

Abstract
There are increased numbers of activated T lymphocytes in the bronchial mucosa of stable chronic obstructive pulmonary disease (COPD) patients. T helper type 17 (Th17) cells release interleukin (IL)-17 as their effector cytokine under the control of IL-22 and IL-23. Furthermore, Th17 numbers are increased in some chronic inflammatory conditions. To investigate the expression of interleukin (IL)-17A, IL-17F, IL-21, IL-22 and IL-23 and of retinoic orphan receptor RORC2, a marker of Th17 cells, in bronchial biopsies from patients with stable COPD of different severity compared with age-matched control subjects. The expression of IL-17A, IL-17F, IL-21, IL-22, IL-23 and RORC2 was measured in the bronchial mucosa using immunohistochemistry and/or quantitative polymerase chain reaction. The number of IL-22+ and IL-23+ immunoreactive cells is increased in the bronchial epithelium of stable COPD compared with control groups. In addition, the number of IL-17A+ and IL-22+ immunoreactive cells is increased in the bronchial submucosa of stable COPD compared with control non-smokers. In all smokers, with and without disease, and in patients with COPD alone, the number of IL-22+ cells correlated significantly with the number of both CD4+ and CD8+ cells in the bronchial mucosa. RORC2 mRNA expression in the bronchial mucosa was not significantly different between smokers with normal lung function and COPD. Further, we report that endothelial cells express high levels of IL-17A and IL-22. Increased expression of the Th17-related cytokines IL-17A, IL-22 and IL-23 in COPD patients may reflect their involvement, and that of specific IL-17-producing cells, in driving the chronic inflammation seen in COPD.
Keywords: autoimmunity, bronchial biopsies, emphysema, neutrophils, pathology
Introduction
The pathological hallmarks of chronic obstructive pulmonary disease (COPD) are destruction of the lung parenchyma, which characterizes pulmonary emphysema; inflammation of the peripheral airways, which characterizes respiratory bronchiolitis; and inflammation of the central airways [1–3]; and are due mainly to cigarette smoking [1]. Airflow obstruction in COPD is caused by small (peripheral) airway lesions [4]. Most patients with COPD have all three pathological conditions (chronic obstructive bronchiolitis, emphysema and mucus plugging), but the relative extent of emphysema and obstructive bronchitis within individual patients can vary widely. Pathological studies show that inflammation in COPD occurs in the central and peripheral airways (bronchioles) and lung parenchyma [3]. Previous studies have emphasized the potential role of many inflammatory cells in the pathogenesis of COPD, mainly alveolar macrophages and T lymphocytes, and particularly CD8 cells [1,2,4,5].
Interleukin (IL)-17, also known as IL-17A (one of the six members of the IL-17 family of cytokines) is a 20–30 kDa glycosylated homodimeric cytokine produced predominantly by CD4 and CD8 T cells of both type 1 and type 2 cytokine profiles, known as Th17 cells [6]. Airway inflammation with a COPD-like phenotype is induced by IL-17A in animal models [7]. IL-17 production can be induced in human T cells in vitro by different combinations of IL-1, IL-2, IL-6, IL-15, IL-18, IL-21 and IL-23 [8]. Interestingly, in vitro, human regulatory T cells (Tregs) can differentiate into IL-17-producing cells when stimulated by monocytes in the presence of IL-2/Il-15 [9]. In contrast, both IL-4 and interferon (IFN)-γ regulated Th17 production of IL-17 negatively [10]. In addition to IL-17A, Th17 cells also release IL-17F, IL-21 and IL-22 [11]. Human Th17 cell differentiation is also regulated by IL-23, which is related structurally to IL-12 and shares some of its biological effects, and is secreted by dendritic cells [12,13]. The transcription factor retinoic orphan receptor (ROR)-gamma t and its human homologue RORC2 has been described recently as a specific marker of Th17 cells [14].
IL-17A induces the release of CXCL1 (GRO-α), CXCL8 (IL-8) and granulocyte–macrophage colony-stimulating factor (GM-CSF) from airway epithelial cells and smooth muscle cells and thereby may orchestrate neutrophilic inflammation [15–17]. Interestingly, IL-17A can induce IL-6 expression in bronchial epithelial cells and fibroblasts [15] and IL-17A, in conjunction with IL-6, is able to induce MUC5AC and MUC5B production in primary human tracheobronchial epithelial cells [18]. Overexpression of IL-17 in murine lung epithelium induces lung inflammation with a COPD-like phenotype involving recruitment of CD4 cells, mucus hypersecretion, small airways fibrosis and induction of the expression of many chemokines (including CXCL1) matrix metalloproteinase-9 and matrix metalloproteinase inhibitor TIMP-1 [10]. IL-17F may also have similar neutrophil-promoting effects [19]. However, in animal models of lung neutrophilia the administration of anti-IL-17F antibodies has no effect on lung neutrophilia [20].
