IFN-γ (limit of detection 15 pg/ml). All samples were processed individually and assayed in duplicate, with plates being read at 450 nm. NO production was estimated by serum nitrite measurement, as described previously [10].
Data and statistical analysis
Statistical comparisons among groups were performed by Kruskal–Wallis test followed by the Mann–Whitney U-test; χ2 and Fisher's exact tests were used to analyse categorical differences. Statistical significance was set at P < 0·05.
Results
Infliximab treatment modifies the features of acute T. cruzi infection and mortality after LPS inoculation in rats
First, we analysed infliximab bioactivity in ‘l’ rats. Antibody capacity to bind and neutralize rat TNF-α was evidenced in acutely infected young rats receiving infliximab (Tc + infliximab). This group showed a significantly increased number of blood-circulating parasites after 10 days p.i. than their physiological saline-treated counterparts [parasites/ml, mean ± standard error of the mean (s.e.m.), n = 5; Tc + infliximab = 630 ± 292; Tc = 120 ± 69; P < 0·05]. Further studies in suckling rats subjected to lethal doses of LPS and treated in parallel with infliximab showed 33% survival after challenge compared with their counterparts [(survival mice/total mice); LPS group = 0/12; LPS + infliximab group = 4/12; P < 0·05]. Both set of results are representative of two experimental rounds each.
Anti-TNF-α treatment during chagasic chronic phase did not produce a noticeable parasite re-emergence
The possibility of reactivation of Chagas' disease during infliximab treatment in chronic infected rats was evaluated by different approaches: (1) detection of trypomastigote forms by direct examination of peripheral blood, blood transference to highly susceptible newborn mice and xenodiagnosis; (2) search for amastigote forms in tissues of different organs by direct microscopy; and (3) molecular detection of kDNA and in blood and heart tissue.
Upon challenge with 1 × 106 parasites of the Tulahuén strain of T. cruzi, all male ‘l’ rats display detectable parasites in circulation until 28 days p.i.; that is, when trypomastigote clearance is produced and the indeterminate phase ensues. All animals survived up to 120 days p.i., the time of killing, and this trend was not modified by anti-TNF-α administration during the chronic phase. No circulating parasites were observed during chronic phase before infliximab treatment (90 days p.i., data not shown) and following its termination (120 days p.i., Table 1), with xenodiagnoses also negative in all infected animals, independently of the treatment (Table 1). In contrast, kDNA in blood and heart tissue was detected pre- and post-infliximab administration, suggesting a very low but persistent infection (Fig. 1). Moreover, no pathological alterations, such as splenomegaly, lymph node enlargement or thymus atrophy, were seen in infected rats given infliximab treatment. A search for the presence of amastigote in diverse tissues was not successful.
Table 1.
Studies on the presence of bloodstream forms of Trypanosoma cruzi or T. cruzi kDNA during acute and chronic infection.
| Treatment | Tc | Tc + infliximab† | ||
|---|---|---|---|---|
| Phase | Acute | Chronic | Acute | Chronic |
| Days p.i. | 10 | 120 | 10 | 120 |
| Test: | ||||
| Direct blood observation | (+) 100% | (−) | (+) 100% | (−) |
| Xenodiagnosis | (−) | (−) | ||
| Inoculation into newborn mice | (−) | (−) | ||
| PCR kDNA (blood) | (+) 100% | (+) 30% | (+) 100% | (+) 35% |
| PCR kDNA (heart) | (+) 100% | (+) 60 % | (+) 100% | (+) 65% |
Data are represented as positive (+) plus the percentage, or negative (−), of each analysed parameter of six to nine rats/group. A representative experiment of two independent rounds.
Treatment with infliximab was applied only during chronic infection. Search of T. cruzi during acute phase [day 10 post-infection (p.i.)] was made solely as control of infection. PCR, polymerase chain reaction.
Fig. 1.
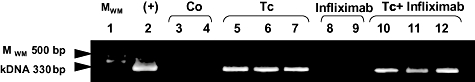
Detection of kDNA in chronic infected heart tissues. Representative agarose gel electrophoresis results for kDNA by heart tissue homogenates from rats after infliximab treatment (120 days post-infection) analysed by polymerase chain reaction. Lane 1: molecular weight marker [MWM 500 base pairs (bp) ladder]; lane 2: heart tissue extracted from a rat with acute infection; lanes 3 and 4: heart tissue from non-infected and physiological saline-treated rats; lanes 5–7: heart tissue from chronic infected rats; lanes 8 and 9: heart tissue from non-infected but infliximab-treated rats; lanes 10–12: heart tissue from infected and infliximab-treated rats. Arrow indicates MWM and Trypanosoma cruzi-specific products of 330 bp. Each line is a sample of a single animal.
