

Abstract
An Ig superfamily cell-adhesion molecule, L1, forms an adhesion complex at the cell membrane containing both signaling molecules and cytoskeletal proteins. This complex mediates the transduction of extracellular signals and generates actin-mediated traction forces, both of which support axon outgrowth. The L1 cytoplasmic region binds ezrin, an adapter protein that interacts with the actin cytoskeleton. In this study, we analyzed L1–ezrin interactions in detail, assessed their role in generating traction forces by L1, and identified potential regulatory mechanisms controlling ezrin–L1 interactions. The FERM domain of ezrin binds to the juxtamembrane region of L1, demonstrated by yeast two-hybrid interaction traps and protein binding analyses in vitro. A lysine-to-leucine substitution in this domain of L1 (K1147L) shows reduced binding to the ezrin FERM domain. Additionally, in ND7 cells, the K1147L mutation inhibits retrograde movement of L1 on the cell surface that has been linked to the generation of the traction forces necessary for axon growth. A membrane-permeable peptide consisting of the juxtamembrane region of L1 that can disrupt endogenous L1–ezrin interactions inhibits neurite extension of cerebellar cells on L1 substrates. Moreover, the L1–ezrin interactions can be modulated by tyrosine phosphorylation of the L1 cytoplasmic region, namely, Y1151, possibly through Src-family kinases. Replacement of this tyrosine together with Y1176 with either aspartate or phenylalanine changes ezrin binding and alters colocalization with ezrin in ND7 cells. Collectively, these data suggest that L1–ezrin interactions mediated by the L1 juxtamembrane region are involved in traction-force generation and can be regulated by the phosphorylation of L1.
Keywords: ERM, Src-family kinase, BRET, cytoskeleton, growth cone, neurite outgrowth
Axon growth depends critically on cell-surface adhesion receptors. When neurons extend axons, growth cones make contact with the substrate through these receptors, forming adhesion points that serve as a mechanical link between the force-generating components of the cytoskeleton and the extracellular environment. Subsequently, the adhesion complex detaches from the substrate, permitting the translocation of the growth cone and the recycling of the receptors to the axon tip. L1 is a cell-adhesion molecule (CAM) of the immunoglobulin superfamily that is involved in neuronal adhesion, axon outgrowth, and guidance. Mutations in L1 lead to severe defects in nervous system development, suggesting it plays a critical role in this process (Fransen et al., 1997). As a receptor, L1 interacts with ligands through its extracellular domain and supports axon outgrowth and cell migration. Ligand-bound L1 forms an adhesion complex on the cell surface that recruits both signaling molecules and cytoskeletal proteins in the cytosol. Thus, L1 mediates transduction of extracellular signals and generation of actin-mediated traction force, both of which contribute to process outgrowth and cell migration (for a review, see Maness and Schachner, 2007).
The L1 cytoplasmic region is a substrate for both tyrosine and serine/threonine kinases (Wong et al., 1996a; Schaefer et al., 2002; Whittard et al., 2006; Nakata and Kamiguchi, 2007). Phosphorylation/dephosphorylation of most of these sites is directly coupled to the regulation of molecular interactions that modulate L1 activity. For example, L1 binding to the clathrin adapter AP2, an interaction that mediates L1 internalization (Kamiguchi et al., 1998), is regulated by Src-family kinase-mediated phosphorylation of the L1 YRSLE motif (Schaefer et al., 2002). The L1 cytoplasmic region also binds to the structural proteins ankyrin B (Davis and Bennett, 1994; Gil et al., 2003; Nishimura et al., 2003) and ERM proteins, including ezrin (Dickson et al., 2002), which are shown to interact with actin cytoskeleton. Ankyrin B binds to the domain terminating in FIGQY, and its interaction is inhibited by the tyrosine phosphorylation of this motif, a process that depends on MAP kinase activity (Whittard et al., 2006). In contrast, ERM proteins bind to two distinct domains (Dickson et al., 2002; Cheng et al., 2005), but it is not known whether ERM-protein binding is modulated by phosphorylation. Nevertheless, these L1 interactions with cytoskeletal components have the potential to support the traction-force generation that plays a role in L1-mediated axon extension.
We have shown previously that L1–ankyrin interactions mediate the static behavior of L1 rather than the actin-mediated retrograde movement of L1 on the cell surface. Perturbation of these interactions increases L1 retrograde movement and enhances outgrowth induced by L1 substrates, suggesting that ankyrin binding inhibits L1 traction-force generation and that molecules other than ankyrin might be involved in linking the L1 receptor to treadmilling actin (Gil et al., 2003). In this study, we analyzed L1–ezrin interactions in this context. Based on structural homology with ICAM-2, a binding motif for ERM proteins was identified in the juxtamembrane region of L1 (Hamada et al., 2003). In this study, ezrin binding to the juxtamembrane region of L1 was confirmed by yeast two-hybrid and protein-binding analyses in vitro. A mutation that reduces ezrin binding to L1 affects L1 diffusion kinetics on the cell surface of ND7 neuroblastoma cells, decreasing L1 retrograde movement that has been linked to traction-force generation through the L1 receptor. Perturbing interactions between the L1 juxtamembrane region and ezrin with a peptide decreases L1-mediated outgrowth from cerebellar granule cells. Furthermore, interactions between L1 and ezrin can be regulated by phosphorylation of this region, possibly through Src-family kinases. Collectively, these data suggest (1) that the L1 juxtamembrane region interacts with ezrin to support traction-force generation and (2) that interactions between L1 CAM and ERM proteins, and by extension, L1 traction-force generation, can be modulated by tyrosine phosphorylation.
Materials and Methods
Reagents and Constructs
Human embryonic kidney (HEK) 293 cells were obtained from American Type Culture Collection. v-Src-transformed and Src-deficient Swiss 3T3 fibroblast cells were kind gifts from Dr. Pamela Schwartzberg (Felsenfeld et al., 1999). Rat L1 cytoplasmic mutants (that also have a Myc-epitope tag in their extracellular region as previously described; Gil et al., 2003) were generated by PCR-based mutagenesis using a QuickChange mutagenesis kit (Stratagene). Modified cDNA was inserted into either pCIneo (Promega) or pIRES2-EGFP (Clontech). cDNA-encoding dominant-negative-ezrin-YFP [ezrin FERM domain, amino acids (aa) 1–406, fused with YFP] in a pEGFP vector backbone was described previously (Dickson et al., 2002). Human ezrin cDNA and Venus cDNA were generous gifts from Dr. Monique Arpin and from Dr. Atsushi Miyawaki, respectively. Ezrin-Venus in pCS2 was made by fusing ezrin cDNA to Venus cDNA by PCR to express ezrin, whose C-terminal is fused with Venus. cDNA encoding chick c-Src in pRK5 was a generous gift from Dr. Jan Sap. The anti-Myc monoclonal antibody 9E10 was from the Developmental Studies Hybridoma Bank (DSHB; maintained at the University of Iowa), anti-GFP was from Molecular Probes, antiphosphotyrosine antibodies were from BD Bioscience (rabbit polyclonal) and from Millipore (mouse monoclonal, 4G10), and rabbit anti-rat L1 polyclonal antibody was described previously (Lustig et al., 2001). Peptides (12–13 aa in length) derived from L1 cytoplasmic region fused with a 16-aa penetratin domain of antennapedia to confer membrane permeability (Derossi et al., 1998) were synthesized at FastMoc Chemistry (Tufts University) and purified by HPLC (Gil et al., 2003). The Ant-1 peptide is SKGGKYSVKDKED, which corresponds to the juxtamembrane region of L1. The RSLE peptide is ETFGEYRSLESD. The control peptide was DETFGEYSDNEE (which is derived from the L1 cytoplasmic region without RSLE). All peptides were biotinylated at the amino terminus. Reagents used in BRET experiments have been described previously (Whittard et al., 2006): genistein, PD-98059, PP1, and PP2 were obtained from BioMol Research Laboratories. Epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) were from Sigma-Aldrich, and the codon humanized pRluc and GFP2 vectors were from Perkin Elmer Life Sciences.
