

Abstract
Kawasaki disease (KD) is the leading cause of acquired heart disease of children in North America. It is characterized by a massive immune activation and multi-system vasculitis, which evolves into a site-specific inflammatory response focused at the coronary arteries. Coronary artery (CA) inflammation leads to elastin breakdown, destruction of the vessel wall and aneurysm formation. We have demonstrated recently the pivotal role of tumour necrosis factor (TNF)-α-mediated matrix metalloproteinase (MMP)-9 activity in the pathogenesis of elastin breakdown in a murine model of KD, Lactobacillus casei cell wall extract-induced coronary arteritis. Using this model, we evaluated the in vitro effects of doxycycline, an antibiotic with MMP inhibitory function, in modulating key pathogenic stages of disease leading to CA damage. Doxycycline inhibits T cell activation and TNF-α production in peripheral immune cells, as assessed by thymidine incorporation and a TNF bioassay respectively. Additionally, doxycycline inhibits directly MMP-9 enzymatic activity derived from TNF-α-stimulated vascular smooth muscle cells as assayed by zymography. More importantly, in vivo treatment of Lactobacillus casei cell wall extract (LCWE)-injected mice with doxycycline reduces significantly the incidence of CA elastin breakdown and reduces loss of elastin. Therefore, doxycycline can mitigate TNF-α-induced MMP-9-mediated coronary elastin breakdown and improve coronary outcome. Agents with the ability to inhibit both inflammation and the downstream effects of inflammation, such as MMP-9 activity, offer a promising therapeutic strategy for the management of children with KD.
Keywords: doxycycline, inflammation, Kawasaki disease, MMP-9, TNF
Introduction
Kawasaki disease (KD) is a vasculitis affecting mainly children, and is characterized by clinical signs and symptoms of massive multi-system inflammation [1,2]. The diagnostic criteria are prolonged fever together with at least four of the following: non-purulent bilateral conjunctivitis, polymorphous skin rash, oral–mucosal changes, extremity changes and cervical lymphadenopathy. KD is a self-limiting syndrome, and clinical features of disease subside even without treatment. However, later in the disease course a significant proportion of patients develop coronary artery (CA) aneurysms [2]. KD is the leading cause of acquired heart disease of children in North America [3]. Accepted treatment protocols for KD differ around the world, but all involve administration of both intravenous immunoglobulin (IVIG) and aspirin. The American Heart Association recommends treatment with a single infusion of high-dose IVIG (2 g/kg) together with high-dose aspirin (80–100 mg/kg/day) in the acute phase of KD [2]. The treatment of KD patients with this therapeutic regimen greatly reduces the incidence of CA damage [2,4]; however, the incidence of CA abnormalities continues to be 5% even in appropriately treated patients. When vessel diameters are adjusted for body surface area CA abnormalities can reach 25% [5], highlighting the need for improvements in therapy to improve coronary outcome. Understanding the pathogenesis of disease will allow for the tailoring of therapeutic intervention for children with KD.
The pathogenesis of KD is not well understood. Using an animal model of KD, Lactobacillus casei cell wall extract (LCWE)-induced coronary arteritis [6,7], we have identified three steps in the pathogenic process leading to CA damage: T cell activation, tumour necrosis factor (TNF)-α production and production of matrix metalloproteinase (MMP)-9. A single intraperitoneal (i.p.) injection of LCWE induces massive peripheral immune activation [6–8], characterized by peripheral T cell activation and proliferation [8]. Secondly, T cell activation leads to the production of TNF-α in the peripheral immune system, followed by local production at the coronary arteries [9]. In situ production of TNF-α occurs throughout all layers of the coronary vessel wall and coincides with the presence of maximal T cell infiltration at the CA [9]. TNF-α activity is absolutely required in the LCWE model of KD, as etanercept-treated or TNF receptor I-deficient mice are protected completely from disease induction [9].
One of the downstream consequences of TNF-α signalling is the expression of the metal-dependent elastolytic protease, MMP-9 [10]. Our recent work has demonstrated that TNF-α-induced MMP-9 enzymatic activity is a key mediator of elastin breakdown in the vasculature in this disease model [11]. Vascular smooth muscle cells (SMCs) produce MMP-9 in response to TNF-α in affected mice [11], which is consistent with human data showing expression of MMP-9 in affected coronary segments from autopsy tissue of fatal cases of KD [12]. MMP-9-deficient animals have a significantly reduced incidence of CA aneurysm formation despite ongoing coronary inflammation, pointing to its role as an important mediator of local vessel degradation and the ability to dissociate CA inflammation from harmful end organ damage by MMP-9 inhibition [11].
Controlling the downstream effects of inflammation such as MMP-9 expression may represent a novel therapeutic strategy for treatment of KD. One well-studied MMP inhibitor is the anti-microbial drug doxycycline. Doxycycline has been successful in treating certain cases of human abdominal aortic aneurysm [13,14], and can prevent abdominal aortic aneurysm formation in experimental models [15]. The efficacy of doxycycline in the treatment of arterial aneurysms is attributed to its anti-MMP activity [16], which occurs through metal chelation and direct ablation of MMP enzymatic function. Using the LCWE model of KD, we investigated the ability of doxycycline to inhibit three key stages in disease progression in vitro: T cell activation, TNF-α production and TNF-α-mediated up-regulation of MMP-9 activity; in addition, we evaluated the impact of drug treatment in vivo on the development of CA aneurysms. Although the use of doxycycline is contraindicated in young children, establishing the mechanisms involved in disease modulation for this family of therapeutic agents and establishing proof of principle are important steps towards developing novel therapies to improve coronary outcome in KD.
Methods
Animals, cell lines and reagents
C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). L-929 cells were purchased from ATCC (Manassas, VA, USA). Mouse vascular smooth muscle cell line (MOVAS) was from Dr M. Husain (Toronto General Hospital Research Institute, Toronto, Canada) [17]. Doxycycline–hyclate was purchased from Sigma-Aldrich (St Louis, MO, USA) and GM6001 was from EMD Biosciences (Gibbstown, NJ, USA). LCWE was produced as described [8].
Proliferation assay
Splenocytes were cultured (0·5 × 106 cells/well, 96-well plate) in triplicate in conditioned Iscove's modified Eagle medium (IMEM) alone, an optimal concentration of LCWE, or LCWE plus various concentrations of doxycycline for 72 h. Thymidine incorporation was performed as described [8].