Infliximab treatment did not modify T. cruzi-specific antibody levels
Because a reactivated infection may co-exist with changes in the T. cruzi-specific antibody levels, we also measured the specific humoral immune response (IgG and IgM) following anti-TNF-α treatment (120 days p.i.). As shown in Fig. 2, detectable amounts of specific IgG were seen in both infected groups (P < 0·001 compared to their non-infected counterparts), with comparisons between Tc + infliximab and Tc animals being insignificant [P = not significant (n.s.)]. While IgM levels were lower and became negative in a >1/20 dilution, antibodies were present in 90% of infected rats (P < 0·05 in comparison to non-infected groups), with no significant differences between the two infected groups.
Fig. 2.
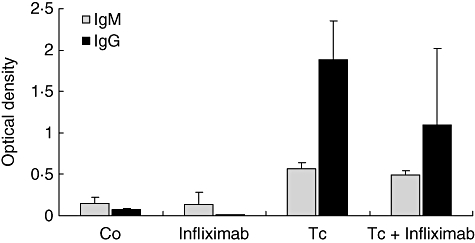
Immunoglobulin (Ig)-specific levels after infliximab therapy. IgM and IgG Trypanosoma cruzi-specific levels were evaluated by enzyme-linked immunosorbent assay after infliximab treatment (120 days post-infection). Detectable amounts of specific IgM (1/20) and IgG (1/200 dilution) were seen in both groups of chronically infected rats, without significant differences between them. Tc and Tc + infliximab groups significantly different from their non-infected counterparts (P < 0·001, both immunoglobulin isotypes). Values represent means ± standard error of the mean of optical density unit of six to nine rats/group. A representative experiment of two independent rounds.
Infliximab treatment increased circulating IL-10 levels and diminished cardiac TNF-α mRNA
Cytokine serum levels after anti-TNF-α administration (120 days p.i.) are shown in Table 2. IFN-γ levels were non-detectable during chronic phase, regardless of treatment. Moreover, TNF-α blocking did not affect levels of NO-derived metabolites in sera. In contrast, increased levels of IL-10 levels were observed in infected and infliximab-treated rats when compared to the infected group undergoing no treatment. At the same time no detectable amounts of systemic TNF-α were seen in any of the study groups. In line with previous studies [26], serum TNF-α was detectable only during the early acute phase (pg/ml, Tc: 142 ± 23, Co: non-detectable, n = 12/group, day 10 p.i.), with no measurable amounts of this cytokine by the time that anti-TNF treatment was initiated. On the other hand, hearts from infected and infliximab-treated rats had a 50% reduction in TNF-α mRNA expression in comparison with the single infected group (Fig. 3). No expression of TNF-α mRNA was seen in either of the non-infected groups.
Table 2.
Serum cytokines in chronically infected rats following anti-tumour necrosis factor (TNF)-α treatment.
| Serum mediator | Co | Infliximab | Tc | Tc + infliximab |
|---|---|---|---|---|
| TNF-α (pg/ml) | n.d. | n.d. | n.d. | n.d. |
| IFN-γ (pg/ml) | n.d. | n.d. | n.d. | n.d. |
| IL-10 (pg/ml) | n.d. | n.d. | 23 ± 8 | 44 ± 12* |
| Total nitrite (nM) | 46 ± 17 | 16 ± 15 | 67 ± 24** | 81 ± 32** |
P < 0·05 Tc + infliximab versus Tc.
P < 0·05 versus the respective control group. Values represent mean ± standard error of the mean of six to nine rats/group. Samples were taken after 120 days post-infection. A representative experiment of two independent rounds. IFN, interferon; IL, interleukin; n.d., non-detectable.
Fig. 3.