Yeast Two-Hybrid Assay
Yeast two-hybrid analysis was carried out as described (Dickson et al., 2002). The cytoplasmic domain of rat L1 (aa K1144-E1257) was fused to the DNA-binding domain of LexA in the plasmid pBTM116-KN-Ade2. This plasmid was used as a template for PCR to obtain other clones. These plasmids were individually cotransformed into the yeast strain L40 with pGADGH plasmids that contained the activation domain of GAL4 fused to either ezrin, ankyrin B, or AP2 (Dickson et al., 2002).
Protein Binding Analysis
Transfection was carried out using liopofectamine (Invitrogen), and cell extracts were prepared 2 days after transfection as described (Sakurai et al., 1997).
One hundred microliters of 1 mg/mL biotinylated peptides was incubated with 20 μL of streptavidin beads (Pierce) at 4°C overnight. The beads were further incubated with 500 μL of 1 mg/mL BSA at 4°C for 2 hr, washed with extraction buffer (Sakurai et al., 1997), and then incubated with 100 μg of extracts prepared from ezrin FERM domain-YFP-transfected 293 cells at 4°C overnight. The beads were further washed with extraction buffer, and bound proteins were subjected to 7% SDS-PAGE followed by Western blotting. Bound ezrin FERM domain was probed with anti-GFP antibody followed by HRP-conjugated anti-rabbit antibody and detected by chemiluminescence (Pierce) and X-ray film (Kodak).
Rat L1 cytoplasmic region was PCR-amplified to add EcoR1 and BamH1 sites and cloned into the EcoR1- and BamH1-cut GST-2K vector (Pharmacia). DH5α cells were transformed by the construct, and 100-mL cultures in midlog phase were induced by the addition of 1 mM IPTG for 3.5 hr. Bacterial cell pellets were extracted by freezing (−80°C) and thawing in 2 mL of extraction buffer (Sakurai et al., 1997), and 500 μL of extracts was incubated with 50 μL of a half slurry of GST–agarose beads (Sigma) preblocked with 1 mg/mL BSA at 4°C overnight. After washing with PBS, the beads were incubated with 100 μg of extracts prepared from ezrin FERM domain-YFP-transfected 293 cells at 4°C overnight. The beads were washed as above, and bound protein was eluted from the beads with 25 μL of 5 mM glutathione. The ezrin FERM domain-YFP in the eluate was detected as above.
Twenty microliters of protein A beads (Pierce) was incubated with 10 μL of 9E10 ascites (2.1 mg/mL, DSHB) and 100 μL of PBS at 4°C for 8 hr and divided into two tubes. Two hundred and fifty micrograms of cell extracts prepared from L1 wild type in pIRES2-EGFP plus ezrin FERM domain-YFP-transfected 293 cells (1:1 molar ratio) or L1 mutants in pIRES2-EGFP plus ezrin FERM domain-YPF-transfected 293 cells (1:1 molar ratio) was added to the tubes and incubated at 4°C overnight. By using IRES2-EGFP constructs for L1 expression, we monitored expression of L1 in the transfected cells by expression of EGFP. Furthermore, EGFP served as control for ezrin-FERM domain-YFP binding to L1. The beads were washed with extraction buffer, and bound protein was subjected to 7% SDS-PAGE followed by Western blotting as above for ezrin FERM domain-YFP or with antiphosphotyrosine antibody for phosphotyrosine. For the experiments using cotransfection of L1 and c-Src, an empty pcDNA3 vector was used when c-Src was not included in order to adjust for an equal amount of DNA in transfection and was analyzed as above.
Single-Particle Analysis
Beads (1 μm latex beads, Polyscience) coated with anti-Myc antibody were prepared as described previously (Gil et al., 2003). ND7 cells were cultured on laminin-coated coverslips (Gil et al., 2003), transfected with L1 constructs in pIRES2-EGFP, and analyzed 2 days after transfection. Video microscopy was performed as described using a Zeiss Axiovert 100TV microscope equipped with a laser trap (Gil et al., 2003). Single-particle displacement was quantified as described (Gil et al., 2003). Quantitative analysis of particle movement was carried out using custom software written for MatLab. Statistical analysis of percentage of trials was performed using the chi-square test as described (Gil et al., 2003).
Neurite Outgrowth Assay
Neurite outgrowth assay was performed as described (Gil et al., 2003) with slight modification. Poly-Lys (5 μg/mL) was spotted on a 35-mm plastic petri dish for 1 hr at room temperature (RT). After washing with PBS, either purified chick NgCAM or laminin (Invitrogen) was spotted as described (Gil et al., 2003). Cerebellar cells were prepared from P4 mouse, and 250 μL of 3 × 105 cells/mL was plated on the dishes. Cultures were incubated at 37°C for 24 hr in 5% CO2. Antennapedia peptides dissolved in HBSS (Invitrogen) were added to the cultures at a final concentration of 20 μg/mL when cells were plated. The control peptide used was DETFGEYSDNEE, described above. Similar results were obtained using scrambled peptide of Ant-1. Cultures were fixed with 4% paraformaldehyde, and neurite outgrowth measurement was performed as described (Gil et al., 2003). P values were determined using t-test analysis.
BRET Analysis
BRET constructs were designed using vectors encoding Renilla luciferase and GFP2 (Sapphire GFP; Biosignal, Perkin Elmer Life Sciences). Coding regions from each individual vector were amplified by PCR with restriction sites added, permitting their ligation into a single, concatenated coding region (GFP2:Rluc) between the Not1 and Xho1 sites in a pcDNA3.1 Hygro (+) vector (Invitrogen). This chimeric construct (CHIM) encodes unique BsrG1 and Asc1 sites in the intervening sequence. To create the reporter constructs from the CHIM construct, complementary oligonucleotides derived from the L1-CAM coding region were synthesized (Sigma Genosys) with the addition of a 5′ overhang designed to generate a sticky end complementary to the BsrG1 and Asc1 sites. Prior to ligation into the CHIM construct, oligonucleotide pairs were mixed in equimolar concentrations, heated to 94°C (4 min), and allowed to cool slowly to room temperature, permitting annealing of the complementary regions.