The aim of the present study was to produce a detailed analysis of the expression of RORC2 and of the Th17-related cytokines IL-17A, IL-17F, IL-21, IL-22 and IL-23 in bronchial biopsies from patients with stable COPD of different severity [Global Obstructive Lung Disease Initiative (GOLD) stages 2–4] and age-matched control groups. To this purpose we have used a combination of immunohistochemistry and real-time quantitative polymerase chain reaction (RT–QPCR) and we have shown that the number of IL-17A+, IL-22+ and IL-23+ immunoreactive cells is increased in the bronchial mucosa of stable COPD compared with control non-smokers and that the number of IL-22+ cells is correlated significantly with the number of both CD4+ and CD8+ cells in the bronchial mucosa. Furthermore, we demonstrate that high levels of IL-17A and IL-22 are expressed in bronchial epithelial cells. These data suggest that Th17-related cytokines may be involved in the pathogenesis of COPD.
Methods
Subjects
All subjects were recruited from the Section of Respiratory Medicine of the Fondazione Salvatore Maugeri, Italy. We examined bronchial biopsies from 47 subjects by immunohistochemistry; 39 were current or ex-smokers with normal lung function (n = 11) or chronic obstructive pulmonary disease (COPD) (n = 28), and eight were lifelong non-smokers with normal lung function (Table 1).
Table 1.
Characteristics of subjects for the immunohistochemical study.
| Control non-smokers | Control smokers | Mild/moderate COPD | Severe COPD | |
|---|---|---|---|---|
| Subjects (n) | 8 | 11 | 14 | 14 |
| Age (years) | 66 ± 2 | 60 ± 3 | 68 ± 2 | 68 ± 3 |
| Sex (male/female) | 4/4 | 9/2 | 12/2 | 11/3 |
| Smoking history pack-years (ex/current) | 0 | 45 ± 9 (2/9) | 43 ± 5 (4/10) | 60 ± 11 (11/3) |
| FEV1(% pred) | 119 ± 4 | 99 ± 4 | 64 ± 3* | 34 ± 3*,** |
| FEV1/FVC (%) | 85 ± 4 | 80 ± 1 | 59 ± 2* | 43 ± 2*,** |
| ΔFEV1 post (% baseline) | – | – | 5 ± 1 | 4 ± 1** |
Analysis of variance (anova)
P < 0·001significantly different from control smokers and control non-smokers;
P < 0·05 significantly different from mild/moderate chronic obstructive pulmonary disease (COPD). Data are presented as mean ± standard error. FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity.
Subjects used in the RT–QPCR (n = 40) study were non-smokers with normal lung function [n = 9; forced expiratory volume in 1 s (FEV1) = 101 ± 4; FEV1/forced vital capacity (FVC) (%) = 82 ± 4), smokers with normal lung function (n = 7; FEV1 = 95·4 ± 5; FEV1/FVC (%) = 80 ± 2; pack-years = 45·7 ± 12), patients with mild/moderate COPD (n = 9; FEV1 = 65·8 ± 3; FEV1/FVC (%) = 56·0 ± 3; pack-years = 44·2 ± 7) and patients with severe COPD (n = 15; FEV1 = 37·0 ± 2; FEV1/FVC (%) = 46·5 ± 2; pack-years = 61·0 ± 11). The severity of the airflow obstruction was staged using the GOLD criteria [1]. All COPD patients were in stable phase with no previous exacerbation in the 6 months prior to bronchoscopy. None of the volunteers had suspected or documented lung cancer. The study conformed to the Declaration of Helsinki, and informed consent was obtained from each subject. Bronchial biopsies were performed according to the local Ethics Committee Guidelines.
Lung function tests and volumes
Pulmonary function tests were performed as described previously [21], according to published guidelines.
Fibreoptic bronchoscopy, collection and processing of bronchial biopsies
A standardized procedure, reported previously [21], was followed for fibreoptic bronchoscopy and collection of bronchial biopsies. Four bronchial biopsy specimens were taken from segmental and subsegmental airways of the right lower and upper lobes. Bronchial biopsies for immunohistochemistry and RT–QPCR were processed as described previously [21]. Briefly, two samples were embedded in Tissue Tek II OCT (Miles Scientific, Naperville, IL, USA), frozen within 15 min in isopentane precooled in liquid nitrogen and stored at −80°C. The best frozen sample was then orientated and 6 mm-thick cryostat sections were cut for immunohistochemical light microscopy analysis and 30 mm-thick cryostat sections were cut for RT–QPCR and processed as described below.
Immunohistochemistry
Serial sections were stained with a panel of primary antibodies applied at optimal dilutions in TRIS-buffered saline (0·15 M saline containing 0·05 M TRIS-hydrochloric acid at pH 7·6) and revealed with the use of appropriate secondary antibodies and fast-red substrate, as described previously [21]. The following panel of primary antibodies was used: goat anti-IL-17A, R&D Systems (http://www.rndsystems.com/), AF-317-NA (1:50); goat anti-IL-17F, R&D Systems, AF-1335 (1:100); goat anti-IL-21, Santa Cruz Biotechnology (http://www.scbt.com), sc-17649 (1:150); goat anti-IL-22, R&D Systems, AF-782 (1:50); goat anti-IL-23, Santa Cruz Biotechnology, sc-21079 (1:100); mouse anti-CD3, CD4, CD8, CD31, CD68 and neutrophil elastase were used as described previously [22]. Control slides were included in each staining run using human tonsil as a positive control. For the negative control slides, normal goat or mouse non-specific immunoglobulins (Santa Cruz Biotechnology) were used at the same protein concentration as the primary antibody. Antibody specificity was confirmed using Western blot analysis of tonsil proteins and the use of purified antigen to block the antibody binding. Double staining for identification of endothelial cells (CD31+ cells) co-expressing IL-17A and IL-22 was performed as described previously [21].