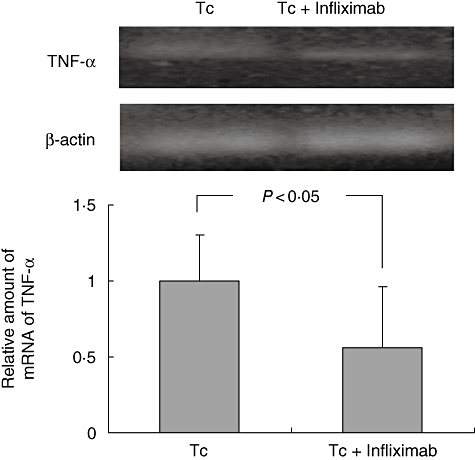
Tumour necrosis factor (TNF)-α mRNA expression in myocardial tissue. TNF-α mRNA expression in myocardial tissue was evaluated by semiquantitative reverse transcription–polymerase chain reaction after infliximab treatment (120 days post-infection). β-actin mRNA was used to normalize TNF-α mRNA among samples. Relative values were calculated considering TNF-α/β-actin ratio as 1 for infected but non-treated animals. No expression of TNF-α mRNA was seen in both non-infected groups, Normalized results correspond to two experimental series of four to six animals/group (media of relative value of sample corresponding to an individual animal ± standard error of the mean).
TNF-α neutralizing antibodies reduce heart lesions during chronic infection
Because probable T. cruzi reactivation following anti-TNF-α treatment may favour tissue damage, microscopic studies in myocardial samples were also carried out. Heart histological analysis from both 120-day infected groups revealed no parasite nests in cardiocytes. The extent of myocarditis was determined as the percentage of normal, mild, moderate or intense damage observed in each group. Among the infected and non-treated rats an easily noticeable myocarditis, with multiple inflammatory foci of 34% intense magnitude, was seen. In contrast, ameliorated lesions were seen in the infected group given infliximab, with only 8·3% of rats showing intense myocarditis (Tc versus Tc + infliximab; P < 0·05, Fig. 4, upper panels). As seen in Table 3, the latter group displayed a lower score of inflammatory foci compared with the non-treated group (P < 0·02). Further immunostaining analysis demonstrated that the reduced chronic myocardial damage of the infected and infliximab-treated group was associated with decreased numbers of CD4+, CD8+ and ED1+ infiltrating cells (Table 4 and bottom panels of Fig. 4).
Fig. 4.
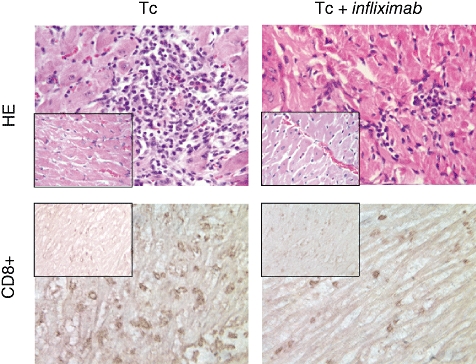
Histopathology evaluation of chronic myocarditis. (a) Intense inflammatory foci in a chronically infected rat; (b) mild inflammatory foci observed in chronically infected and infliximab-treated rat; (c) CD8+ cells in heart tissue of infected animals (mild foci); (d) CD8+ cells in the myocardium of chronically infected and infliximab-treated animals (mild foci). Magnification panels 20×. In all cases, the respective non-infected control groups are showed in the small cages.
Table 3.
Score of myocarditis severity after infliximab treatment.
| Co | Infliximab | Tc | Tc + infliximab | |
|---|---|---|---|---|
| Score of inflammatory foci | 0·6 ± 1·2 | 0·5 ± 0·2 | 13 ± 5·4 | 5 ± 2·6* |
P < 0·02 Tc + infliximab versus Tc. Heart samples were obtained after the treatment with the tumour necrosis factor (TNF)-α blocker (120 days post-infection) and fixed in buffered formalin. Paraffin-embedded 5 mm sections were stained with haematoxylin and eosin (five serial sections of each animal). A score was also calculated on the basis of the number and severity of chronic inflammatory foci: normal tissue (score 0); mild foci (score 1); moderate-sized foci (score 2); intense foci (score 3). In this way a rat showing three foci of chronic myocarditis (two mild, one moderate) had a score of 4. Values represent the mean ± standard error of the mean of 11–12 animals/group. A representative experiment of two independent rounds.
Table 4.
CD4+, CD8+ and ED1+ cells in myocardial tissue after infliximab treatment (day 120 post-infection).
| Co | Infliximab | Tc | Tc + infliximab | |
|---|---|---|---|---|
| CD4+ cells | 13 (3–24) | 16 (2–19) | 107 (76–165) | 78 (21–111)* |
| CD8+ cells | 44 (13–73) | 35 (7–45) | 228 (169–334) | 150 (125–197)* |
| ED1+ cells | 15 (3–47) | 12 (2–33) | 148 (96–198) | 94 (64–134)* |
P < 0·05 Tc + infliximab versus Tc. Fifty fields in five sections were examined for each animal. The results represent count cells as median (range) of data from each group (six to nine rats/group). A representative experiment of two independent rounds.