Near-confluent cultures of HEK 293 or Swiss 3T3 cells were harvested with trypsin-EDTA (0.05% trypsin, 0.53 mM EDTA; Invitrogen) and resuspended to a density of 2.5 × 105 cells/mL. Aliquots (200 μL) of cell suspensions were added to white 96-well culture plates (CulturPlate™ PerkinElmer Life Sciences) and incubated for 12 hr at 37°C. The 293 cells were transfected with 0.1 μg of the KGGKY BRET construct/well using Lipofectamine reagents (Invitrogen) according to the manufacturer's instructions. After incubation of plates for 48 hr at 37°C, the cells were washed once with warm D-MEM without phenol red (Invitrogen), supplemented with 25 mM HEPES (Invitrogen). Transfected 293 or 3T3 cells were treated with either EGF or bFGF for 15 min and inhibitors for 1 hr (PD-98059, PP1, and PP2) or 4 hr (genistein). To each well, 10 μL of DeepBlueC™ substrate (final concentration of 5 μM; PerkinElmer Life Sciences) diluted in Dulbecco's PBS containing 0.1% (wt/vol) CaCl2, 0.1% (wt/vol) d-glucose, 0.1% (wt/vol) MgCl2, and 10 μg/mL aprotinin was added. The plates were immediately counted using a Fusion Universal Microplate Analyzer (PerkinElmer Life Sciences). Bioluminescence resulting from Rluc emission was counted at 410 nm using a 370- to 450-nm bandpass filter, and the energy transferred to GFP2 was counted at 515 nm using a 500- to 530-nm bandpass filter. The efficiency of energy transfer between Rluc and GFP2 was determined by dividing acceptor emission intensity (GFP2) by donor emission intensity (Rluc). The result reflects the proximity of GFP2 to Rluc and is referred to as the BRET ratio.
Immunofluorescence
ND7 cells were transfected with Myc-tagged L1 constructs in pCIneo plus ezrin-Venus in pCS2. Cells were live-stained with 9E10, anti-Myc antibody (DSHB), at 37°C, followed by Cy5-conjugated anti-mouse antibody (Jackson Immunological) at RT, and then fixed with 4% paraformaldehyde. Images were collected by a Zeiss meta 540 confocal microscope housed in the Microscopy Shared Research Facility at Mount Sinai School of Medicine (supported by NSF grant DBI-9724504 and NIH grants 5R24CA095823 and 1S10RR09145). One plane of sections was collected for each cell, which was randomly chosen (∼30 cells were imaged for each condition). Colocalization was evaluated by Image J with the colocalization finder plug-in. For colocalization analysis, we first chose cells that expressed L1 at similar levels and then checked the expression of ezrin-Venus. For each cell, we obtained a correlation value (r). The average value for approximately 10 cells for each construct is given.
Results
Juxtamembrane Region of L1 Contains a Binding Site for Ezrin FERM Domain
ERM proteins recognize the cytoplasmic region of transmembrane glycoproteins to form a bridge between the membrane and the cytoskeleton. ERM proteins contain an amino-terminal FERM (4.1-Ezrin-Radixin-Moesin) domain that mediates interactions with both cell-surface glycoproteins and PIP2 on the inner leaflet of the membrane. Structural analysis of ICAM-2 together with the FERM domain of radixin identified the amino acids in the ICAM-2 cytoplasmic domain responsible for this interaction (Hamada et al., 2003). This analysis further identified a general binding motif that can be found in several adhesion molecules including L1 (1147–1153, KxxKYxV; Fig. 1). This sequence motif is in the juxtamembrane region of L1.
Fig. 1.
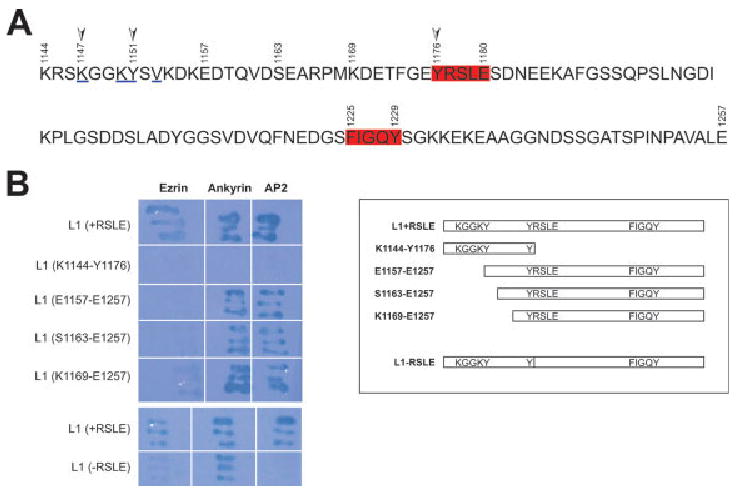
Cytoplasmic region of L1 (1144–1257) contains binding sites for ERM, ankyrin, and AP2. A: Structural analysis of ICAM-2 and FERM domain of radixin identified amino acids responsible for interactions between these two (Hamada et al., 2003). This analysis further identified a binding motif that can be found in several adhesion molecules including L1, which is KxxKYxV, underlined. YRSLE (1176–1180), which binds to AP2, and FIGQY (1225–1229), which binds to ankyrin B, are boxed. Arrows indicate K1147 and Y1151 in the binding motif of FERM domain and Y1176 in YRSLE, all of which were mutated for interaction analysis in this study. Y1176 is phosphorylated by Src-family kinases (Schaefer et al., 2002), and Y1151 can be phosphorylated by Src-family kinases (this study). Note that the L1 cytoplasmic region contains four tyrosines including Y1151 and Y1176. B: In yeast two-hybrid analysis, RSLE in L1 is necessary for ezrin interactions (compare +RSLE and −RSLE). Similarly, a truncation mutant that ends right before RSLE does not bind to ezrin (see K1144-Y1176 and compare with K1144-E1180 in Dickson et al., 2002). Several deletion mutants that do not have the juxtamembrane region but do have RSLE (e.g., E1157-E1257, S1163-E1257, and K1169-E1257) do not bind to ezrin, suggesting that both the juxtamembrane region and YRSLE are important for ezrin binding to L1.
Previously, we have demonstrated that both Y1176 and the RSLE miniexon (1177–1180) are required for the binding of L1 to the ezrin FERM domain (aa 1–406) in yeast two-hybrid studies and in vitro protein binding assays (+RSLE lane vs. −RSLE lane; see Dickson et al., 2002; Fig. 1B). In the present study, by using yeast two-hybrid studies, we confirmed that the K1144-E1180 (YRSLE) region (which contains both the juxtamembrane region and Y1176-E1180) in L1 is necessary for ezrin binding, with data showing that truncation mutants encoding stop codons after D1158, D1162, or Y1176 (YRSLE) did not bind to the FERM domain of ezrin (K1144-Y1176 lane and data not shown; Fig. 1B).
To clarify involvement of the juxtamembrane region of L1 in the interaction, we constructed a series of plasmids having LexA fused to the L1 cytoplasmic domain, each having progressively less of the L1 juxtamembrane domain: E1157-E1259, S1163-E1259, and K1169-E1259 (all contained both the RSLE miniexon and Y1176 intact). None of these LexA fusion proteins interacted with the ezrin FERM domain, whereas all interacted with AP2 and with ankyrin B (Fig. 1B). Additionally, the entire juxtamembrane region lacking RSLE (K1144-Y1176 lane; Fig. 1B) was not sufficient for FERM domain binding in yeast two-hybrid analysis. This suggests that both the juxtamembrane region and YRSLE are important for ezrin FERM domain interactions in the L1 cytoplasmic region. Our result is consistent with previously-published work showing that either mutations in the juxtamembrane region or deletion of RSLE affected ezrin binding to L1 in yeast two-hybrid analysis (Cheng et al., 2005).
