

Lipid-laden macrophages are the predominant cell type in the formative stages of atherosclerosis in most animal models and humans.1-3 Lipid deposition appears as large numbers of intracellular droplets that have a foam-like appearance when paraffin-embedded tissues sections are viewed at high magnification. Consequently, the descriptive name of foam cells is applied to these lipid-laden macrophages. Originating from recruited monocytes, it has been hypothesized that foam cells remove lipoproteins that have been retained and modified in the subendothelial space.4 For this function to be beneficial, lipid-laden macrophages would subsequently egress from the area of the forming lesion. However, the system frequently goes awry. Macrophages recruited to the arterial wall become grossly engorged with lipid, presumably due to an imbalance in lipid metabolism. In this greatly hypertrophied state, macrophages are unable to transit through the endothelium and be transferred back to the blood compartment. Therefore, instead of exiting the artery, these cells are retained and accumulate. In addition to these cells forming the mass of the evolving lesions, there is also the potential for secretion of many bioactive molecules that may perpetuate and modify the atherogenic process.
There have been many approaches to modify the development of foam cells by manipulating intracellular transport, intracellular storage, or efflux of lipids. The transport of extracellular lipid to form intracellular droplets is presumed to occur via endocytosis through lipoprotein receptors that are not downregulated by increased cholesterol content. There are many classes of scavenger receptors that transport modified lipoproteins into macrophages, of which the most intensely studied has been class A scavenger receptor (SR-A) and CD36.5 However, genetic manipulation of SR-A and CD36 has generated an inconsistent literature for effects on atherosclerosis.6-8 Once inside the cell, lipoprotein-derived cholesterol ester is cleaved in lysosomes by an acidic cholesterol ester hydrolase. Unesterified cholesterol is transported to the cytosol for re-esterification by acyl-coenzyme A: cholesterol acyltransferase (ACAT) to generate lipid droplets that are protein coated. Once stored in lipid droplets, neutral cholesterol hydrolase can convert the core content back to unesterified cholesterol. Unesterified cholesterol may also partition to the plasma membrane and transfer to extracellular acceptors. Several pathways have been proposed for cholesterol efflux including the ABC transporters and SR-B1. Therefore, cholesterol homeostasis in macrophages has many levels of regulation involved in their conversion to foam cells (Figure 1).
Figure 1. Lipoprotein-cholesterol ester trafficking in macrophages.
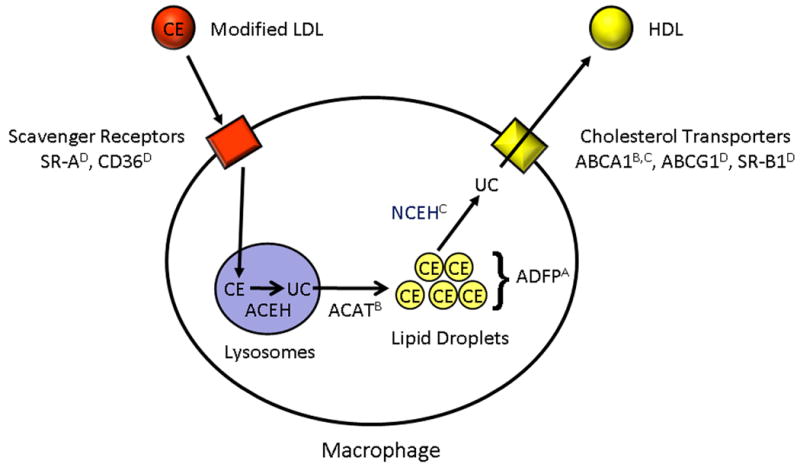
Modified lipoproteins are transported into macrophages through scavenger receptors (including SR-A and CD36). In lysosomes, cholesterol esters (CE) are cleaved to unesterified cholesterol (UC) by acid cholesterol ester hydrolase (ACEH). Unesterified cholesterol is transported out of lysosomes and re-esterified by acyl CoA: cholesterol acyltransferase (ACAT) to form cholesterol esters that are stored in lipid droplets. Cholesterol esters are cleaved to cholesterol by neutral cholesterol ester hydrolase (NCEH). Unesterified cholesterol can be transferred to the extracellular space by several potential transporters (including ABCA1, ABCG1, and SR-B1) to acceptor molecules such as HDL. The lipid droplets are associated with ADFP. Paul et al. demonstrated that absence of ADFP prevented lipid engorgement, both in vitro and in vivo. The effects of macrophage-specific regulation of receptors, enzymes, and transporters on lipoprotein-cholesterol ester trafficking are denoted as: A. Absence decreases atherosclerosis. B. Absence increases atherosclerosis. C. Overexpression decreases atherosclerosis. D. Absence has inconsistent effects on atherosclerosis.