Discussion
The relevance of TNF-α in acute T. cruzi infection has been supported by studies of cytokine neutralization or in TNF receptor knock-out mice, showing serious deficiencies in host resistance to this protozoan [11,12,27–29]. In contrast, evidence on the role of TNF-α during the chronic chagasic phase is scarce, although its presence might contribute to disease immunopathology. It is now accepted that T. cruzi persists during the chronic period, as it can be detected sporadically in chronic chagasic patients [30]. As TNF-α contributes to parasite control [6,7] TNF-α blocking might favour T. cruzi reactivation, in view of the increased susceptibility to diseases caused by intracellular pathogens after anti-TNF-α therapies, i.e. tuberculosis [31], malaria [32], toxoplasmosis [33] and leishmaniasis [34]. Our studies in chronically infected rats given infliximab failed to detect parasites either by direct blood examination, xenodiagnosis or blood inoculation into highly susceptible mice, implying that TNF-α might be less relevant for T. cruzi control during the chronic phase. However, as tissue TNF-α mRNA after infliximab treatment was reduced but still detected, the question remains of whether a more prolonged blockade of TNF-α will result in parasite reactivation. So far, cases reported of Chagas' reactivation have occurred in immunocompromised hosts such as acquired immune deficiency syndrome (AIDS) patients, subjects undergoing chemotherapy or transplanted individuals [35–37]. This may imply that T. cruzi reactivation occurs only when cell-mediated immune response is affected profoundly, precluding an effective anti-parasite response.
Human and experimental studies reveal considerable amounts of anti-T. cruzi antibodies during chronic infection, suggesting that humoral response may control circulating parasites rapidly in case of reactivation. In this sense, our previous study showed that parasitaemia in T. cruzi-reinfected rats was of lesser magnitude than that seen in the initial infection, but with abundant amounts of specific antibodies [22]. Castaños-Velez et al. suggested that susceptibility to acute T. cruzi infection in TNF-R1 knock-out mice might be related to a deficiency in antibody production [12]. In fact, TNF-R1 participates in lymph node structure maintenance, germinal centre formation and the switch and affinity maturation of immunoglobulins [38,39], although these effects seem to be mediated by lymphotoxin-alpha, which also interacts with TNF-R1 [38]. Some authors suggest that TNF-α blockade prevents the generation and maintenance of adaptive immune responses by inhibiting lymphoid hyperplasia [40]. However, there is consensus that in adult animals, TNF-α neutralization has no apparent effect on lymphoid structure or germinal centre formation [41]. In our hands, anti-TNF-α treatment did not reduce anti-T. cruzi antibodies levels, because an important humoral response against T. cruzi was maintained in the absence of detectable circulating parasite forms. In addition, the absence of increased levels of specific IgM favours the view of no reactivation, as IgM is regarded as a sign of parasite re-emergence in chronically infected pregnant women [42,43]. Moreover, congenital cases of Chagas' disease are associated with measurable maternal IgM, whereas infected mothers not transmitting parasites during pregnancy show a non-detectable IgM response [44].
Chronic myocardial damage in Chagas' disease may be promoted by the parasite and/or by the immune response against parasite antigens or mimetic epitopes. TNF-α is likely to contribute to the development of chronic myocarditis, as it was detected in sera [15,16] and myocardial biopsies from chronic chagasic patients [45], also related to myocarditis severity [46]. A consistent feature of our rat model is the development of focal myocarditis, characterized by lymphocyte and monocyte infiltration, destruction of the myocardial fibre and fibrosis. Treatment with TNF-α antagonist in chronically infected rats reduced TNF-α mRNA, the number and the severity of lesions and the presence of CD8+, CD4+ and ED1+ inflammatory cells. Because TNF-α is related to the ability of T cells and macrophages to migrate to target tissues, infliximab treatment probably altered leucocyte recruitment to the heart. These results agree with our previous studies, where mice deficient in both TNF-α receptors showed an absence of inflammatory infiltration and fibre destruction in myocardial tissue, contrasting with the severe injury observed in acutely infected wild-type mice [26]. Nevertheless, our data are at variance with a report in chronically infected hamsters, in which the TNF-α antagonist etanercept was implied in heart dysfunction, but without variations in the severity of myocarditis [47]. Differences in TNF-α antagonists and experimental conditions may account for such discrepancy. While infliximab and etanercept both neutralize the soluble form of TNF-α, infliximab binds more powerfully to the membrane form of TNF-α (mTNF) than does etanercept. Etanercept is a recombinant soluble receptor TNF-R2-IgG1, whereas infliximab is a chimeric monoclonal antibody [48]. As mTNF expression may be up-regulated on T lymphocytes activated by T. cruzi antigens, treatment with anti-TNF-α monoclonal antibodies may alter migratory properties of activated anti-parasite T lymphocytes more than does etanercept. Also, etanercept can bind and neutralize LT-α, which can be produced by macrophages and T cells [49].