We further analyzed FERM domain interactions with the L1 cytoplasmic region using a biochemical approach. First, we made a GST fusion protein containing the entire L1 cytoplasmic region. GST alone or GST fused to the L1 cytoplasmic region was immobilized on glutathione agarose beads and incubated with cell extract prepared from HEK293 cells transfected with a construct encoding the entire FERM domain of ezrin fused to YFP (FERM-YFP). As shown in Figure 2A, the GST-L1 cytoplasmic region coprecipitated the ezrin FERM domain, showing that this interaction can be detected under these conditions. To identify the binding site for ezrin, we prepared several biotinylated peptides (12–13 aa each) derived from the L1 cytoplasmic region. Equal amounts of the peptides were mixed with avidin-agarose beads and incubated with cell extract prepared from 293 cells expressing FERM-YFP. The peptide containing the juxtamembrane region of L1 (Ant-1) coprecipitated the ezrin FERM domain, as did the peptide containing the RSLE region of L1 (RSLE), to a much greater extent that did a control L1 peptide corresponding to the L1 isoform without the RSLE sequence. However, binding to the juxtamembrane peptide appeared to be greater than the RSLE region (Fig. 2B). These data suggest that the L1 cytoplasmic region may have two binding sites for ezrin: the juxtamembrane region and the region surrounding RSLE, with the former having a higher affinity. Although individual L1 peptide fragments can bind to the FERM domain, the native L1 cytoplasmic tail seems to require both regions (and perhaps surrounding sequences) for FERM domain binding based on the yeast two-hybrid interaction trap analysis, above.
Fig. 2.
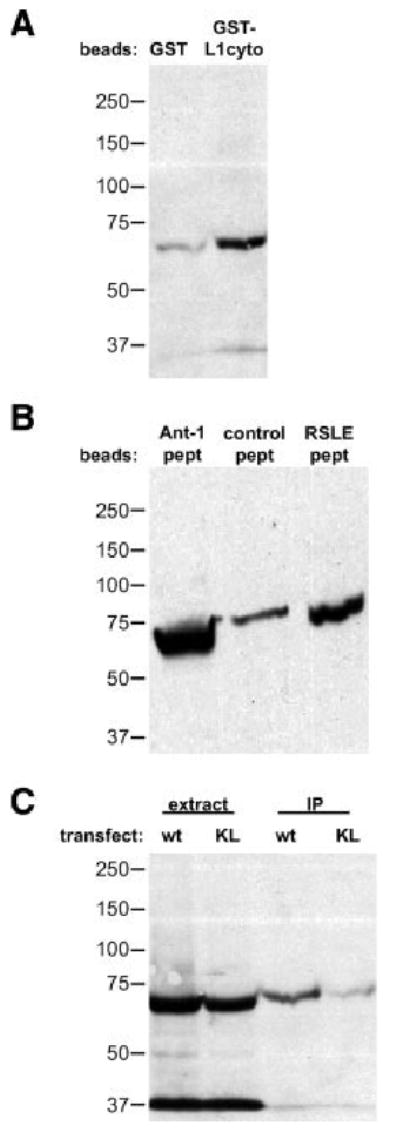
Juxtamembrane region of L1 contains a binding site for the FERM domain of ezrin. A: GST alone or GST fused with the L1 cytoplasmic region was immobilized on glutathione agarose beads and incubated with cell extract prepared from 293 cells transfected with a construct encoding the FERM domain of ezrin fused with YFP. After extensive washing, bound proteins were subjected to SDS-PAGE and analyzed by Western blotting with an anti-GFP antibody. B: We prepared several biotinylated peptides (∼10 amino acids each) derived from the L1 cytoplasmic region. Equal amounts of these peptides were mixed with avidin agarose beads and incubated with cell extract prepared from 293 cells expressing the FERM domain of ezrin fused with YFP. Bound proteins were analyzed by Western blotting with an anti-GFP antibody. C: We expressed wild-type and K1147L mutant of L1 with the FERM domain of ezrin-YFP fusion protein in 293 cells and performed immunoprecipitation of the L1 molecules. Coprecipitated proteins were subjected to Western blotting with an anti-GFP antibody. Note that in extract lanes, EGFP band was detected at around 27 kDa, whereas no EGFP band was detected in IP lanes (see Materials and Methods section).
It has been shown previously that a K1147L mutation in the juxtamembrane FERM-binding region of L1 disrupts localization of L1 to actin stress fibers in glioma cells (Dahlin-Huppe et al., 1997). We coexpressed either wild-type or the K1147L mutant of full-length L1 with the ezrin FERM domain–YFP fusion protein in 293 cells and performed immunoprecipitation of L1. Although L1 and the ezrin FERM domain showed similar expression, the K1147L mutant of L1 brought down less FERM domain than did the wild type (Fig. 2C). These results suggest that the juxtamembrane region of L1 is involved in interactions with the FERM domain of ezrin and that lysine 1147 of L1 is important for interactions between the ezrin FERM domain and L1 in intact cells.
K1147L Mutation Disrupts Retrograde Movement of L1 on the Cell Surface
We have shown previously that L1 interacts through its cytoplasmic domain with treadmilling actin in the cytosol, resulting in directed, retrograde movement of L1 on the cell surface (Gil et al., 2003): the generation of the traction forces at adhesion sites during cell migration has been suggested to depend on the linking process between cell-surface molecules and treadmilling actin (Sheetz et al., 1998). As ERM proteins have the capacity to bind both L1 and the actin cytoskeleton, it is possible that ezrin interactions might be involved in the traction-force generation mediated by the L1 receptor. To investigate this possibility, we quantified the diffusion characteristics of cell-surface L1 by single-particle analysis (Felsenfeld et al., 1996; Choquet et al., 1997). A wild-type or K1147L mutant of L1 with an amino-terminal Myc-epitope tag was expressed in ND7 neuroblastoma cells (Dunn et al., 1991). We recorded the movement of beads bound by anti-Myc (9E10) antibody to cell-surface L1 expressed on the upper surface of the lamella near the leading edge of polarized cells. In previous studies, we showed that wild-type L1 displays at least three types of behavior on the cell surface: these are random diffusion (high diffusion in the absence of directed movement), stationary behavior (low diffusion in the absence of directed movement) and retrograde movement (low diffusion coupled with directed movement away from the leading edge of the cell; Gil et al., 2003; Fig. 3A). There three classes of protein movement are present in a characteristic ratio where retrograde movement is observed in approximately 60% of the trials (Fig. 3; Gil et al., 2003). Whereas the stationary behavior has been shown to depend on ankyrin binding (Gil et al., 2003), the interactions mediating retrograde movement have not been characterized. When the K1147L mutant was analyzed, the proportion of beads displaying retrograde movement was decreased, whereas the proportion of beads remaining stationary had increased (Fig. 3A). A similar reduction in retrograde-moving beads was observed when the FERM domain of ezrin (aa 1–406), a dominant-negative form that can bind to L1 but not actin, was coexpressed with wild-type L1 (Fig. 3B). However, in contrast to our findings with the K1147L mutant L1, the expression of dominant-negative ezrin did not result in an increase in the percentage of beads in the low-diffusion stationary state. Instead, the decrease in retrograde movement was balanced by a relative increase in beads undergoing free diffusion on the cell surface. As L1 stationary behavior depends on binding to ankyrin, this result raises the possibility that FERM domain binding to L1, either by wild-type or dominant-negative ezrin, may inhibit ankyrin binding despite the physical separation of their binding sites. Nevertheless, these results suggest that ezrin interactions mediate the retrograde movement of L1, involved in traction-force generation.