TNF bioassay
Splenocytes were cultured (7·5 × 106 cells/ml) in conditioned IMEM alone, an optimal concentration of LCWE or LCWE plus various concentrations of doxycycline for 3 h. An L-929-based TNF bioassay was performed as described [18] using an MTT assay according to the manufacturer's protocol (Roche Applied Science, Laval, QC, Canada).
Real-time polymerase chain reaction (PCR)
MOVAS cells were seeded (0·5 × 106 cells/well, 12-well plate) in triplicate in Dulbecco's modified Eagle medium (DMEM) for 18 h. Medium was removed and replaced with fresh medium alone, medium containing 20 ng/ml recombinant TNF (rTNF)-α or 20 ng/ml rTNF-α plus various concentrations of doxycycline for 6 h. Real-time PCR for MMP-9 gene expression was performed as described [11].
Immunoblotting
MOVAS cells were seeded as above in DMEM for 18 h. Medium was removed and replaced with serum-free DMEM for 48 h. Serum-free medium was removed and replaced with fresh serum-free medium alone, serum-free medium containing 20 ng/ml rTNF-α or 20 ng/ml rTNF-α plus various concentrations of doxycycline for 18 h. Culture supernatants were assayed for MMP-9 protein by immunoblotting as described [11].
Enzymatic assays
MOVAS cells were seeded (10 × 106 cells/flask, 75 cm2 flask) and serum-starved as above, and left untreated or treated with 75 ng/ml rTNF-α for 18 h. Supernatants were harvested and concentrated (10 kDa molecular weight cut-off, Amicon Ultra-15; Millipore, Billerica, MA, USA). Equal amounts of total protein were subjected to gelatin zymography as described [11]. Replicate gels were incubated in the presence of 25 µg/ml doxycycline during the proteolysis step. In other experiments, equal amounts of total protein were incubated with 1 mM 4-aminophenylmercuric acetate (APMA) to activate MMPs for 3 h at 37°C. Samples were then assayed for gelatinolytic activity in the presence or absence of 25 µg/ml doxycycline using 0·1 mg/ml of a dye-quenched fluorescent substrate according to the manufacturer's protocol (EnzChek Gelatinase Assay Kit; Invitrogen, Burlington, ON, Canada).
In vivo treatment
C57BL/6 mice (4–5 weeks) were injected i.p. with 1·0 mg LCWE or 0·5 ml phosphate-buffered saline (PBS) as a negative control. Mice were left untreated or treated with doxycycline to 30–50 mg/kg/day in the drinking water beginning at the time of injection. This dose is within the therapeutic range of aortic aneurysm patients undergoing doxycycline treatment [14,15]. Three days post-injection, splenocytes were harvested and cultured (7·5 × 106 cells/ml) in conditioned IMEM in the presence of 0·1 µM phorbol myristate acetate (PMA) and 0·5 µM ionomycin for 3 h. Supernatants were assayed for TNF-α protein using an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's protocol (eBiosciences, San Diego, CA, USA). In other experiments, hearts were collected 42 days post-injection to determine the effect of doxycycline on coronary outcome. Tissues were fixed in 10% formalin, paraffinized, and sections were stained with haematoxylin and eosin (H&E) or elastin van Gieson (EVG) to identify the presence of CA inflammation or elastin layer integrity, respectively. Incidence of inflammation or elastin breakdown was evaluated by two investigators blinded to the experimental conditions. Quantification of EVG staining was performed using threshold analysis (Photoshop CS3; Adobe Systems, San Jose, CA, USA) and elastin content per animal was defined as the amount of EVG+ elastin normalized to the total medial area of the CA (Image Processing Tool Kit; Reindeer Graphics, Asheville, NC, USA). All animal experiments were approved by the Animal Care Committee at the Hospital for Sick Children Research Institute.
Statistical analysis
Statistical analysis was performed using the t-test or the χ2-test where appropriate. Significance was taken at P < 0·05.
Results
Doxycycline inhibits lymphocyte activation
To determine whether doxycycline affects the peripheral immune response, we assayed lymphocyte proliferation as a marker for lymphocyte activation. LCWE contains a novel superantigen that induces characteristically a marked proliferation of naive T cells [8]. Splenocytes were cultured in the presence of LCWE alone or LCWE containing various concentrations of doxycycline, and assessed for thymidine incorporation in proliferating cells. Concentrations of doxycycline used correspond to therapeutic serum levels found in other vascular diseases, including abdominal aortic aneurysm [14]. LCWE induced a rapid and intense primary activation of naive T cells resulting in a greater than 10-fold increase by day 3 (Fig. 1). The addition of doxycycline inhibited LCWE-induced lymphocyte proliferation significantly in a dose-dependent manner from 5–20 µg/ml. To demonstrate that doxycycline inhibited LCWE-induced proliferation separate from its metal-chelating anti-MMP activity, we performed the same experiment using the metal-chelating drug, GM6001 [19]. This drug is a broad-spectrum MMP inhibitor. As shown in the Supplementary information (Fig. S1A, at end), GM6001 was not able to inhibit LCWE-induced proliferation at any concentration examined.
Fig. 1.
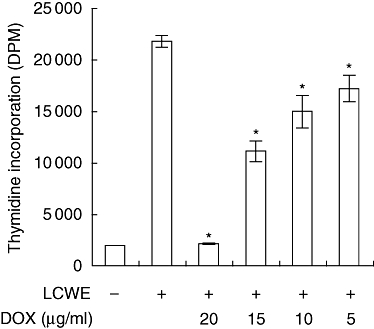
Doxycycline inhibits lymphocyte proliferation. Ex vivo splenocytes were cultured in medium alone, medium containing an optimal concentration of Lactobacillus casei cell wall extract (LCWE) or LCWE plus various concentrations of doxycycline for 72 h. Cells were pulsed with [3H]-thymidine for the last 18 h of culture. Thymidine incorporation was assessed using a scintillation counter. Data are presented as the mean DPM ± standard error of the mean of triplicate cultures (representative of three independent experiments; *P < 0·05 compared to LCWE alone). DPM, disintegrations per minute.