An alternative and mutually non-exclusive possibility for decreased cellular infiltration deals with the presence of anti-inflammatory cytokine milieu. Increased systemic levels of IL-10 observed in infected and treated rats lend some support to this assumption. Raised levels of IL-10 in anti-TNF-α treated rats is in line with our previous findings of cytokine dysregulation in TNF-Rs knock-out mice infected with T. cruzi[28].
Infliximab therapy in humans has been reported in a variety of off-label uses, mainly autoimmune and inflammatory diseases [49]. As chronic chagasic myocarditis may be the result of several concomitant pathogenic mechanisms, such as immune responses to parasite antigens [50], autoimmune processes [51,52] and the inflammation accompanying these responses [53], the potential effects of infliximab on chagasic myocarditis were worthy of study.
The present results suggest that blocking TNF-α activity is not critical for causing T. cruzi reactivation during the chronic phase, and at the same time highlight the key role of TNF-α in the development of T. cruzi-associated immunoinflammatory pathology. The findings also provide a stimulating background for assessing whether longer treatment periods continue to be safe and beneficial, as a basic requirement for further human studies.
Acknowledgments
We thank Dr Marcos Rosemffet for providing infliximab antibodies and Vanina Tartalini for her technical assistance, respectively. Chagatest recombinant 3·0, Wiener Laboratories, was generously provided by Gustavo Capriotti (Wiener Laboratories, Rosario, Argentina.). Triatoma infestans used in xenodiagnosis was kindly provided by Laura Fichera from Instituto Fatala Chabén, Buenos Aires. This work was supported by grants from Secretariat for Science and Technology (SECYT-UNR), Josefina Prats Foundation and Fellowship of Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET).
Disclosure
The authors declare that there is no conflict of interest that could prejudice the impartiality of this scientific work.
References
- 1.World Health Organization (WHO) Expert Committee. Control of Chagas' disease. World Health Organ Tech Rep. 2002;905:1–109. [PubMed] [Google Scholar]
- 2.Prata A. Clinical and epidemiological aspects of Chagas disease. Lancet Infect Dis. 2001;1:92–100. doi: 10.1016/S1473-3099(01)00065-2. [DOI] [PubMed] [Google Scholar]
- 3.Umezawa ES, Stolf AM, Corbett CE, Shikanai-Yasuda MA. Chagas' disease. Lancet. 2000;357:797–9. doi: 10.1016/S0140-6736(00)04174-X. [DOI] [PubMed] [Google Scholar]
- 4.Brener Z, Gazzinelli RT. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas' disease. Int Arch Allergy Immunol. 1997;114:103–10. doi: 10.1159/000237653. [DOI] [PubMed] [Google Scholar]
- 5.Savino W, Villa-Verde DM, Mendes-da-Cruz DA, et al. Cytokines and cell adhesion receptors in the regulation of immunity to Trypanosoma cruzi. Cytokine Growth Factor Rev. 2007;18:107–24. doi: 10.1016/j.cytogfr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Silva JS, Vespa GN, Cardoso MA, Aliberti JC, Cunha FG. Tumor necrosis factor alpha mediates resistance to Trypanosoma cruzi infection in mice by inducing nitric oxide production in infected gamma interferon-activated macrophages. Infect Immun. 1995;63:4862–7. doi: 10.1128/iai.63.12.4862-4867.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos Lima EC, Garcia I, Vicentelli MH, Vassalli P, Minoprio P. Evidence for a protective role of tumor necrosis factor in the acute phase of Trypanosoma cruzi infection in mice. Infect Immun. 1997;65:457–65. doi: 10.1128/iai.65.2.457-465.