Fig. 3.
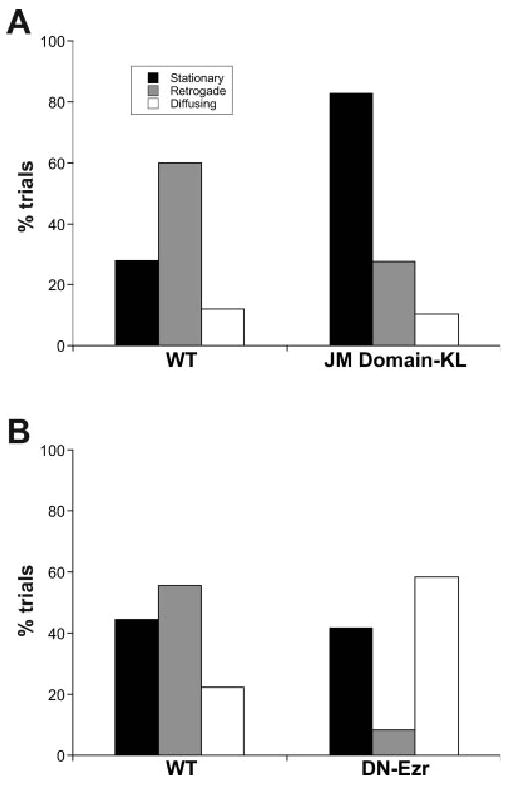
A mutation in the juxtamembrane region of L1 changed L1 behavior on the cell surface. A: Wild-type or K1147L mutant L1 was expressed in ND7 neuroblastoma cells. We recorded the movement of beads bound by antibody to cell-surface L1 (n = 25 for wild type, n = 29 for KL mutant). The behavior was categorized into three types: random diffusion, stationary behavior, and retrograde movement. K1147L mutation increased stationary behavior and reduced retrograde movement (P < 0.0001). B: Wild-type L1 or wild-type L1 with dominant-negative ezrin was expressed in ND7 cells, and the movement of beads bound by antibody to cell-surface L1 was recorded (n = 12 for control, n = 18 for dominant-negative ezrin, P < 0.0001).
A Membrane-Permeable Peptide Derived from Juxtamembrane Region of L1 Partially Inhibits L1-dependent Axon Outgrowth from Cerebellar Cells
Receptor-mediated traction-force generation is believed to play a critical role in the migration of adherent cells, including growth cone translocation during axon extension (Sheetz et al., 1998). Therefore, if ezrin interactions are involved in traction-force generation mediated by the L1 receptor, they would be expected to play a role in axon outgrowth mediated by L1. As we showed previously, membrane-permeable peptides derived from the L1 cytoplasmic region containing FIGQY, the binding site for ankyrin, inhibit L1–ankyrin interactions and static behavior of L1 on the cell surface and as a result stimulate axon outgrowth (Gil et al., 2003). We prepared a membrane-permeable peptide derived from the juxtamembrane region of L1 and treated cerebellar cells prepared from P4 mouse cultured on Ng-CAM (chick L1) or laminin substrate (Fig. 4). The peptide inhibited outgrowth induced by Ng-CAM by 22% (P = 0.019) but not by laminin (P = 0.25). These results suggest that ezrin interactions through the juxtamembrane region of L1 play a role in L1-mediated traction-force generation and axon outgrowth. We also analyzed the effect on branching of axons but did not observe a statistically significant difference (the number of total branch points/total number of cells; control treated, 0.831 ± 0.032, vs. AP-1-treated, 0.736 ± 0.068, P = 0.11).
Fig. 4.
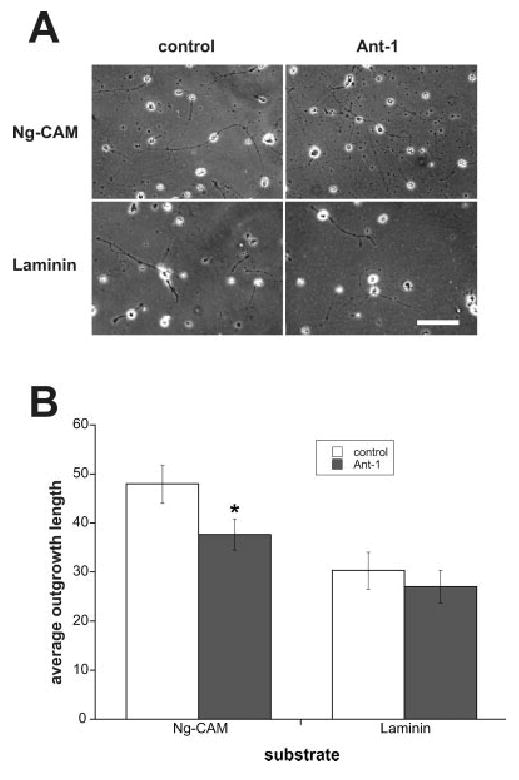
A membrane-permeable peptide derived from the juxtamembrane region of L1 inhibited axon outgrowth from cerebellar granule cells mediated by the L1 receptor. A: We prepared membrane-permeable peptide derived from the juxtamembrane region of L1 (Ant-1) and control peptide and treated cerebellar cells plated on NgCAM or laminin. Neurite outgrowth length was measured after 24 hr in vitro. Scale bar = 100 μm. B: In the presence of Ant-1 peptide, average length on NgCAM was 37.7 ± 3.1 μm (n = 195), significantly shorter than the average length of outgrowth in the presence of control peptide (48.0 ± 3.9 μm; n = 181, P = 0.019). Average length on laminin in the presence of Ant-1 and that in the presence of control peptide were not significantly different [30.3 ± 3.8 μm (n = 129) versus 27.0 ± 3.3 μm (n = 146), P = 0.25].
Juxtamembrane Region of L1 Can Be Phosphorylated by Tyrosine Kinases
Ezrin localization to the membrane is regulated by phosphorylation of ezrin itself and by PIP2 binding to a site on the ezrin FERM domain (Fievet et al., 2004). However, once ezrin is activated, its ability to bind selectively to distinct proteins at the membrane must be independently regulated. L1 is phosphorylated on residue S1152 in the juxtamembrane region by p90rsk in vitro and in vivo; this phosphorylation is important for axon outgrowth mediated by L1 (Wong et al., 1996b). This serine is in the middle of the ERM binding motif in the L1 cytoplasmic region (Fig. 1), raising the possibility that phosphorylation of S1152 might be involved in regulation of ezrin binding to L1.