Scoring system for immunohistochemistry
Light-microscopic analysis was performed at a magnification of 630. Immunostained cells in the bronchial submucosa were quantified as described previously and the final result was expressed as the number of positive cells/mm2[21]. The immunostaining was also scored [range: 0 (absence of immunostaining) to 3 (extensive intense immunostaining)] in the bronchial epithelium [22]. A mean ± standard deviation (s.d.) of 0·620 ± 0·220 mm of epithelium was analysed in COPD patients and control subjects.
Quantification of RORC2, IL-17A, IL-17F, IL-21, IL-22 and IL-23 mRNA levels in bronchial biopsies using QPCR
Total RNA was extracted (Micro RNeasy Kit; Qiagen, Milan, Italy) from 30 mm-thick cryostat sections of bronchial biopsies and 1 µg used for cDNA synthesis. Primer pairs for human ROR2 (NR1F3, Cat. no. QT01007685), IL-17A (Cat. no. QT00009233), IL-17F (Cat. no. QT00032151), IL-21 (Cat. no. QT00038612), IL-22 (Cat. no. QT00034853) and IL-23A (Cat. no. QT00204078) were purchased from Qiagen. QPCR was performed using Sybr-Green (QuantiFast Sybr Green PCR kit Cat. no. 204054; Qiagen) following the manufacturer's protocol. Relative levels of mRNAs were expressed as the ratio of the Ct value for the gene of interest Ct/housekeeping gene Ct.
Data analysis
Group data were expressed as mean ± standard error (s.e.) for functional data or median (range) for morphological data. Differences between groups were analysed using analysis of variance (anova) for functional data. anova was followed by the unpaired t-test for comparison between groups. Kruskal–Wallis analysis of morphological data was followed, when significant, by the Mann–Whitney U-test for between-group comparison. Correlation coefficients were calculated using the Spearman's rank method. Probability values of P < 0·05 were considered significant. Data analysis was performed by using the StatView SE Graphics program (Abacus Concepts Inc., Berkeley, CA, USA).
Results
Clinical findings
The clinical characteristics of the subjects studied are reported in Table 1. The four groups of subjects examined were similar with regard to age. Smoking history was similar in mild/moderate, severe COPD patients and healthy control smokers with normal lung function. As expected from the selection criteria, the values of FEV1 (% predicted) and FEV1/FVC (%) were significantly different in the groups with mild/moderate and severe COPD compared to both healthy smokers and control non-smokers. Severe COPD patients also differed significantly from mild/moderate disease [for overall groups, anova test: P < 0·0001 for FEV1% and FEV1/FVC (%) values].
Immunohistochemistry
The results of the immunohistochemical study are summarized in Table 2.
Table 2.
Quantification of interleukin (IL)-17A, IL-17F, IL-21, IL-22 and IL-23 cytokines and inflammatory cells in chronic obstructive pulmonary disease (COPD) patients stratified by severity and in control subjects.
| Control non-smokers | Control healthy smokers | Mild/moderate COPD | Severe COPD | Kruskal–Wallis P-value | |
|---|---|---|---|---|---|
| Epithelium (score 0–3): | |||||
| IL-17 | 0·37 (0·0–0·5) | 1·0 (0·0–1·5) | 0·37 (0·0–1·5) | 0·25 (0·25–1·0) | 0·53 |
| IL-17F | 0·62 (0·0–1·5) | 0·62 (0·0–1·25) | 0·25 (0·0–1·5) | 0·62 (0·25–1·75) | 0·41 |
| IL-21 | 0·75 (0·0–1·0) | 1·25 (0·25–1·5) | 1·0 (0·25–2·5) | 1·5 (0·25–2·25) | 0·39 |
| IL-22 | 0·5 (0·0–1·25) | 1·0 (0·25–2·5) | 1·5 (0·5–2·0)† | 1·5 (0·5–2·5)† | <0·03 |
| IL-23 | 0·25 (0·0–1·0) | 0·0 (0·0–0·25) | 0·12 (0·0–1·0) | 0·38 (0·0–1·5)‡ | <0·025 |
| Submucosa (cells/mm2) | |||||
| IL-17 | 30·0 (16·0–55·0) | 83·0 (18·0–150·0)† | 61·0 (21·0–245·0)† | 74·0 (11·0–297·0)† | <0·04 |
| IL-17F | 53·0 (7·0–105) | 58·0 (8·0–142·0) | 54·0 (23·0–125·0) | 24·0 (8·0–297·0) | 0·23 |
| IL-21 | 90·0 (16·0–151·0) | 103·0 (32·0–219·0) | 87·0 (21·0–280·0) | 81·0 (48·0–274·0) | 0·53 |
| IL-22 | 84·0 (21·0–177·0) | 207·0 (43·0–328·0) | 226·0 (75·0–361·0)† | 197·0 (97·0–502·0)† | <0·05 |
| IL-23 | 64·0 (40·0–97·0) | 89·0 (38·0–253·0) | 79·5 (32·0–155·0) | 103·0 (6·0–168·0)† | 0·10 |
| CD68+ | 259·0 (110–487) | 645·0 (457–860)† | 722·0 (399–1500)† | 690·0 (272–1306)† | <0·001 |
| CD3+ | 327·0 (47–523) | 490·0 (242–1139)† | 580·0 (323–1105)† | 436·0 (115–753)§ | <0·05 |
| CD4+ | 172·0 (88–378) | 290·0 (135–444) | 347·0 (94–731) | 289·0 (66–581) | 0·10 |
| CD8+ | 154·0 (15–286) | 394·0 (135–562)† | 230·0 (102–523)† | 250·0 (111–665)† | <0·015 |
| Neutrophil Elastase+ | 101·0 (59–179) | 180·0 (59–355) | 249·0 (78·0–610)† | 290·0 (47–599)†‡ | <0·01 |
Results expressed as median (range). Mann–Whitney U-test:
significantly different from control non-smokers;
significantly different from control smokers;
significantly different from mild/moderate COPD.