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hölscher C, Mohrs M, Dai WJ, et al. Tumor necrosis factor alpha-mediated toxic shock in Trypanosoma cruzi-infected interleukin 10-deficient mice. Infect Immun. 2000;68:4075–83. doi: 10.1128/iai.68.7.4075-4083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roggero E, Pérez AR, Tamae-Kakazu M, et al. Differential susceptibility to acute Trypanosoma cruzi infection in BALB/c and C57BL/6 mice is not associated with a distinct parasite load but cytokine abnormalities. Clin Exp Immunol. 2002;128:421–8. doi: 10.1046/j.1365-2249.2002.01874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pérez AR, Tamae-Kakazu M, Pascutti MF, et al. Deficient control of Trypanosoma cruzi infection in C57BL/6 mice is related to a delayed specific IgG response and increased macrophage production of pro-inflammatory cytokines. Life Sci. 2005;77:1945–59. doi: 10.1016/j.lfs.2005.01.025. [DOI] [PubMed] [Google Scholar]
- 11.Truyens C, Torrico F, Angelo-Barrios A, et al. The cachexia associated with Trypanosoma cruzi acute infection in mice is attenuated by anti-TNF-α, but not by anti-IL-6 or anti-IFN-γ antibodies. Parasite Immunol. 1995;17:561–8. doi: 10.1111/j.1365-3024.1995.tb00999.x. [DOI] [PubMed] [Google Scholar]
- 12.Castaños-Velez E, Maerlan S, Osorio LM, et al. Trypanosoma cruzi infection in tumor necrosis factor receptor p55-deficient mice. Infect Immun. 1998;66:2960–8. doi: 10.1128/iai.66.6.2960-2968.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Tarleton RL. Parasite persistence correlates with disease severity and localization in chronic Chagas' disease. J Infect Dis. 1999;180:480–6. doi: 10.1086/314889. [DOI] [PubMed] [Google Scholar]
- 14.Añez N, Carrasco H, Parada H, et al. Myocardial parasite persistence in chronic chagasic patients. Am J Trop Med Hyg. 1999;60:726–32. doi: 10.4269/ajtmh.1999.60.726. [DOI] [PubMed] [Google Scholar]
- 15.Ferreira RC, Ianni BM, Abel LC, et al. Increased plasma levels of tumor necrosis factor-alpha in asymptomatic/‘indeterminate’ and Chagas disease cardiomyopathy patients. Mem Inst Oswaldo Cruz. 2003;98:407–11. doi: 10.1590/s0074-02762003000300021. [DOI] [PubMed] [Google Scholar]
- 16.Talvani A, Rocha MO, Barcelos LS, Gomes YM, Ribeiro AL. Teixeira MM Elevated concentrations of CCL2 and tumor necrosis factor-alpha in chagasic cardiomyopathy. Clin Infect Dis. 2004;38:943–50. doi: 10.1086/381892. [DOI] [PubMed] [Google Scholar]
- 17.Bonney KM, Engman DM. Chagas heart disease pathogenesis: one mechanism or many? Curr Mol Med. 2008;8:510–18. doi: 10.2174/156652408785748004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Revelli SS, Amerio N, Moreno HS, Valenti JL, Balbarrey H, Morini J. Enfermedad de Chagas crónica en la rata. Características serológicas, electrocardiográficas e histopatológicas. Medicina (B Aires) 1980;40:69–76. [PubMed] [Google Scholar]
- 19.Revelli S, Davila H, Ferro ME, et al. Experimental. Trypanosoma cruzi infection in the rat. Response to systemic treatment with recombinant rat interferon gamma. Microbiol Immunol. 1995;39:275–82. doi: 10.1111/j.1348-0421.1995.tb02201.x. [DOI] [PubMed] [Google Scholar]
- 20.Bottasso OA, Revelli SS, Dávila H, et al. Enhanced myocardial lesions in chronically Trypanosoma cruzi-infected rats subjected to adult thymectomy. Immunol Lett. 1993;37:175–80. doi: 10.1016/0165-2478(93)90028-z. [DOI] [PubMed] [Google Scholar]
- 21.Davila HO, Revelli S, Moreno HS, et al. Infection with Trypanosoma cruzi during pregnancy in rats and a decreased in chronic myocardial lesions in their infected offspring. Am J Trop Med Hyg. 1994;50:506–11. doi: 10.4269/ajtmh.1994.50.506. [DOI] [PubMed] [Google Scholar]