In addition, there is a tyrosine residue that is directly involved in ERM binding to the adhesion molecule (Y1151; Cheng et al., 2005; Fig. 1). This tyrosine residue is highly conserved in the L1 molecule among species from flies to mammals. Interestingly, Y1176, which is also involved in ERM binding, has been shown to be phosphorylated by Src-family kinases, important for regulation of AP-2 binding to L1 and L1 internalization (Schaefer et al., 2002). The localization and functional role of these tyrosines in the ERM binding site suggest that L1–ERM interactions may depend on L1 tyrosine phosphorylation in a manner similar to other L1–cytoskeleton interactions.
To identify the kinase or kinases that phosphorylate Y1151, we designed a reporter construct based on intramolecular bioluminescence resonance energy transfer (BRET) as described recently by our group (Whittard et al., 2006). This BRET reporter is composed of a short peptide fragment containing a candidate phosphorylation site concatenated between a Renilla luciferase donor and a modified GFP acceptor (Whittard et al., 2006). Small changes in substrate peptide conformation that result from phosphorylation of the target residue lead to quantifiable differences in the efficiency of energy transfer, measured as a change in the emission spectrum of the reporter. By using this reporter construct, we can screen for kinases that phosphorylate this target sequence in transfected cells. Unless phosphorylation site-specific antiphosphotyrosine antibodies (e.g., Jenkins et al., 2001; Schaefer et al., 2002) are available, analysis of phosphorylation of the L1 cytoplasmic region using traditional methods is challenging for two major reasons. First, the L1 cytoplasmic region contains four tyrosines that can be potentially phosphorylated, and mutations that eliminate one or more phosphorylation sites may affect the phosphorylation of the remaining tyrosines. Second, phosphorylation of L1 molecules is predicted to be local and transient. The BRET reporter construct permits a sensitive and quantitative method for measuring kinase activity responsible for phosphorylation of individual tyrosines in the L1 cytoplasmic region in situ.
We made a BRET construct, KGGKY, that contains 12 aa of the juxtamembrane region of L1 as the reporter insert and transfected it into 293 cells. Previously, we have shown that another construct containing a reporter derived from the FIGQY domain responded to EGF stimulation in 293 cells, displaying a significant, quantifiable change in the BRET ratio caused by phosphorylation of the FIGQY construct by MAPK pathways (Whittard et al., 2006). When we applied EGF to these cells transfected with the KGGKY construct, the BRET ratio was significantly reduced, suggesting that the phosphorylation of KGGKY occurs in response to growth factor stimulation (Whittard et al., 2006; Fig. 5A). We confirmed this by treating the cells with genistein, a tyrosine kinase inhibitor that significantly increases the BRET ratio (Fig. 5A). However, the MEK inhibitor, PD-98059 had no effect on the BRET ratio suggesting that unlike the FIGQY site (Whittard et al., 2006; Fig. 5), MEK is not responsible for phosphorylation of the KGGKY site. We further screened several tyrosine kinase inhibitors and found that the Src-family kinase inhibitor PP1, but not PP2, increased the BRET ratio of the KGGKY construct in a dose-dependent manner (Fig. 5B). To demonstrate further that Src-family kinases are involved in phosphorylation of KGGKY, we took advantage of Src−/− fibroblasts (Soriano et al., 1991; Kaplan et al., 1995). We transfected the BRET construct into Src+/+ and Src−/− fibroblasts. Whereas FGF treatment reduced the BRET ratio in Src+/+ cells, in Src−/− cells, FGF treatment had no effect on the BRET ratio, suggesting that Src plays critical role in the phosphorylation of this site (Fig. 5C). Together, these results suggest that Y1151 in the juxtamembrane region of L1 can be phosphorylated by Src-family kinases.
Fig. 5.
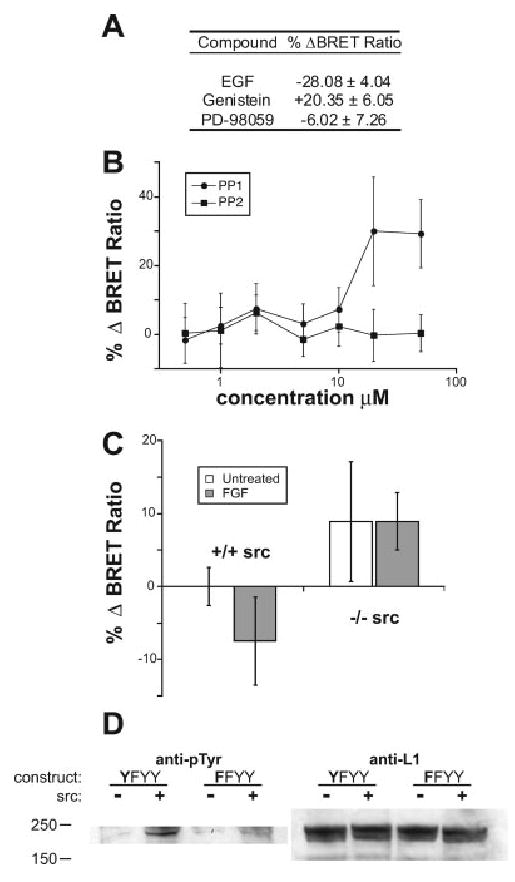
The Src family of tyrosine kinases can phosphorylate Y1151 of L1. A: Application of 100 ng/mL EGF significantly reduced the BRET ratio of the KGGKY construct transiently transfected in 293 cells. The tyrosine kinase inhibitor genistein (100 μM) significantly increased the BRET ratio of the KGGKY construct. The MEK inhibitor PD-98059 (100 μM) had no effect on the BRET ratio. B: The Src-family kinase inhibitor PP1, but not PP2, increased the BRET ratio of the KGGKY construct in a dose-dependent manner. C: Application of 100 ng/mL bFGF significantly reduced the BRET ratio of the KGGKY construct in Src+/+ Swiss 3T3 fibroblasts but not in Src−/− cells. In A–C, results shown are means ± SDs, n = 5; *P < 0.01 (t test). D: L1 YFYY (Y1151, Y1176F, and the other two downstream Ys in the cytoplasmic region intact) and FFYY (Y1151F, Y1176F, and the other two downstream Ys intact) constructs were transfected with or without the c-Src construct into 293 cells. L1 was immunoprecipitated by anti-Myc antibody from extracts of these cells and subjected to Western blotting with an antiphosphotyrosine antibody (lef panel). Note that in the presence of c-Src, L1 phosphorylation increased, but FFYY had reduced phosphorylation compared with YFYY, suggesting that Y1151 is phosphorylated by c-Src. Expression of L1 was similar in all conditions, shown by anti-L1 blot on extracts (right).
To determine if Y1151 can be phosphorylated by Src-family kinases in intact L1 molecules in cells, we cotransfected L1 and c-Src into 293 cells and analyzed the phosphorylation status of L1 by antiphosphotyrosine (Fig. 5D). For this experiment, we used Y1176F mutants to exclude the possibility of Src-dependent phosphorylation of Y1176. When we cotransfected c-Src with the L1 YF (Y1151Y, Y1176F) form, which has intact Y1151, tyrosine phospho-rylation of L1 YF was drastically increased. However, when we coexpressed the L1 FF (Y1151F, Y1176F) mutant with c-Src, tyrosine phosphorylation of L1 FF was substantially reduced, suggesting that phosphorylation by c-Src is responsible for phosphorylation of Y1151 in live cells.