Number of inflammatory cells in bronchial submucosa
The number of neutrophils in the bronchial submucosa was significantly higher in severe COPD [290 (47–599)] compared with control smokers [180 (59–355), P = 0·008] and non-smokers [101 (59–179) P = 0·009]. Mild/moderate COPD [249 (78–610), P = 0·04] also differed significantly from control non-smokers. Compared with control non-smokers [327 (47–523)], the number of total (CD3+) lymphocytes was increased significantly in mild/moderate COPD [580 (323–1105), P = 0·004] and control smokers [490 (242–1139), P = 0·008]. Mild/moderate COPD also tended to differ from severe COPD patients [436 (115–753), P = 0·050]. Compared with control non-smokers [259 (110–487)]), the number of CD68+ macrophages was significantly higher in severe COPD [690 (272–1306), P = 0·0018], mild/moderate COPD [722 (399–1500), P = 0·0005] and control smokers [645·0 (457–860), P = 0·004] (Table 2).
IL-17 and other Th17-related cytokines
Immunohistochemistry in the bronchial epithelium
The number of IL-23+ immunoreactive cells was increased significantly in the epithelium of severe COPD patients compared to control healthy smokers, but did not differ in comparison with control non-smokers. The number of IL-22+ immunoreactive cells was increased significantly in the bronchial epithelium of severe and mild/moderate COPD compared to control non-smokers (P < 0·05). No significant differences in bronchial epithelial expression of IL-17A, IL-17F and IL-21 were observed between groups (Table 2).
Immunohistochemistry in the bronchial submucosa
IL-17A (Fig. 1) and IL-17F staining were observed in endothelial cells and in inflammatory cells and fibroblasts. The number of IL-17A+ cells was significantly higher in severe COPD [74 (11–297); P = 0·013], mild/moderate COPD [61 (21–245); P = 0·049] and in control smokers [83 (18–150); P = 0·041] compared to control non-smokers [30 (16–55)]). No significant differences were observed for IL-17F and IL-21 immunostaining in the four groups of subjects examined.
Fig. 1.
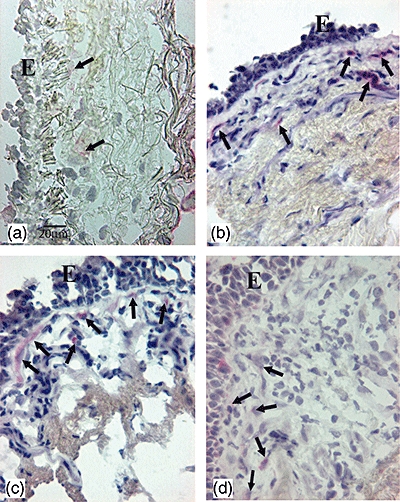
Photomicrographs showing the bronchial mucosa from (a) control non-smoker, (b) control healthy smoker with normal lung function, (c) mild/moderate stable chronic obstructive pulmonary disease (COPD) and (d) severe stable COPD immunostained for identification of interleukin (IL)-17A+ cells (arrows) in the bronchial submucosa. Results are representative of those from eight non-smokers, 11 healthy smokers, 14 mild/moderate COPD and 14 with severe COPD. E, epithelium; bar, 20 micron.