- 22.Yang MX, Shenoy B, Disttler M, et al. Crystalline monoclonal antibodies for subcutaneous delivery. Proc Natl Acad Sci USA. 2003;100:6934–49. doi: 10.1073/pnas.1131899100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Revelli S, Berra H, Valenti J, et al. Efecto de la re-infección sobre la evolución de ratas infectadas con T. cruzi. Rev Inst Med Trop (Sao Paulo) 1990;32:260–8. doi: 10.1590/s0036-46651990000400005. [DOI] [PubMed] [Google Scholar]
- 24.Wincker P, Britto C, Pereira JB, Cardoso MA, Oelemann W, Morel CM. Use of a simplified polymerase chain reaction procedure to detect Trypanosoma cruzi in blood samples from chronic chagasic patients in a rural endemic area. Am J Trop Med Hyg. 1994;51:771–7. doi: 10.4269/ajtmh.1994.51.771. [DOI] [PubMed] [Google Scholar]
- 25.Pascutti MF, Bottasso OA, Hourquescos MC, Wietzerbin J, Revelli S. Age-related increase in resistance to acute Trypanosoma cruzi infection in rats is associated with an appropriate antibody response. Scand J Immunol. 2003;58:173–9. doi: 10.1046/j.1365-3083.2003.01262.x. [DOI] [PubMed] [Google Scholar]
- 26.Piaggio E, Roggero E, Pitashny M, Wietzerbin J, Bottasso OA, Revelli SS. Treatment with benznidazole and its immunomodulating effects on experimentally Trypanosoma cruzi-infected rats. Parasitol Res. 2001;87:539–47. doi: 10.1007/s004360000357. [DOI] [PubMed] [Google Scholar]
- 27.Kroll-Palhares K, Silvério JC, Silva AA, et al. TNF/TNFR1 signaling up-regulates CCR5 expression by CD8+ T lymphocytes and promotes heart tissue damage during Trypanosoma cruzi infection: beneficial effects of TNF-alpha blockade. Mem Inst Oswaldo Cruz. 2008;103:375–85. doi: 10.1590/s0074-02762008000400011. [DOI] [PubMed] [Google Scholar]
- 28.Pérez AR, Roggero E, Nicora A, et al. Thymus atrophy during Trypanosoma cruzi infection is caused by an immuno-endocrine imbalance. Brain Behav Immun. 2007;21:890–900. doi: 10.1016/j.bbi.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Roggero E, Piazzon I, Nepomnaschy I, et al. Thymocyte depletion during acute Trypanosoma cruzi infection in C57BL/6 mice is partly reverted by lipopolysaccharide pretreatment. FEMS Immunol Med Microbiol. 2004;41:123–31. doi: 10.1016/j.femsim.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Monteón-Padilla V, Hernández-Becerril N, Ballinas-Verdugo MA, Aranda-Fraustro A, Reyes PA. Persistence of Trypanosoma cruzi in chronic chagasic cardiopathy patients. Arch Med Res. 2001;32:39–43. doi: 10.1016/s0188-4409(00)00261-7. [DOI] [PubMed] [Google Scholar]
- 31.Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 32.Geraghty EM, Ristow B, Gordon SM, Aronowitz P. Overwhelming parasitemia with Plasmodium falciparum infection in a patient receiving infliximab therapy for rheumatoid arthritis. Clin Infect Dis. 2007;44:82–4. doi: 10.1086/515402. [DOI] [PubMed] [Google Scholar]
- 33.Young JD, McGwire BS. Infliximab and reactivation of cerebral toxoplasmosis. N Engl J Med. 2005;353:1530–1. doi: 10.1056/NEJMc051556. [DOI] [PubMed] [Google Scholar]
- 34.Fabre S, Gibert C, Lechiche C, Dereure J, Jorgensen C, Sany J. Visceral leishmaniasis infection in a rheumatoid arthritis patient treated with infliximab. Clin Exp Rheumatol. 2005;23:891–2. [PubMed] [Google Scholar]
- 35.Sartori AM, Caiaffa-Filho HH, Bezerra RC, do S Guilherme C, Lopes MH, Shikanai-Yasuda MA. Exacerbation of HIV viral load simultaneous with asymptomatic reactivation of chronic Chagas' disease. Am J Trop Med Hyg. 2002;67:521–3. doi: 10.4269/ajtmh.2002.67.521. [DOI] [PubMed] [Google Scholar]