Doxycycline inhibits TNF-α production
To examine whether doxycycline is able to inhibit TNF-α production, splenocytes were activated with LCWE alone or cultured with LCWE and various concentrations of doxycycline, and TNF bioactivity assayed in culture supernatant using L-929 cells. LCWE induced bioactive TNF secretion into culture supernatant within hours, typical to that of a superantigen (Fig. 2a). The addition of doxycycline inhibited TNF activity derived from LCWE-activated immune cells in a dose-dependent fashion similar to those able to inhibit the proliferative response. Using a TNF-α-specific ELISA or a TNF-α-specific neutralizing antibody, we confirmed that > 95% TNF activity produced by LCWE-activated splenocytes was attributable to TNF-α (data not shown). Interestingly, GM6001 also inhibited TNF-α production in LCWE-activated splenocytes in a dose-dependent manner (see Supplementary information, Fig. S1b). This is probably attributable to generalized metalloproteinase inhibition, with the ability to inhibition the TNF-α-converting enzyme (TACE/ADAM17) [20], a metalloproteinase which regulates shedding of processed TNF-α.
Fig. 2.
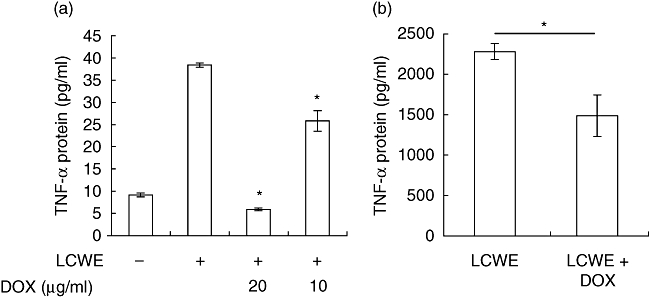
Doxycycline inhibits tumour necrosis factor (TNF)-α production. (a) Ex vivo splenocytes were cultured in medium alone, medium containing an optimal concentration of Lactobacillus casei cell wall extract (LCWE) or LCWE plus various concentrations of doxycycline for 3 h. Supernatants were assayed for TNF activity using an L-929-based bioassay. Data are presented as the mean TNF activity ± standard error of the mean (s.e.m.) of triplicate wells (representative of five independent experiments; *P < 0·05 compared to LCWE alone). (b) C57BL/6 mice were injected with LCWE and left untreated (n = 3) or treated with doxycycline in the drinking water to 30–50 mg/kg/day (n = 4) for 72 h. Ex vivo splenocytes were cultured in the presence of phorbol myristate acetate (PMA)/ionomycin for 3 h, and supernatants were assayed for TNF-α protein by enzyme-linked immunosorbent assay (data are presented as the mean TNF-α protein ± s.e.m.; *P = 0·035).
To determine whether the anti-inflammatory effects of doxycycline are operant during disease evolution in the complex milieu of the whole animal, coronary disease was induced in C57BL/6 mice using LCWE in the presence or absence of doxycycline treatment. Three days post-disease induction, we harvested splenocytes from experimental animals and stimulated them in culture with PMA/ionomycin for 3 h. TNF-α production was determined by assaying culture supernatant using ELISA. Consistent with our in vitro data, immune cells from mice treated with doxycycline responded significantly less to mitogenic rechallenge (*P = 0·035). Notably, the impaired ability to produce TNF-α was evident even without the addition of exogenous doxycycline to culture media (Fig. 2b). Taken together, doxycycline is able to inhibit production of the TNF-α both in tissue culture systems and in the context of the whole animal, and the principles demonstrated in vitro were echoed by in vivo results.
Doxycycline does not affect TNF-α-induced MMP-9 expression in SMCs
TNF-α is required for disease development in this model and is a key regulator of MMP-9 expression in SMCs [9–11]. To determine whether doxycycline can modulate TNF-α-mediated MMP-9 expression in SMCs, MOVAS cells (a murine vascular SMC line) were stimulated with TNF-α alone, or TNF-α plus various concentrations of doxycycline, and MMP-9 gene expression was assayed by real-time quantitative reverse transcription–polymerase chain reaction (RT–PCR) 6 h later. TNF-α induced marked expression of MMP-9 by 6 h (Fig. 3a), with the amount used (20 ng/ml) established previously to be in the linear range of the dose–response curve for MMP-9 production. However, doxycycline was not able to reduce MMP-9 gene expression induced by TNF-α at all concentrations of doxycycline examined, between 5–50 µg/ml. Even concentrations up to fivefold beyond therapeutic doses were not capable of affecting TNF-α-induced MMP-9 gene expression in this system.
Fig. 3.
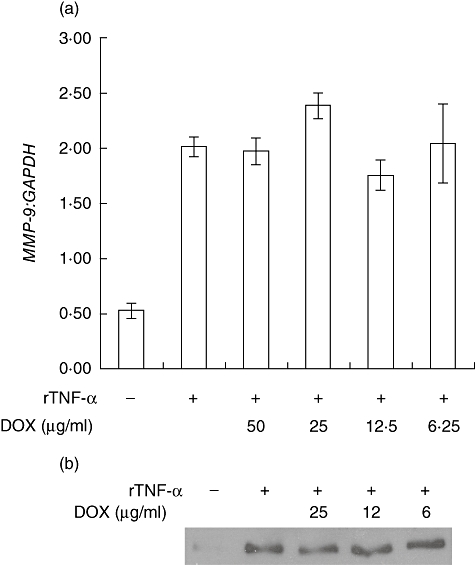
Doxycycline does not affect tumour necrosis factor (TNF)-α-induced matrix metalloproteinase (MMP)-9 expression. (a) Mouse vascular smooth muscle cell line (MOVAS) cells were cultured in medium alone, medium containing 20 ng/ml TNF-α, or TNF-α plus various concentrations of doxycycline for 6 h. MMP-9 gene expression was determined by real-time polymerase chain reaction (PCR) and normalized to GAPDH. Data is presented as the mean ratio ± standard error of the mean of triplicate cultures (representative of six independent experiments). (b) MOVAS cells were serum-starved for 48 h prior to culture in serum-free medium alone, medium containing 20 ng/ml TNF-α, or TNF-α plus various concentrations of doxycycline for 18 h. Supernatants were collected and immunoblotted for MMP-9 protein (representative of two independent experiments).