Ezrin Binding to L1 Receptor Can Be Regulated by Tyrosine Phosphorylation of Juxtamembrane Region
The above results suggest that Y1151 can be phosphorylated and raise the possibility that tyrosine phosphorylation may modulate ezrin binding to L1. Therefore, we mutated this residue to aspartate (to mimic phosphorylated tyrosine) or phenylalanine (to mimic unphosphorylated tyrosine) and analyzed their abilities to immunoprecipitate the ezrin FERM domain. However, we did not observe significant differences between the wild-type and mutant forms (data not shown). Because the RSLE region of L1 also seems to be involved in ezrin binding, we mutated tyrosine 1176 (YRSLE, shown to be directly phosphorylated by Src kinase; Schaefer et al., 2002) to aspartate or phenylalanine in addition to the mutations in tyrosine 1151 in the juxtamembrane region. We found that double mutants that have both tyrosines changed to aspartate (Y1151D, Y1176D) have decreased ability to bind the ezrin FERM domain (Fig. 6A). This supports the idea that there are two binding sites for ezrin on L1 and that ezrin binding to L1 may be regulated by tyrosine phosphorylation of both sites. However, ezrin binding to cell-surface proteins in situ can be modulated by multiple molecular components, including PIP2 at the membrane.
Fig. 6.
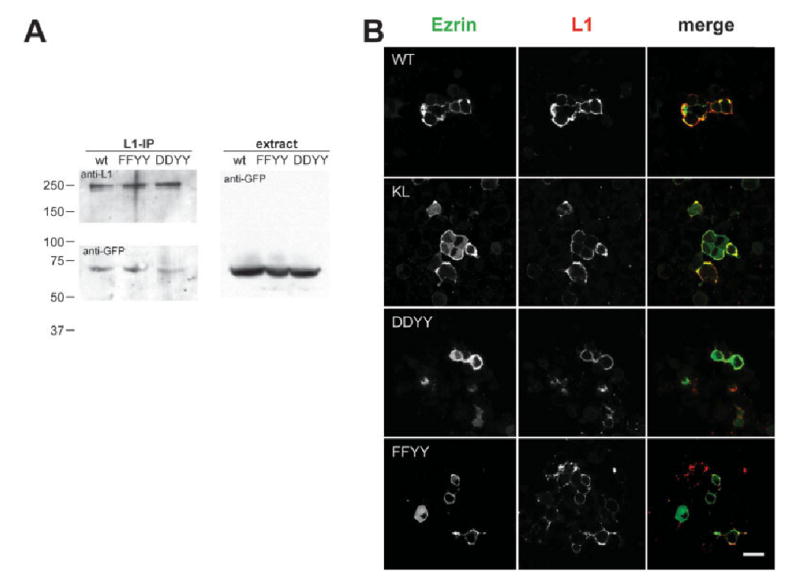
Ezrin binding to the L1 receptor might be regulated by tyrosine phosphorylation on Y1151. A: L1 wild-type or mutants (FFYY: Y1151F, Y1176F; DDYY: Y1151D, Y1176D) were cotransfected with the ezrin FERM domain fused to YFP into 293 cells. L1 was immunoprecipitated by anti-Myc antibody, and precipitated proteins were blotted with anti-GFP antibody. The DDYY mutant coprecipitated less ezrin FERM domain than the wild-type or FFYY mutant (average ∼40% of intensity of wild-type IP band judged by densitometry of 3 independent experiments). B: Myc-tagged L1 wild-type or mutants were cotransfected with ezrin-Venus into ND7 cells. Cells were immunostained with an anti-Myc antibody and secondary antibody prior to fixation to visualize L1 (red channel), ezrin-Venus (green channel), and their colocalization (yellow). K1147L or DDYY mutants showed significantly less colocalization with ezrin-Venus compared with either the wild-type or the FFYY mutant, as reflected by the high levels of ezrin-Venus diffusely distributed in the cytosol. Colocalization (Pearson's correlation) was quantified using Image J with the colocalization finder plugin (see text for details). Scale bar = 10 μm.
To explore the above possibility directly, we examined the function of these mutants in live cells. If phosphorylation affects interactions between L1 and ezrin, ezrin localization in the cells when cotransfected with wild-type L1 or tyrosine phosphorylation mutants may be different. We cotransfected Myc-tagged L1, either wild-type, DD, or FF (Y1151, Y1176 to D, Y1176 to F, respectively), with full-length ezrin-Venus into ND7 cells and analyzed localization on the membrane and cytosol by confocal microscopy (Fig. 6B). We also used K1147L mutants, which are likely to be deficient in ezrin binding, for comparison. Wild-type L1 and ezrin-Venus were colocalized in some areas of the membrane, whereas other regions showed discrete patches of each protein (Pearson's correlation, r = 0.10 ± 0.01). When we expressed the K1147L mutant with ezrin-Venus, however, most of the ezrin-Venus showed diffuse distribution in the cytosol, suggesting a decreased degree of binding to L1 at the membrane (r = 0.04 ± 0.01), consistent with our biochemical analysis. When the FF mutant was expressed with ezrin-Venus, the degree of colocalization increased compared with the wild type (r = 0.24 ± 0.01), whereas when the DD mutant was expressed, colocalization was reduced, similar to the K1147L mutant (r = 0.05 ± 0.01). These data are consistent with biochemical analysis and suggest that tyrosine phosphorylation in the juxtamembrane region of L1 may affect ezrin binding to L1 (i.e., the dephosphorylated form of L1 binds to ezrin, whereas the phosphorylated form of L1 does not).
Together, these results suggest that phosphorylation of Y1151 and Y1176 may inhibit ezrin binding to L1 and therefore may decrease retrograde movement mediated by actin cytoskeleton through ezrin binding.
Discussion
L1 CAM plays a role in neuronal adhesion, axon outgrowth, and guidance. During these processes, the activity of the L1 adhesion complex is modulated through molecular interactions mediated by the L1 cytoplasmic region that is a kinase substrate. The L1 adhesion complex generates actin-mediated traction forces that support axon outgrowth. In this study, we have shown that an ERM protein, ezrin, which interacts with the cytoskeleton, binds to the juxtamembrane region of L1 and may support traction-force generation by L1. Furthermore, we have shown that this interaction may be regulated by phosphorylation of Y1151 mediated by Src-family kinases.
L1 Cytoskeletal Interactions and Traction-Force Generation
The cytoplasmic region of L1 family CAMs has been shown to bind ankyrin through its FIGQY sequence (Davis and Bennett, 1994). We have shown previously that L1–ankyrin interactions are involved in the static behavior of L1 rather than retrograde movement of L1 on the cell surface: perturbation of these interactions enhances outgrowth induced by L1 substrates, suggesting that molecules other than ankyrin must be involved in retrograde movement of L1 to support the actin-mediated traction-force generation by the L1 receptor (Gil et al., 2003). We proposed that interactions between L1 and cytoskeletal proteins such as ankyrin regulate the activity of L1 on the cell surface, supporting either cell adhesion (stationary behavior) or axon extension (retrograde movement/traction-force generation; Gil et al., 2003). In this study, we have demonstrated that ezrin, which interacts with a distinct region of the L1 cytoplasmic tail from ankyrin, may mediate the retrograde movement of the L1 receptor on the cell surface. Although we cannot completely exclude the possibility that proteins other than ezrin interact with L1 through the juxtamembrane region of L1 and mediate the retrograde movement of the L1 receptor, we think that ezrin is a strong candidate based on our studies shown here. It should be noted that when L1 was coexpressed with the dominant-negative form of ezrin, which can still interact with L1 but cannot link L1 to the actin cytoskeleton, an increase in L1 diffusion rather than ankyrin-mediated stationary behavior was observed, suggesting that ezrin and ankyrin binding may be mutually inhibitory. This is consistent with the inhibition of L1 retrograde movement by ankyrin binding, which we have observed previously (Gil et al., 2003). However, in light of the lack of overlap between the ezrin- and ankyrin-binding sites, we would postulate that the cross-inhibition is unlikely to involve direct binding competition.