- 36.Kohl S, Pickering LK, Frankel LS, Yaeger RG. Reactivation of Chagas' disease during therapy of acute lymphocytic leukemia. Cancer. 1982;50:827–8. doi: 10.1002/1097-0142(19820901)50:5<827::aid-cncr2820500503>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 37.Riarte A, Luna C, Sabatiello R, et al. Chagas' disease in patients with kidney transplants: 7 years of experience 1989–1996. Clin Infect Dis. 1999;29:561–7. doi: 10.1086/598634. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto M, Fu YX, Molina H, Chaplin DD. Lymphotoxin-alpha-deficient and TNF receptor-I-deficient mice define developmental and functional characteristics of germinal centers. Immunol Rev. 1997;156:137–44. doi: 10.1111/j.1600-065x.1997.tb00965.x. [DOI] [PubMed] [Google Scholar]
- 39.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choo-Kang BS, Hutchison S, Nickdel MB, et al. TNF-blocking therapies: an alternative mode of action? Trends Immunol. 2005;26:518–22. doi: 10.1016/j.it.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 41.Hofmann SR, Ettinger R, Zhou YJ, et al. Cytokines and their role in lymphoid development, differentiation and homeostasis. Curr Opin Allergy Clin Immunol. 2002;2:495–506. doi: 10.1097/00130832-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 42.Szarfman A, Urman J, Otalora A, Larguia A, Yanovsky JF. Specific agglutinins and immunoglobulin levels in congenital Chagas infection. Medicina (B Aires) 1975;35:245–50. [PubMed] [Google Scholar]
- 43.Sorni P, Bolsi FL. Embarazo y parasitismo por Trypanosoma cruzi. Medicina (B Aires) 1979;39:193–7. [PubMed] [Google Scholar]
- 44.Reyes MB, Lorca M, Muñoz P, Frasch AC. Fetal IgG specificities against Trypanosoma cruzi antigens in infected newborns. Proc Natl Acad Sci USA. 1990;87:2846–50. doi: 10.1073/pnas.87.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cunha-Neto E, Rizzo LV, Albuquerque F, et al. Cytokine production profile of heart-infiltrating T cells in Chagas' disease cardiomyopathy. Braz J Med Biol Res. 1998;31:133–7. doi: 10.1590/s0100-879x1998000100018. [DOI] [PubMed] [Google Scholar]
- 46.Drigo SA, Cunha-Neto E, Ianni B, et al. TNF gene polymorphisms are associated with reduced survival in severe Chagas' disease cardiomyopathy patients. Microbes Infect. 2006;8:598–603. doi: 10.1016/j.micinf.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Bilate AM, Salemi VM, Ramires FJ, et al. TNF blockade aggravates experimental chronic Chagas disease cardiomyopathy. Microbes Infect. 2007;9:1104–13. doi: 10.1016/j.micinf.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 48.Wallis RS. Tumour necrosis factor antagonists: structure, function, and tuberculosis risks. Lancet Infect Dis. 2008;8:601–11. doi: 10.1016/S1473-3099(08)70227-5. [DOI] [PubMed] [Google Scholar]
- 49.Lin J, Ziring D, Desai S, et al. TNF-alpha blockade in human diseases: an overview of efficacy and safety. Clin Immunol. 2008;126:13–30. doi: 10.1016/j.clim.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reis MM, Higuchi ML, Benvenuti LA, et al. An in situ quantitative immunohistochemical study of cytokines and IL-2R+ in chronic human chagasic myocarditis: correlation with the presence of myocardial Trypanosoma cruzi antigens. Clin Immunol Immunopathol. 1997;83:165–72. doi: 10.1006/clin.1997.4335. [DOI] [PubMed] [Google Scholar]
- 51.Girones N, Fresno M. Etiology of Chagas disease myocarditis: autoimmunity, parasite persistence, or both? Trends Parasitol. 2003;19:19–22. doi: 10.1016/s1471-4922(02)00006-5. [DOI] [PubMed] [Google Scholar]
- 52.Kalil J, Cunha-Neto E. Autoimmunity in Chagas disease cardiomyopathy: fulfilling the criteria at last? Parasitol Today. 1996;12:396–9. doi: 10.1016/0169-4758(96)10058-2. [DOI] [PubMed] [Google Scholar]
- 53.Brener Z, Gazzinelli RT. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas' disease. Int Arch Allergy Immunol. 1997;114:103–10. doi: 10.1159/000237653. [DOI] [PubMed] [Google Scholar]