To extend these findings at the protein level, MOVAS cells were cultured in a similar fashion and assayed for MMP-9 protein secretion into culture supernatant. As shown in Fig. 3b, TNF-α induced a marked up-regulation of MMP-9 protein, following the up-regulation of MMP-9 mRNA (Fig. 3a). Consistent with our gene expression findings, therapeutic levels of doxycycline did not affect TNF-α-induced MMP-9 expression at the protein level.
Doxycycline inhibits TNF-α-induced MMP-9 enzymatic activity
Doxycycline possesses metal-chelating properties and has been demonstrated to inhibit MMP activity by this mechanism [21]. To determine the MMP inhibitory activity of doxycycline, we collected TNF-α-stimulated SMC supernatant and assayed MMP-9 enzymatic activity using gelatin zymography. Supernatants from TNF-α-stimulated SMCs produced a bright enzymatically digested band in zymography (Fig. 4a). When replicate gels were incubated in the presence of optimal concentrations of doxycycline, however, these bands disappeared. Similar findings were obtained when replicate gels were treated with the metal-chelating compounds ethylenediamine tetraacetic acid (EDTA) (data not shown) or GM6001 (see Supplementary information, Fig. S1c), but corresponding protein levels of MMP-9 were not affected, as demonstrated by immunoblotting (Fig. 3b). To confirm these results, we prepared supernatants from untreated or TNF-α-treated SMCs, and activated MMPs using APMA. These supernatants were then incubated with or without doxycycline in the presence of a gelatin substrate, of which enzymatic cleavage results in the release of fluorescence. As shown in Fig. 4b, TNF-α-treated SMCs induced a marked increase in gelatinolytic activity, which was reduced significantly in the presence of doxycycline. These results demonstrate that doxycycline cannot affect MMP-9 expression at the message or protein level, but is able to directly inhibit MMP-9 enzymatic activity.
Fig. 4.
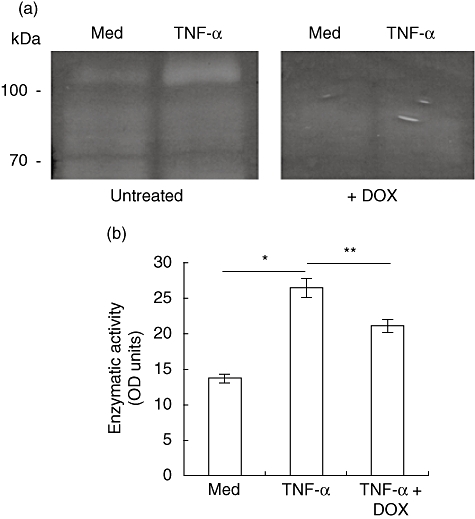
Doxycycline inhibits tumour necrosis factor (TNF)-α-induced matrix metalloproteinase (MMP)-9 enzymatic activity. (a) Mouse vascular smooth muscle cell line (MOVAS) cells were serum-starved for 48 h prior to culture in serum-free medium alone, or medium containing 75 ng/ml TNF-α for 18 h. Supernatants were concentrated by size-exclusion centrifugation and assayed for MMP-9 enzymatic activity by gelatin zymography in replicate. One gel was treated with 25 µg/ml doxycycline during the proteolysis step. Areas of enzymatic activity appear as white against a dark background (representative of three independent experiments). (b) MOVAS supernatants were prepared as described above and incubated with 1 mM 4-aminophenylmercuric acetate (APMA) for 3 h to activate MMPs. Supernatants were incubated with a fluorescent gelatin substrate for 18 h in the presence or absence of 25 µg/ml doxycycline. Digestion of substrate was quantified using a fluorescent microplate reader. Data are presented as the mean optical density ± s.e.m. of triplicate wells (representative of four independent experiments; *P = 0·001, **P = 0·029).
Treatment with doxycycline improves coronary outcome
To examine whether doxycycline can improve coronary outcome, we determined the presence of inflammation and elastin breakdown in coronary vessels from experimental mice treated with or without doxycycline. There was a significantly reduced incidence of LCWE-induced coronary arteritis in experimental animals treated with doxycycline, 46% versus 75% in untreated mice (P = 0·0275, Table 1, Fig. 5a). Serial sections were stained with EVG to evaluate the integrity of elastin layers. In mice injected with LCWE, elastin breakdown was present in 46% of animals, evidenced by less intense EVG staining (indicating less elastin content), a decrease in the number of elastin layers and, in certain cases, discontinuation and/or breakage of the elastic lamina (Fig. 5a, Table 1). Conversely, in animals treated with doxycycline, elastin layers were intact and black-stained intensely with EVG, with only 17% of treated animals demonstrating signs of elastin breakdown, and a significantly reduced (P = 0·0144) incidence of CA elastin breakdown compared to untreated animals. Incidences of coronary arteritis and elastin breakdown in the untreated animals were consistent with previous findings, both by our group [9,11] and others [6,7].
Table 1.
Incidence of coronary artery inflammation and elastin breakdown in untreated and doxycycline-treated mice.
| LCWE-injected | Incidence of inflammation† | Elastin breakdown‡ |
|---|---|---|
| Untreated | 21 / 28 | 13 / 28 |
| Doxycycline-treated§ | 14 / 30* | 5 / 30** |
P = 0·0275, compared to untreated controls;
P = 0·0144, compared to untreated controls.
Incidence of inflammation is presented as the number of mice with coronary arteritis over the total number of mice injected with Lactobacillus casei cell wall extract (LCWE) at day 42.
Incidence of elastin breakdown is presented as the number of mice with elastin breakdown over the total number of mice injected with LCWE at day 42.
Wild-type mice were treated with doxycycline in the drinking water to 30–50 mg/kg/day.
Fig. 5.