In this current edition of Circulation Research, Paul et al.9 have studied the role of adipose differentiation-related protein (also known as adipophilin, ADRP, or ADFP) on macrophage foam cell formation and atherosclerosis. ADFP, as it is referred to in this article, is a member of the PAT-domain family of proteins that are named from the founding 3 members of this group: perilipin, adipophilin, and tail-interacting protein of 47 kDa. ADFP is probably expressed in the majority of cell types, although unlike some other PAT family members, it is relatively sparsely expressed in mature adipocytes.10 Mice that are deficient in ADFP have a modest phenotype that includes reductions in liver triglyceride content and resistance to diet-induced fatty liver. However, they have no difference in body weight, plasma triglyceride and cholesterol concentrations, fat mass, or adipocyte differentiation.11
Although mice deficient in ADFP have a mild phenotype, the expression of this protein alters lipid metabolism in cultured macrophages. Overexpression of ADFP increased the storage of triglycerides and cholesterol following incubation with acetylated LDL (AcLDL) in THP-1 cells, while depletion of the protein using small interfering RNAs reduced lipid accumulation. This increase occurred without affecting transport of AcLDL particles or regulating many proteins involved in cholesterol efflux.12 While these cell culture studies with manipulated ADFP expression portend a protection against atherosclerosis, the field has been led astray in the past by such expectations. Perhaps one of the best examples of this involves studies on ACAT. The development of many pharmacological inhibitors of this enzyme demonstrated that they decreased lipid deposition in macrophages. In contrast, repopulation of irradiated LDL receptor deficient mice with bone marrow derived stem cells from ACAT1 deficient mice led to a dramatic increase in macrophage lipid deposition in atherosclerotic lesions.13 Conversely, macrophage-specific overexpression of neutral cholesterol hydrolase reduced intracellular lipid deposition and atherosclerotic lesion formation.14 Since the net effect of ACAT inhibition and excess neutral cholesterol hydrolase should both increase unesterified cholesterol, it is quandary that they produce such divergent results. Despite the several decades of research, it is clear that the we have not resolved all the mechanisms of macrophage lipid engorgement and its effects on atherosclerosis.
Therefore, while the cell culture studies on ADFP may be consistent with an effect in reducing atherosclerosis, this needs to be tested experimentally in vivo. In this impressive study performed by Paul et al.9, the authors have demonstrated ADFP is upregulated in atherosclerotic tissue from apoE-/- mice, compared to small changes in other PAT-domain proteins. Its direct role in the disease process was demonstrated in ADFP-/- mice that were compared to littermate controls of ADFP +/+ mice. Analysis of lesion size was performed by sectioning throughout the aortic sinus. These studies demonstrated that ADFP deficiency causes large decreases in lesion size in reasonable sized groups of both male and female mice. The use of the bone marrow transplantation technique in female mice provided data that was consistent with the reduced atherosclerosis being due to ablation of ADFP activity in macrophages. The interpretation of ADFP deficiency was not confounded by compensatory changes in mRNA or protein abundance of other common lipid droplet proteins. Although lipid engorgement has the potential to alter cytokine secretion, no changes were discernable in cultured cells or in vivo. The only discernable changes noted by ADFP deficiency were differences in macrophage lipid metabolism. No changes were noted on transport of modified lipoproteins (AcLDL and oxidized LDL) into macrophages. Also, ADFP deficiency had no effect on the mRNA abundance in cultured macrophages of a wide range of molecules involved in cholesterol entry and efflux. However, cholesterol efflux to apoAI from AcLDL loaded cultured macrophages was enhanced by ADFP deficiency. This was associated with reduced esterification of cholesterol during AcLDL incubation that was unrelated to changes in activities of ACAT or neutral cholesterol hydrolase. Interestingly, the absence of ADFP did not influence the number of macrophages in lesions or the size distribution of lipid droplets in these cells. However, ADFP deficiency reduced the number of lipid droplets. This comprehensive study clearly demonstrates ADFP deficiency in macrophages reduces atherosclerosis through a mechanism that is solely based on inhibition of lipid deposition rather than secondary consequences on inflammatory processes.