As with IL-17, the immunostaining for IL-22 (Fig. 2) and IL-23 (Fig. 3) was localized in endothelial cells and in inflammatory cells and fibroblasts. The number of IL-22+ cells was significantly higher in severe COPD [197·0 (97·0–502·0)] and mild/moderate COPD [226·0 (75·0–361·0), P < 0·05] compared to control non-smokers [84·0 (21·0–177·0)] but did not differ in comparison with control smokers. The number of IL-23+ cells was significantly higher in severe COPD [103 (6–168); P = 0·039] compared to control non-smokers [64 (40–97)], but did not differ in comparison with control smokers or mild/moderate COPD. IL-23 expression in mild/moderate COPD did not differ from the other groups, nor did the two control groups differ significantly from each other (Table 2). Double staining for identification of endothelial (CD31+) cells co-expressing IL-17A (Fig. 4a) and IL-22 (Fig. 4b) was performed in three representative healthy smokers with normal lung function and in three patients with COPD. We found no difference in the percentage (mean ± s.e.) of CD31+ IL-17+ double-stained cells between control smokers (40 ± 5%) and COPD patients (37 ± 16%). Similarly, there was no difference between the percentage of CD31+ IL-22+ double-stained cells between control smokers (82 ± 1%) and COPD patients (70 ± 13%).
Fig. 2.
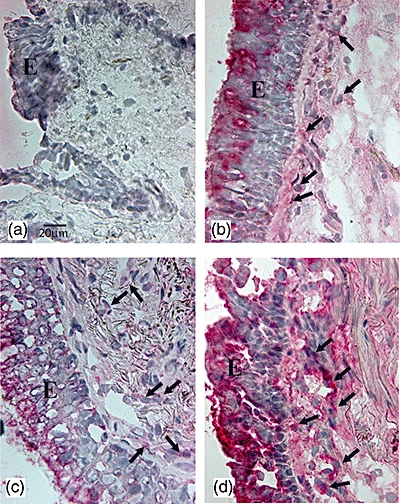
Photomicrographs showing the bronchial mucosa from (a) control non-smoker, (b) control healthy smoker with normal lung function, (c) mild/moderate stable chronic obstructive pulmonary disease (COPD) and (d) severe stable COPD immunostained for identification of interleukin (IL)-22+ cells (arrows) in the bronchial submucosa. Results are representative of those from eight non-smokers, 11 healthy smokers, 14 mild/moderate COPD and 14 with severe COPD. E, epithelium; bar, 20 micron.
Fig. 3.

Photomicrographs showing the bronchial mucosa from (a) control non-smoker, (b) control healthy smoker with normal lung function, (c) mild/moderate stable chronic obstructive pulmonary disease (COPD) and (d) severe stable COPD immunostained for identification of interleukin (IL)-23+ cells (arrows) in the bronchial epithelium. Results are representative of those from eight non-smokers, 11 healthy smokers, 14 mild/moderate COPD and 14 with severe COPD. E, epithelium; bar, 20 micron.
Fig. 4.
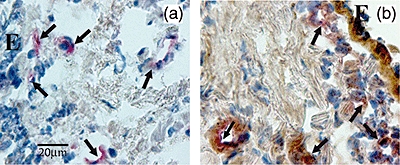
Photomicrographs showing the bronchial mucosa from a patient with severe chronic obstructive pulmonary disease (COPD) double immunostained for identification of endothelial (CD31+) cells (coloured red) co-expressing interleukin (IL)-17A (a) (coloured brown) or IL-22 (b) (coloured brown) in the bronchial submucosa. IL-17A and IL-22 were revealed by diaminobenzidine substrate, whereas endothelial (CD31+) cells were revealed using fast red substrate. Arrows indicate double-stained bronchial vessels. E, epithelium; bar, 20 micron.
RORC2, IL-17A, IL-17F, IL-21, IL-22 and IL-23A mRNA expression
Both IL-22 and IL-23 mRNA expression, but not that for mRNA of other Th17 markers, were increased significantly in COPD patients compared with control non-smokers. However, the absolute and relative mRNA levels of RORC2, IL-17A, IL-17F, IL-21, IL-22 and IL-23A were not significantly different between smokers with or without COPD or between COPD of different severity (Table 3).
Table 3.
Expression of cytokines as determined by real time quantitative polymerase chain reaction.
| Gene | Non-smokers with normal lung function (n = 9) | Smokers with normal lung function (n = 7) | Mild to moderate COPD (n = 9) | Severe COPD (n = 15) |
|---|---|---|---|---|
| RORC2 | 0·80 ± 0·03 | 0·74 ± 0·04 | 0·83 ± 0·05 | 0·77 ± 0·04 |
| IL-17A | 0·76 ± 0·03 | 0·79 ± 0·02 | 0·74 ± 0·03 | 0·78 ± 0·04 |
| IL-17F | 0·95 ± 0·03 | 0·97 ± 0·01 | 0·98 ± 0·03 | 1·00 ± 0·01 |
| IL-21 | 0·53 ± 0·03 | 0·51 ± 0·03 | 0·57 ± 0·07 | 0·55 ± 0·04 |
| IL-22 | 0·50 ± 0·03* | 0·55 ± 0·06 | 0·63 ± 0·04 | 0·69 ± 0·09 |
| IL-23A | 0·78 ± 0·03** | 0·91 ± 0·01 | 0·93 ± 0·01 | 0·91 ± 0·02 |
P < 0·01 versus mild to moderate and severe chronic obstructive pulmonary disease (COPD) groups;
P < 0·001 versus all other groups. No other comparisons between groups were significant. Relative levels of mRNAs expressed as the ratio Ct gene of interest/Ct housekeeping Ct [guanine nucleotide binding protein (G protein) – GNB2L]. IL, interleukin.