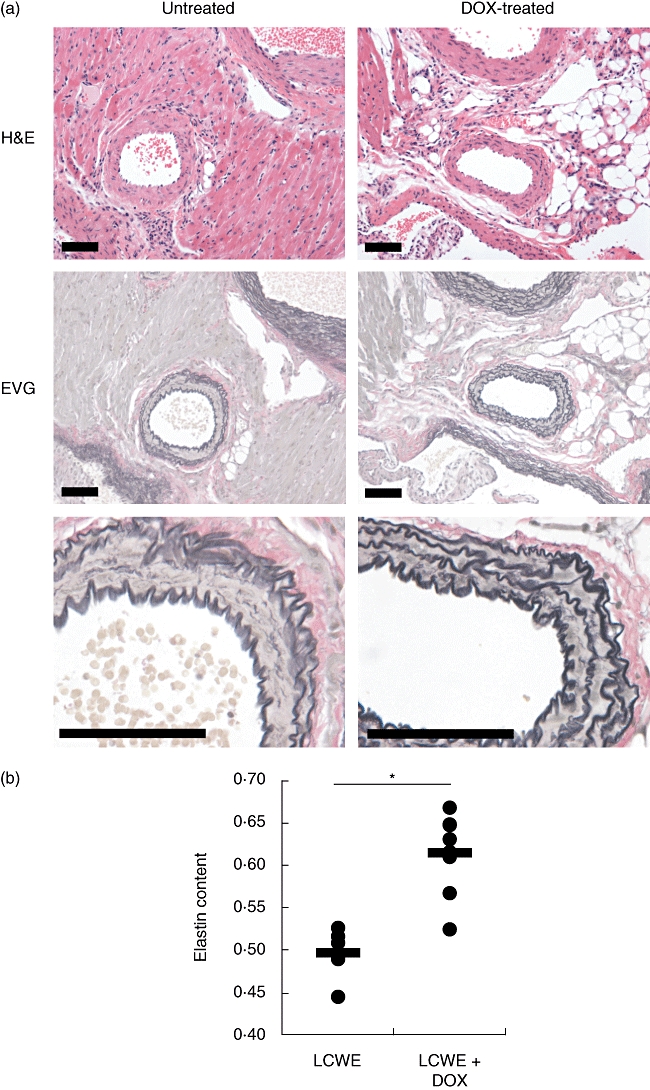
Treatment with doxycycline improves coronary outcome. (a) C57BL/6 mice were injected with Lactobacillus casei cell wall extract (LCWE) and left untreated or treated with doxycycline in the drinking water to 30–50 mg/kg/day. Heart sections were prepared 42 days later and stained with haematoxylin and eosin or elastin van Gieson (EVG) to evaluate the presence of coronary artery inflammation or elastin breakdown, respectively (bar, 50 µm). (b) Digital images of LCWE-responsive (n = 5) or doxycycline-treated LCWE-injected (n = 8) mice were subjected to measurement of EVG+ elastin by colour threshold and arterial area. Elastin content is defined as the ratio of elastin to artery area (*P = 0·0005).
To quantitate the impact of doxycycline on preventing elastin loss in LCWE-injected animals, we determined elastin content in the CA by morphometric analysis. Using digitized histological images, we defined elastin using EVG+ colour threshold analysis, and the area of the artery as the region bordered by the lumen and the external elastic lamina. Elastin content in the CA was then calculated as the area of elastin divided by the total area of the artery. Interestingly, the elastin content in doxycycline-treated LCWE-injected mice (0·614 ± 0·017) was significantly higher compared to untreated LCWE-injected mice (0·497 ± 0·014, P = 0·0005, Fig. 5b), and was similar to PBS-injected control animals (0·596 ± 0·057, data not shown). Taken together, these data suggest that treatment of mice with doxycycline, an anti-MMP drug, can decrease elastin breakdown significantly, thus improving coronary outcome.
Discussion
KD is characterized by multi-system inflammation followed by CA damage in a significant proportion of patients [2]. Despite optimal therapeutic management with IVIG and aspirin, 5% of affected children continue to develop coronary aneurysms [2,4,22,23]. When vessel diameters are adjusted for body surface area, and not just adult-sized definitions of aneurysms are used, up to 25% of appropriately treated patients have evidence of CA lesions [5]. Anti-inflammatory agents alone may not be sufficient to prevent coronary damage. Additionally, inflammation is long-standing when a diagnosis of KD is usually entertained (by definition 5 days of fever), initiation of therapy is days after initiation of inflammation. Therefore, pharmacological targeting of the downstream effects of inflammation may be of benefit to KD patients.
Using an animal model of KD, LCWE-induced coronary arteritis in mice [6,7], we have previously identified key pathogenic events leading to end organ damage. Immune activation leading to TNF-α activity is absolutely necessary for the development of CA inflammation and elastin breakdown [9], which is mediated by MMP-9 activity localized to coronary vascular SMCs [11,24]. MMP-9 appears to be the major MMP family member with increased activity at the coronary arteries, as elevated levels of other elastases (namely MMP-1, 2, 3, 7 or 12) were not detected in response to LCWE in this model [11]. Furthermore, MMP-9 elastolytic activity is critical to the development of coronary artery aneurysms, as MMP-9-deficient mice continue to develop coronary inflammation but have a significantly reduced incidence of elastin breakdown [11]. Therefore, the ablation of MMP-9 activity holds the promise of dissociating inflammation from end organ damage. One well-studied MMP inhibitor is doxycycline, a tetracycline family member. Doxycycline is a classic anti-microbial drug with the ability to prevent bacterial polypeptide synthesis [25], but was discovered later to have the ability to chelate metals independently of its anti-microbial functions [21,26]. As a result, doxycycline can inactivate metal-dependent proteases such as the MMPs, but not others, such as the serine proteases. We therefore examined the effects of doxycycline on the inflammatory response leading to MMP-9 expression, as well as on MMP-9 enzymatic activity, in our disease model both in vitro and in vivo.
TNF-α is the central and key mediator of CA damage [9]. We show that doxycycline is able to inhibit the expression and activity of TNF-α in response to LCWE in both in vitro and in vivo systems (Fig. 2). Other immunomodulatory actions of doxycycline are the inhibition of lymphocyte proliferation in response to LCWE in a dose-dependent manner (Fig. 1). Although the exact mechanisms of action are not clear, doxycycline has also been demonstrated to inhibit components of the immune system. For example, it inhibits superantigen-induced T cell activation, cytokine production and chemokine production in human peripheral blood mononuclear cells (PBMCs) [27]. Treatment of mice with tetracycline family compounds also protects against toxic shock through a similar down-regulation of proinflammatory mediators [28]. It is notable that tetracyclines can inhibit the activation of protein kinase C [29], an important mediator of T cell activation and subsequent development of inflammation. This mechanism of action may also be related to its metal-chelating properties as chemically modified tetracyclines, which lack anti-microbial functions but retain anti-MMP properties, are capable of inhibiting protein kinase C and have other anti-inflammatory properties [30]. Along these lines, drugs such as GM6001, conventionally known as anti-MMP agents with metal-chelating properties, can block the secretion of TNF-α via inhibiting the metal-dependent TACE [20].