Although macrophages sequester cholesterol esters in membrane limited droplets associated with PAT-domain proteins, macrophages store intracellular lipids in diverse structures within evolving atherosclerotic lesions. In addition to the lipid droplets described in this study, there are also complex lipid structures that are encased by acid phosphatase positive orgenelles.15 These structures are likely to arise from the phagocytic engulfment of modified lipoproteins by macrophages coupled with an inability of the cholesterol esters to be hydrolyzed and exit this domain. The presence of excessive lipid storage in acid phosphatase organelles versus lipid droplet changes as a function of time and complexity of atherosclerotic lesions. Currently, it is unclear what the consequences are of excessive storage in these two venues. In addition, the relative ability to remove intracellular lipid located in these two regions has not been defined. Therefore, the availability of mice that can change the distribution of these lipid stores will provide important insight into the progression of lesions into advanced stages. Furthermore, it would be of interest to determine whether the efficacy of ADFP inhibition on reducing the initiation of atherosclerosis would be matched by inhibiting this enzyme in established atherosclerotic lesions.
While there are many factors that modulate atherogenesis, regulation of lipid metabolism is still the mainstay of targets for atherosclerosis therapies.16 This publication by Paul et al.9 invokes ADFP as a new target to reduce macrophage lipid deposition as an approach to decreasing atherosclerosis.
Acknowledgments
Sources of funding: The authors are supported by grants from NIH (HL80100)
Footnotes
Disclosures: None
References
- 1.Rosenfeld ME, Tsukada T, Gown AM, Ross R. Fatty streak initiation in Watanabe heritable hyperlipidemic and comparably hypercholesterolemic fat-fed rabbits. Arteriosclerosis. 1987;7:9–23. doi: 10.1161/01.atv.7.1.9. [DOI] [PubMed] [Google Scholar]
- 2.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 3.Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis - A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. 1994;14:840–856. doi: 10.1161/01.atv.14.5.840. [DOI] [PubMed] [Google Scholar]
- 4.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du BH, Fu CZ, Kent KC, Bush H, Schulick AH, Kreiger K, Collins T, McCaffrey TA. Elevated Egr-1 in human atherosclerotic cells transcriptionally represses the transforming growth factor-beta type II receptor. J Biol Chem. 2000;275:39039–39047. doi: 10.1074/jbc.M005159200. [DOI] [PubMed] [Google Scholar]
- 6.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daugherty A, Rateri DL, Whitman SC. Class A scavenger receptors: Recent advances in elucidation of structure-function relationships and their role in atherosclerosis. Curr Opin Cardiovasc Pulm Ren Invest Drugs. 2000;2:223–232. [Google Scholar]
- 9.Paul A, Chang BHJ, Li L, Yechoor VK, Chan L. Deficiency of adipose differentiation-related protein impairs foam cell formation and protects against atherosclerosis. Circ Res. 2008;102:XXX–XXX. doi: 10.1161/CIRCRESAHA.107.168070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ducharme NA, Bickel PE. Lipid droplets in lipogenesis and lipolysis. Endocrinology. 2008;149:942–949. doi: 10.1210/en.2007-1713. [DOI] [PubMed] [Google Scholar]
- 11.Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, Chan L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol. 2006;26:1063–1076. doi: 10.1128/MCB.26.3.1063-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larigauderie G, Furman C, Jaye M, Lasselin C, Copin C, Fruchart JC, Castro G, Rouis M. Adipophilin enhances lipid accumulation and prevents lipid efflux from THP-1 macrophages: potential role in atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:504–510. doi: 10.1161/01.ATV.0000115638.27381.97. [DOI] [PubMed] [Google Scholar]
- 13.Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest. 2001;107:163–171. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao B, Song J, Chow WN, St Clair RW, Rudel LL, Ghosh S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J Clin Invest. 2007;117:2983–2992. doi: 10.1172/JCI30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jerome WG, Yancey PG. The role of microscopy in understanding atherosclerotic lysosomal lipid metabolism. Microscopy Microanalysis. 2003;9:54–67. doi: 10.1017/S1431927603030010. [DOI] [PubMed] [Google Scholar]
- 16.Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–913. doi: 10.1038/nature06796. [DOI] [PubMed] [Google Scholar]