Correlations between inflammatory cell counts, Th17 cells and their related cytokines and clinical parameters
In all smokers there is a positive correlation between the number of IL-22+ immunoreactive cells and the number of CD4+ (r = 0·46, P = 0·009) and CD8+ (r = 0·39, P = 0·025) cells in the bronchial submucosa. Also, the number of IL-23+ immunoreactive cells correlates significantly with the number of IL-17A+ (r = 0·40, P = 0·016) cells in the bronchial submucosa of all smokers. When correlations were restricted to patients with COPD alone the number of IL-22+ cells again correlated significantly with the number of CD4+ (r = 0·49, P = 0·021) and CD8+ (r = 0·43, P = 0·045) cells and the number of IL-23+ cells again correlated significantly with the number of IL-17A+ (r = 0·40, P = 0·038) cells (Fig. 5). No other significant correlations were found between inflammatory cells, Th17 cells and their related cytokines and any clinical parameters.
Fig. 5.
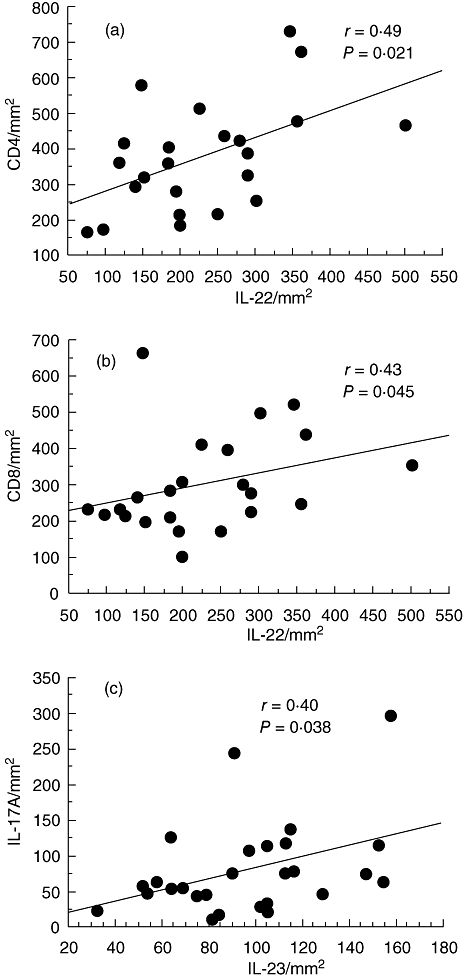
Regression analysis between numbers of interleukin (IL)-22+ and CD4+ (a), IL-22+ and CD8+ (b) and IL-23+ and IL-17A+ (c) cells in the submucosa of all patients with chronic obstructive pulmonary disease (COPD). Correlation coefficients were calculated by using the Spearman's rank method.
Discussion
In our present study we report a significant increase of the number of IL-17A+ immunoreactive cells in the bronchial submucosa of mild/moderate and severe COPD patients compared to control non-smokers. In addition, there is a significant increase in the number of IL-22+ and IL-23+ cells in the bronchial epithelium and submucosa of COPD patients compared with non-smokers. The increased number of IL-22+ immunoreactive cells was correlated significantly with the number of CD4+ and CD8+ cells in all smokers and patients with COPD. These results suggest that increased expression of IL-17A, IL-22 and IL-23 in the bronchial mucosa of stable COPD patients may be involved in the pathogenesis of COPD. However, the increase in ILl-17A+ immunoreactive cells was also observed in control smokers with normal lung function, suggesting that smoking per se may be responsible for this increase of IL-17A+ cells in the bronchial submucosa. IL-17A is also increased in the lungs in asthmatic patients [22], suggesting further that IL-17A expression in the airway may be a marker of airway inflammation.
Despite IL-17A and IL-17F being implicated in the expression of neutrophil chemoattractants, the administration of anti-IL-17F antibodies has no effect on lung neutrophilia in animal models [20]. Our data are in keeping with these animal models, because we found no significant differences in the expression of IL-17F in any the groups of subjects examined. The lack of a direct correlation between numbers of IL-17A+ cells and numbers of neutrophils in the submucosa of our patients may suggests that the mechanism of IL-17A induction of neutrophilia could be mediated by T cell (CD4+ and CD8+) and structural (endothelial) cell activation. This, in turn, could induce the production of specific mediators, such as chemokines and cytokines, capable of inducing neutrophil recruitment and activation in the bronchial mucosa. Our present observations of significant correlations between IL-22+ and CD4+ and CD8+ cells and between IL-23+ and IL-17A+ cells populating the bronchial mucosa are in line with this hypothesis. A cascade of events involving increased IL-23, IL-22 and IL-17A protein expression by T cell subsets and endothelial cells in the bronchi to enhance neutrophilic chemokine expression can develop, accounting for the neutrophilia seen in COPD.