In the context of KD, doxycycline's effects as a broad-spectrum inhibitor of MMP enzymatic activity may reflect another level of beneficial activity in addition to its role in immune suppression [31]. Doxycycline been demonstrated to inhibit gene transcription of several MMPs [32,33], as well as decrease MMP-2 mRNA stability [34]. Indirectly, doxycycline can modulate the expression of MMPs by inhibiting the growth and migration of SMCs [35]. In KD, immune activation results in the production of the critical cytokine, TNF-α, leading to pathogenic events such as increased local MMP-9 expression and activity [9,11]. In particular, MMP-9 is the critical link between inflammation and end organ damage in our model of KD [11].We therefore examined the ability of doxycycline to affect both TNF-α-induced MMP-9 gene expression as well as enzymatic activity in cultured vascular SMCs, as this cell type produces MMP-9 in the CA during disease development [11]. Doxycycline was not able to affect MMP-9 synthesis at the message or protein level induced by TNF-α (Fig. 3), however, was able to ablate MMP-9 functional activity at the post-transcriptional level (Fig. 4). The presence of doxycycline did not affect MMP-9 protein levels (Fig. 3b), suggesting that a direct inhibition of MMP-9 enzymatic activity was the mechanism responsible for this finding. The similarities between doxycycline-treated enzymatic assays and treatment with other metal chelators such as GM6001 provided further evidence to support this notion (Supplementary information, Fig. S1). An additional explanation may be that doxycycline affects processing of pro-MMP-9 to its active enzymatic form, especially if upstream activators are metal-dependent. Regardless of the exact molecular mechanism by which doxycycline acts, the end result is that it effectively inhibits MMP-9 enzymatic activity and reduces coronary damage. Therefore, these results taken together provide evidence that doxycycline can affect disease progression at several stages. It can inhibit MMP-9 production indirectly through modulating lymphocyte activation/proliferation and secretion of TNF-α by its anti-inflammatory actions. Additionally, it can directly inhibit MMP-9 enzymatic activity produced by vascular SMCs, the major producer of MMP-9 during disease evolution. Therefore, pharmacological agents with the ability to inhibit both inflammation and the downstream consequences of inflammation such as matrix remodelling may be of therapeutic value in the treatment of KD.
Consistent with our in vitro results, treatment of affected mice with doxycycline reduced the incidence of CA inflammation as well as elastin breakdown (Fig. 5, Table 1). A return to baseline levels of elastin in the CA was seen in doxycycline-treated animals compared with untreated animals (P = 0·0005, Fig. 5b). In addition, immune cells from doxycycline-treated mice produced less TNF-α in response to PMA/ionomycin, a potent mitogen (Fig. 2b). Coupled with our previous findings, that TNF-α is the central mediator of inflammation and subsequent CA damage in this model of KD [9,11], this highlights the ability of this family of drugs to modulate both inflammation and its downstream effects including matrix breakdown. Furthermore, five of 14 (36%) doxycycline-treated animals with coronary arteritis developed vessel damage as measured by histological evidence of elastin breakdown, compared with 13 of 21 (62%) untreated mice (Table 1). Although this did not reach statistical significance (P = 0·13), a trend towards a reduction in vessel damage in mice with arteritis was seen. This provides further support that, in addition to the ability of doxycycline to inhibit inflammation, it is also capable of inhibiting MMP activity, because coronary damage was reduced further in doxycycline-treated mice. These results demonstrate clearly that treatment with doxycycline affects multiple stages of disease. While immunomodulatory agents such as IVIG and aspirin are certainly efficacious in decreasing the inflammatory response, they do not protect fully from coronary sequelae. Treatment of LCWE-injected animals with the MMP inhibitor doxycycline leads to a significant improvement in coronary disease, characterized by a marked reduction in elastin breakdown. Doxycycline treatment has been demonstrated previously to ameliorate aortic aneurysm formation, both in animal models and clinical trials in human diseases [13–15], and may be even more efficacious than traditional therapies [16].
Given the known potential adverse effects of doxycycline in the developing child, this family of compounds is not indicated for use in children. Our results demonstrate clearly that the principle of MMP inhibition, either directly or indirectly through TNF-α, improves coronary outcome in a model of KD. Interestingly ulinastatin, a serine protease inhibitor, has been used successfully in the treatment of recalcitrant KD [36], and also modulates MMP-9 [37]. Similar to doxycycline, ulinastatin belongs to a family of pharmacological agents with dual anti-inflammatory and anti-MMP functions. The results of this study suggest that combating both inflammation and matrix breakdown together may be a beneficial therapeutic strategy. Furthermore, MMP inhibition in patients who continue to develop coronary damage, even after successful IVIG treatment, may be a promising therapeutic option in KD. Commonly used anti-MMP drugs such as GM6001 exploit the metal-dependent aspect of this family of enzymes and may continue to pose similar risks to children as doxycycline [19]. The recent development of agents targeting specific MMPs (e.g. monoclonal antibodies [38,39]) and their use in combination with proven anti-inflammatory agents may also be an interesting management option. Our results highlight the need to develop novel agents, strategies and regimens to target both immune and inflammation-driven pathogenic events leading to coronary damage in KD while minimizing potential side effects to developing children.
We examined both early and late pathogenic events that are critical to the pathogenesis of coronary disease in the LCWE model of KD. Immune activation and TNF-α production leads to up-regulation of MMP-9 activity, matrix degradation and CA damage. Doxycycline inhibits immune activation, TNF-α production and matrix-degrading enzymatic activity. Inhibition of MMP-9 production is indirect, occurring via inhibition of TNF-α production, but when MMP-9 protein is produced doxycycline is able to inhibit its enzymatic activity directly. Importantly, coronary outcome is improved significantly by treating affected mice with doxycycline. Drugs which modulate both the inflammatory response as well as inhibit MMP activity are promising strategies for the treatment of diseases involving inflammation-driven matrix breakdown. Understanding the pathogenesis of disease will allow for the tailoring of therapeutic interventions to improve coronary outcome in KD.