We have also demonstrated increased expression of IL-22 in the bronchial epithelium and submucosa of COPD patients compared with non-smoking subjects. IL-22 is expressed predominantly in Th1 cells and Th17 cells, particularly in the presence of IL-23 [23], which was also reported to be increased in the epithelium and submucosa of COPD patients. In animal models IL-22 is a crucial effector molecule in host defence against Gram-negative bacterial pneumonia [24], and we reported IL-22 as the most abundant cytokine, between two and four times more highly expressed in comparison with IL-17, IL-21 and IL-23 in the bronchial mucosa of our patients with COPD. These differences are absent, however, when comparison is made for the same cytokines in the group of control non-smoking subjects. Human bronchial epithelial cells also express IL-22R and IL-17, and IL-22 increases the expression of anti-microbial proteins such as lipocalin-2 in airway epithelial cells [25] even though, in animal models, neither administration of IL-22 nor of IL-22 blocking antibodies has any effect on lung neutrophilia [20]. In contrast, administration of an IL-17A-specific antibody completely prevented Th17 cell-induced neutrophilia and CXCL5 expression in mouse [20]. Our data suggest that in COPD IL-22 may act upon bronchial epithelial cells to induce anti-microbial proteins involved in the host defence against Gram-negative bacteria.
IL-23 induces the proliferation of memory T cells and the secretion of IFN-γ, and in an animal model cigarette smoking increases the lung expression of IL-23 [26]. However, its role in the pathogenesis of COPD lung remains, in part, unknown. In this study we have reported increased epithelial and submucosal expression of IL-23 in comparison with control groups. This finding is in line with our previous demonstration of an increased expression of IFN-γ and phosphorylated signal transducer and activator of transcription-4 (STAT-4), its downstream transcription factor, in bronchial biopsies from patients with stable COPD [3]. IL-17A production by Th17 cells is induced by IL-23 [12,27] with potential positive-feedback loops. Blocking antibodies to IL-17 and IL-23 are effective against neutrophilic inflammation in several diseases and in animal models [20], and are now in clinical development [28].
IL-21 belongs to a family of cytokines that includes IL-2, IL-4, IL-7, IL-9 and IL-15, all of which bind to private (or shared) receptors as well as the common cytokine receptor γ-chain as a component [29]. In the mouse, IL-21 inhibits Tregs and stimulates Th17 cell differentiation [30]. IL-21 can also increase in vitro production of IL-17 by activated human T cells [8], showing a potential role for IL-21 in the pathogenesis of COPD. However, our data showing no change in IL-21 expression in COPD fits with animal models where Th17 differentiation is unaffected by deletion of IL-21 or IL-21 receptor [31]. Furthermore, although RORC2 expression is induced by IL-21 during the in vitro differentiation of human Th17 cells from naive CD4+ T cells [30], failure to observe changes in RORC2 mRNA expression in COPD reflects the lack of IL-21 induction.
Both IL-22 and IL-23 mRNA expression, but not that of other Th17 markers, were increased significantly in smokers with COPD compared with control non-smokers, in agreement with our immunohistochemical findings. However, the lack of significant changes of RORC2, IL-17A and IL-17F mRNA expression in our COPD patients of different severity and control subjects, compared with the changes seen in protein expression, indicates possibly that protein expression for these mRNAs is regulated mainly at the post-transcriptional level, or the fact that mRNA changes in the Th17 populations are overwhelmed by the total amount of mRNA obtained from the bronchial biopsies.
Interestingly, we observed that many bronchial endothelial cells can express IL-17A and IL-22, suggesting a potential immunomodulatory role for these ‘structural’ cells. This finding is in agreement with previous observations showing that IL-17A and IL-22 can be expressed by endothelial cells in different tissues and that this expression increases further in pro-atherogenic conditions [32–34]. The present study is the first report showing immunostaining for IL-17A and IL-22 in bronchial endothelial cells. Further in vitro studies are needed to characterize more clearly the functional role of IL-17 and IL-22 production from endothelial cells.
In summary, our results showing increased expression of IL-17A, IL-22 and IL-23 in the bronchial mucosa of stable COPD patients indicate that these Th17-related cytokines may be involved in the T cell and endothelial cell activation reported in patients with COPD. This, in turn, can play a role in inducing neutrophilia and tissue remodelling in the bronchi of these patients. However, their true relevance will await clinical studies with selective antagonists or monoclonal antibodies, some of which are already in clinical development.
Acknowledgments
This work was supported by Fondazione Salvatore Maugeri, IRCCS, Ricerca Corrente and Regione Piemonte, Ricerca Sanitaria Finalizzata, FAR 2007 of the University of Ferrara (to GC), Associazione Italiana per la Ricerca dell'Asma (Padova, Italy) and the Wellcome Trust.
Disclosure
None of the authors has any conflict of interest to declare.
References
- 1.Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease. NHLBI/WHO workshop report. NIH Publication no. 2701:1-100. Bethesda, MD: National Heart, Lung and Blood Institute, April. Last update 2008. Available at: http://www.goldcopd.com (accessed 12 March 2009.
- 2.Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–80. doi: 10.1056/NEJM200007273430407. [DOI] [PubMed] [Google Scholar]
- 3.Di Stefano A, Caramori G, Ricciardolo FLM, Capelli A, Adcock IM, Donner CF. Cellular and molecular mechanisms in chronic obstructive pulmonary disease: an overview. Clin Exp Allergy. 2004;34:1156–67. doi: 10.1111/j.1365-2222.2004.02030.x. [DOI] [PubMed] [Google Scholar]
- 4.Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–53. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 5.Di Stefano A, Capelli A, Lusuardi M, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158:1277–85. doi: 10.1164/ajrccm.158.4.9802078. [DOI] [PubMed] [Google Scholar]
- 6.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–76. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 7.Linden A, Hoshino H, Laan M. Airway neutrophils and interleukin-17. Eur Respir J. 2000;15:973–7. doi: 10.1034/j.1399-3003.2000.15e28.x. [DOI] [PubMed] [Google Scholar]
- 8.Hoeve MA, Savage ND, de Boer T, et al. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol. 2006;36:661–70. doi: 10.1002/eji.200535239. [DOI] [PubMed] [Google Scholar]
- 9.Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T-cells differentiate into IL-17 producing cells. Blood. 2008;112:2340–52. doi: 10.1182/blood-2008-01-133967. [DOI] [PubMed] [Google Scholar]
- 10.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 13.Laurence A, O'Shea JJ. T(H)-17 differentiation: of mice and men. Nat Immunol. 2007;8:903–5. doi: 10.1038/ni0907-903. [DOI] [PubMed] [Google Scholar]
- 14.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 15.Jones CE, Chan K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-α, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am J Respir Cell Mol Biol. 2002;26:748–53. doi: 10.1165/ajrcmb.26.6.4757. [DOI] [PubMed] [Google Scholar]
- 16.Rahman MS, Yang J, Shan LY, et al. IL-17R activation of human airway smooth muscle cells induces CXCL-8 production via a transcriptional-dependent mechanism. Clin Immunol. 2005;115:268–76. doi: 10.1016/j.clim.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 17.Vanaudenaerde BM, Wuyts WA, Dupont LJ, Van Raemdonck DE, Demedts MM, Verleden GM. Interleukin-17 stimulates release of interleukin-8 by human airway smooth muscle cells in vitro: a potential role for interleukin-17 and airway smooth muscle cells in bronchiolitis obliterans syndrome. J Heart Lung Transplant. 2003;22:1280–3. doi: 10.1016/s1053-2498(02)01234-2. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278:17036–43. doi: 10.1074/jbc.M210429200. [DOI] [PubMed] [Google Scholar]
- 19.Hizawa N, Kawaguchi M, Huang SK, Nishimura M. Role of interleukin-17F in chronic inflammatory and allergic lung disease. Clin Exp Allergy. 2006;36:1109–14. doi: 10.1111/j.1365-2222.2006.02550.x. [DOI] [PubMed] [Google Scholar]
- 20.Liang SC, Long AJ, Bennett F, et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179:7791–9. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- 21.Ricciardolo FLM, Caramori G, Ito K, et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2005;116:1028–35. doi: 10.1016/j.jaci.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 22.Molet S, Hamid Q, Davoine F, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–8. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 23.Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)-22 and IL-17 are coexpressed by TH17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–9. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aujla SJ, Chan YR, Zheng M, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–81. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aujla SJ, Dubin PJ, Kolls JK. Interleukin-17 in pulmonary host defense. Exp Lung Res. 2007;33:507–18. doi: 10.1080/01902140701756604. [DOI] [PubMed] [Google Scholar]
- 26.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–22. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 28.Bowman EP, Chackerian AA, Cua DJ. Rationale and safety of anti-interleukin-23 and anti-interleukin-17A therapy. Curr Opin Infect Dis. 2006;19:245–52. doi: 10.1097/01.qco.0000224818.42729.67. [DOI] [PubMed] [Google Scholar]
- 29.Ettinger R, Kuchen S, Lipsky PE. The role of IL-21 in regulating B-cell function in health and disease. Immunol Rev. 2008;223:60–86. doi: 10.1111/j.1600-065X.2008.00631.x. [DOI] [PubMed] [Google Scholar]
- 30.Deenick EK, Tangye SG. Autoimmunity: IL-21: a new player in Th17-cell differentiation. Immunol Cell Biol. 2007;85:503–5. doi: 10.1038/sj.icb.7100114. [DOI] [PubMed] [Google Scholar]
- 31.Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are not required for development of Th17 cells and autoimmunity in vivo. Eur J Immunol. 2008;38:1833–8. doi: 10.1002/eji.200838511. [DOI] [PubMed] [Google Scholar]
- 32.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in rat coronary arteries. FASEB J. 2003;17:1183–5. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- 33.Brooks AR, Lelkes PI, Rubanyi GM. Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol Genomics. 2002;9:27–41. doi: 10.1152/physiolgenomics.00075.2001. [DOI] [PubMed] [Google Scholar]
- 34.Chang H, Hanawa H, Liu H, et al. Hydrodynamic-based delivery of an interleukin-22-Ig fusion gene ameliorates experimental autoimmune myocarditis in rats. J Immunol. 2006;177:3635–43. doi: 10.4049/jimmunol.177.6.3635. [DOI] [PubMed] [Google Scholar]