Acknowledgments
The authors are grateful to Lily Morikawa for histological expertise, Kamila Bertlik for LCWE production, Devina Ramsaroop for technical assistance, Peter Wilson, Mohammad Eskandarian and Sue Omar for assistance with elastin measurements and Dr Mansoor Husain for MOVAS cells. This study was funded by operating grants from the Canadian Institutes of Health Research (FRN 53245) and the Arthritis Society of Canada. R. S. M. Y. is a recipient of an Investigator Award from the Arthritis Society of Canada.
Disclosure
All authors have no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. GM6001 affects TNF production and MMP-9 activity, but not lymphocyte proliferation. (a) Splenocytes were activated with LCWE and indicated concentrations of GM6001 for 72 h. Thymidine incorporation was used to determine proliferative response. (b) Splenocytes were activated with LCWE and indicated concentrations of GM6001 for 3 h. TNF bioactivity was assessed in culture supernatants using L-929 cells. (c) MOVAS cells were stimulated with TNF-α for 18 h and supernatants were assessed for MMP-9 enzymatic activity using zymography. Replicate gels were treated in the presence of GM6001.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children [in Japanese. Arerugi. 1967;16:178–222. [PubMed] [Google Scholar]
- 2.Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747–71. doi: 10.1161/01.CIR.0000145143.19711.78. [DOI] [PubMed] [Google Scholar]
- 3.Taubert KA, Rowley AH, Shulman ST. Nationwide survey of Kawasaki disease and acute rheumatic fever. J Pediatr. 1991;119:279–82. doi: 10.1016/s0022-3476(05)80742-5. [DOI] [PubMed] [Google Scholar]
- 4.Newburger JW, Takahashi M, Beiser AS, et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med. 1991;324:1633–9. doi: 10.1056/NEJM199106063242305. [DOI] [PubMed] [Google Scholar]
- 5.de Zorzi A, Colan SD, Gauvreau K, Baker AL, Sundel RP, Newburger JW. Coronary artery dimensions may be misclassified as normal in Kawasaki disease. J Pediatr. 1998;133:254–8. doi: 10.1016/s0022-3476(98)70229-x. [DOI] [PubMed] [Google Scholar]
- 6.Lehman TJ, Walker SM, Mahnovski V, McCurdy D. Coronary arteritis in mice following the systemic injection of group B Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum. 1985;28:652–9. doi: 10.1002/art.1780280609. [DOI] [PubMed] [Google Scholar]
- 7.Lehman TJ, Warren R, Gietl D, Mahnovski V, Prescott M. Variable expression of Lactobacillus casei cell wall-induced artertis: an animal model of Kawasaki's disease in selected inbred mouse strains. Clin Immunol Immunopathol. 1988;48:108–18. doi: 10.1016/0090-1229(88)90161-4. [DOI] [PubMed] [Google Scholar]
- 8.Duong TT, Silverman ED, Bissessar MV, Yeung RS. Superantigenic activity is responsible for induction of coronary arteritis in mice: an animal model of Kawasaki disease. Int Immunol. 2003;15:79–89. doi: 10.1093/intimm/dxg007. [DOI] [PubMed] [Google Scholar]
- 9.Hui-Yuen JS, Duong TT, Yeung RS. TNF-α is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. J Immunol. 2006;176:6294–301. doi: 10.4049/jimmunol.176.10.6294. [DOI] [PubMed] [Google Scholar]
- 10.Moon SK, Cha BY, Kim CH. ERK1/2 mediates TNF-α-induced matrix metalloproteinase-9 expression in human vascular smooth muscle cells via the regulation of NF-κB and AP-1: involvement of the ras dependent pathway. J Cell Physiol. 2004;198:417–27. doi: 10.1002/jcp.10435. [DOI] [PubMed] [Google Scholar]
- 11.Lau AC, Duong TT, Ito S, Yeung RSM. Matrix metalloproteinase-9 activity leads to elastin breakdown in an animal model of Kawasaki disease. Arthritis Rheum. 2008;58:854–63. doi: 10.1002/art.23225. [DOI] [PubMed] [Google Scholar]
- 12.Gavin PJ, Crawford SE, Shulman ST, Garcia FL, Rowley AH. Systemic arterial expression of matrix metalloproteinases 2 and 9 in acute Kawasaki disease. Arterioscler Thromb Vasc Biol. 2003;23:576–81. doi: 10.1161/01.ATV.0000065385.47152.FD. [DOI] [PubMed] [Google Scholar]
- 13.Curci JA, Mao D, Bohner DG, et al. Preoperative treatment with doxycycline reduces aortic wall expression and activation of matrix metalloproteinases in patients with abdominal aortic aneurysms. J Vasc Surg. 2000;31:325–42. doi: 10.1016/s0741-5214(00)90163-0. [DOI] [PubMed] [Google Scholar]
- 14.Prall AK, Longo GM, Mayhan WG, et al. Doxycycline in patients with abdominal aortic aneurysms and in mice: comparison of serum levels and effect on aneurysm growth in mice. J Vasc Surg. 2002;35:923–9. doi: 10.1067/mva.2002.123757. [DOI] [PubMed] [Google Scholar]
- 15.Pyo R, Lee JK, Shipley JM, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–9. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung AW, Yang HH, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ Res. 2008;102:e73–85. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- 17.Afroze T, Yang LL, Wang C, et al. Calcineurin-independent regulation of plasma membrane Ca2+ ATPase-4 in the vascular smooth muscle cell cycle. Am J Physiol Cell Physiol. 2003;285:C88–95. doi: 10.1152/ajpcell.00518.2002. [DOI] [PubMed] [Google Scholar]
- 18.Branch DR, Shah A, Guilbert LJ. A specific and reliable bioassay for the detection of femtomolar levels of human and murine tumor necrosis factors. J Immunol Methods. 1991;143:251–61. doi: 10.1016/0022-1759(91)90050-p. [DOI] [PubMed] [Google Scholar]
- 19.Grobelny D, Poncz L, Galardy RE. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry. 1992;31:7152–4. doi: 10.1021/bi00146a017. [DOI] [PubMed] [Google Scholar]
- 20.Solorzano CC, Ksontini R, Pruitt JH, et al. A matrix metalloproteinase inhibitor prevents processing of tumor necrosis factor α (TNF α) and abrogates endotoxin-induced lethality. Shock. 1997;7:427–31. doi: 10.1097/00024382-199706000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Golub LM, Lee HM, Lehrer G, et al. Minocycline reduces gingival collagenolytic activity during diabetes. Preliminary observations and a proposed new mechanism of action. J Periodontal Res. 1983;18:516–26. doi: 10.1111/j.1600-0765.1983.tb00388.x. [DOI] [PubMed] [Google Scholar]
- 22.Terai M, Shulman ST. Prevalence of coronary artery abnormalities in Kawasaki disease is highly dependent on gamma globulin dose but independent of salicylate dose. J Pediatr. 1997;131:888–93. doi: 10.1016/s0022-3476(97)70038-6. [DOI] [PubMed] [Google Scholar]
- 23.Durongpisitkul K, Gururaj VJ, Park JM, Martin CF. The prevention of coronary artery aneurysm in Kawasaki disease: a meta-analysis on the efficacy of aspirin and immunoglobulin treatment. Pediatrics. 1995;96:1057–61. [PubMed] [Google Scholar]
- 24.Lau AC, Rosenberg H, Duong TT, McCrindle BW, Yeung RS. Elastolytic matrix metalloproteinases and coronary outcome in children with Kawasaki disease. Pediatr Res. 2007;61:710–15. doi: 10.1203/pdr.0b013e318053418b. [DOI] [PubMed] [Google Scholar]
- 25.Goldman RA, Hasan T, Hall CC, Strycharz WA, Cooperman BS. Photoincorporation of tetracycline into Escherichia coli ribosomes. Identification of the major proteins photolabeled by native tetracycline and tetracycline photoproducts and implications for the inhibitory action of tetracycline on protein synthesis. Biochemistry. 1983;22:359–68. doi: 10.1021/bi00271a020. [DOI] [PubMed] [Google Scholar]
- 26.Golub LM, McNamara TF, d'Angelo G, Greenwald RA, Ramamurthy NS. A non-antibacterial chemically-modified tetracycline inhibits mammalian collagenase activity. J Dent Res. 1987;66:1310–14. doi: 10.1177/00220345870660080401. [DOI] [PubMed] [Google Scholar]
- 27.Krakauer T, Buckley M. Doxycycline is anti-inflammatory and inhibits staphylococcal exotoxin-induced cytokines and chemokines. Antimicrob Agents Chemother. 2003;47:3630–3. doi: 10.1128/AAC.47.11.3630-3633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shapira L, Barak V, Soskolne WA, Halabi A, Stabholz A. Effects of tetracyclines on the pathologic activity of endotoxin: in vitro and in vivo studies. Adv Dent Res. 1998;12:119–22. doi: 10.1177/08959374980120010401. [DOI] [PubMed] [Google Scholar]
- 29.Nikodemova M, Watters JJ, Jackson SJ, Yang SK, Duncan ID. Minocycline down-regulates MHC II expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC)alpha/betaII. J Biol Chem. 2007;282:15208–16. doi: 10.1074/jbc.M611907200. [DOI] [PubMed] [Google Scholar]
- 30.Sandler C, Ekokoski E, Lindstedt KA, et al. Chemically modified tetracycline (CMT)-3 inhibits histamine release and cytokine production in mast cells: possible involvement of protein kinase C. Inflamm Res. 2005;54:304–12. doi: 10.1007/s00011-005-1358-5. [DOI] [PubMed] [Google Scholar]
- 31.Golub LM, Ramamurthy NS, McNamara TF, Greenwald RA, Rifkin BR. Tetracyclines inhibit connective tissue breakdown: new therapeutic implications for an old family of drugs. Crit Rev Oral Biol Med. 1991;2:297–321. doi: 10.1177/10454411910020030201. [DOI] [PubMed] [Google Scholar]
- 32.Jonat C, Chung FZ, Baragi VM. Transcriptional downregulation of stromelysin by tetracycline. J Cell Biochem. 1996;60:341–7. doi: 10.1002/(sici)1097-4644(19960301)60:3<341::aid-jcb6>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 33.Hanemaaijer R, Visser H, Koolwijk P, et al. Inhibition of MMP synthesis by doxycycline and chemically modified tetracyclines (CMTs) in human endothelial cells. Adv Dent Res. 1998;12:114–18. doi: 10.1177/08959374980120010301. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Xiong W, Baca-Regen L, Nagase H, Baxter BT. Mechanism of inhibition of matrix metalloproteinase-2 expression by doxycycline in human aortic smooth muscle cells. J Vasc Surg. 2003;38:1376–83. doi: 10.1016/s0741-5214(03)01022-x. [DOI] [PubMed] [Google Scholar]
- 35.Bendeck MP, Conte M, Zhang M, Nili N, Strauss BH, Farwell SM. Doxycycline modulates smooth muscle cell growth, migration, and matrix remodeling after arterial injury. Am J Pathol. 2002;160:1089–95. doi: 10.1016/S0002-9440(10)64929-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zaitsu M, Hamasaki Y, Tashiro K, et al. Ulinastatin, an elastase inhibitor, inhibits the increased mRNA expression of prostaglandin H2 synthase-type 2 in Kawasaki disease. J Infect Dis. 2000;181:1101–9. doi: 10.1086/315332. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto K, Nagao Y, Kato K, Mori Y, Ito A. Human urinary trypsin inhibitor inhibits the activation of pro-matrix metalloproteinases and proteoglycans release in rabbit articular cartilage. Life Sci. 1998;63:205–13. doi: 10.1016/s0024-3205(98)00261-6. [DOI] [PubMed] [Google Scholar]
- 38.King AG, Horowitz D, Dillon SB, et al. Rapid mobilization of murine hematopoietic stem cells with enhanced engraftment properties and evaluation of hematopoietic progenitor cell mobilization in rhesus monkeys by a single injection of SB-251353, a specific truncated form of the human CXC chemokine GROβ. Blood. 2001;97:1534–42. doi: 10.1182/blood.v97.6.1534. [DOI] [PubMed] [Google Scholar]
- 39.Martens E, Leyssen A, Van Aelst I, et al. A monoclonal antibody inhibits gelatinase B/MMP-9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim Biophys Acta. 2007;1770:178–86. doi: 10.1016/j.bbagen.2006.10.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.