Nishimura et al. recently showed that L1 interactions with ankyrin are involved in the initial extension of neurites from the membranous protrusions surrounding the soma (Nishimura et al., 2003). Bead-tracking experiments on primary neurons demonstrated that ankyrin mediates L1 coupling with retrograde F-actin flow in these perisomatic structures. Interestingly, ligation of the L1 ectodomain by an immobile substrate induces L1–ankyrin binding and the formation of stationary ankyrin clusters where neurite initiation preferentially occurs. Furthermore, they also found that ankyrin is involved neither in L1 coupling with F-actin flow in growth cones nor in L1-based neurite elongation. Collectively, their results and our results suggest that during axon extension, ezrin interactions may mediate retrograde movement of L1, linking L1 to F-actin flow. More importantly, these results also suggest that the L1 receptor may use different molecular mechanisms to link L1 to F-actin flow in support of distinct biological phenomena, (i.e., axon initiation and axon extension). Characterizing where and when interactions of L1 with cytoskeletal proteins such as ankyrin and ezrin take place in neurons and how these interactions are regulated in situ will be crucial to understanding L1 function and its regulation in neurons.
Cheng and coworkers showed that ezrin interacts with two motifs in the L1 cytoplasmic region, the RSLE region and a juxtamembrane region (Cheng et al., 2005), consistent with the results shown here. They also showed that L1 cytoplasmic deletion mutants that would be expected to diminish ezrin binding could still rescue axon extension of L1-null neurons on an L1 substrate but that axons branched less in these neurons, suggesting that (1) the L1 cytoplasmic region is not necessary for L1-mediated outgrowth, and (2) L1–ezrin interactions are important for neurite branchping. When we treated cerebellar neurons with a membrane-permeable peptide that could inhibit ezrin interactions with L1, we observed a modest reduction in outgrowth induced by an L1 substrate without any significant effects on branching. However, we did not detect effects on branching, perhaps reflecting that we analyzed our cultures after 24 hr of incubation, before most branches were formed (we discuss L1 involvement in branching below). Neurite outgrowth mediated by the L1 receptor complex involves L1 and other coreceptors such as integrins and GPI-linked CAMs on the cell surface (Felsenfeld et al., 1994; Buchstaller et al., 1996; also see Cheng et al., 2005). It is possible that even though this outgrowth requires the presence of L1 on the cell surface, induction of outgrowth could be mediated through several different pathways originating from the receptor complex. In their study, Cheng et al. rescued L1-mediated outgrowth in L1-null neurons by introducing L1 mutants that were unable to bind ezrin, suggesting that the neurons may employ other pathways that can compensate for the lack of L1–ezrin interactions (Cheng et al., 2005). It should be noted that in the Cheng et al. study, when L1–ezrin interactions were perturbed, even though outgrowth was intact, the morphology of neurites was different, and total axon length was reduced. This supports the idea that there may be several different pathways that can induce outgrowth. Our previous study showed that inhibition of L1-stimulated nerve growth by inhibitors of the MAP kinase pathway has both an ankyrin-dependent and an ankyrin-independent component (Gil et al., 2003), suggesting that structural interactions mediated by L1 are acting in parallel with traditional signaling pathways (Schaefer et al., 1999; Schmid et al., 2000). Our observation that membrane-permeable peptides, which disrupt ERM binding, showed modest effects on outgrowth is also consistent with this hypothesis.
Finally, L1–ezrin interactions may be involved in neurite branching, as demonstrated by the study of Cheng et al. Interestingly, Y1151, the most critical residue for L1-mediated branching, can be a target of phosphorylation that affects ezrin binding (see below). Nevertheless, because L1 activity would be dynamically modulated in the processes of neurite branching, realtime analyses of L1–ezrin interactions in growing neurons will provide critical insights into L1 involvement in neurite branching.
L1 Regulation by Phosphorylation of Its Cytoplasmic Tail
The L1 cytoplasmic region has been shown to be phosphorylated by several kinases that modulate L1 activity. For example, the unphosphorylated FIGQY sequence (1225–1229) in the L1 cytoplasmic region binds to ankyrin, whereas the phosphorylated form does not (Garver et al., 1997). Our recent studies have shown that MAPK is involved in L1 phosphorylation on Y1229 (Whittard et al., 2006 and unpublished). Similarly, Y1176, which comprises a binding site for AP2 together with the RSLE sequence (1177–1180; Kamiguchi et al., 1998), is phosphorylated by Src-family kinases modulating L1 endocytosis (Schaefer et al., 2002). Furthermore, S1181, immediately after RSLE, is phosphorylated by casein kinase II–modulating endocytic L1 trafficking (Nakata and Kamiguchi, 2007). Finally, in vitro, S1204 and S1248 can be phosphorylated by ERK2, although the functional relevance of these phosphorylation events is not clear (Schaefer et al., 1999).
In the juxtamembrane region, it has been shown that S1152 can be phosphorylated by p90rsk, and treatment with a peptide containing the amino acid sequence encompassing S1152 inhibited L1-mediated outgrowth in cultured neurons (Wong et al., 1996b). This serine residue is highly conserved in the mammalian L1 sequences as well as L1-related molecules in vertebrates from fish to birds (Wong et al., 1996b). We found that Y1151 can be phosphorylated by Src-family kinases and that this affects interactions with ezrin, intracellular localization of ezrin, and L1 behavior on the cell surface. Previously, Maness's group showed that Src-null cerebellar neurons show 50% reduction in neurite length on L1 substrates (Ignelzi et al., 1994). In subsequent studies, they further demonstrated that Src is essential in regulating endocytosis of L1 in MAPK-dependent neurite outgrowth on an L1 substrate (Schmid et al., 2000). Our studies postulate that in addition to L1 endocytosis, Src-family kinases may also regulate ezrin interactions with the L1 juxtamembrane region.
Regulation of L1 Adhesion
It should be noted that both the juxtamembrane region and the RSLE sequence are important for ezrin binding and that both Y1151 and Y1176 can be phosphorylated by Src-family kinases. Our studies also suggest that unphosphorylated Y1151 and Y1176 can support ezrin binding to the L1 cytoplasmic region. It has also been shown that unphosphorylated Y1176 binds to AP2, potentially inducing internalization of L1 from the cell surface (Schaefer et al., 2002). This suggests that ezrin and AP2 binding could compete with one another at the unphosphorylated Y1176 on L1. How are these interactions regulated? It is possible that phosphorylation of both tyrosines is modulated by the same kinase and phosphatase in concert or alternatively that their phosphorylation status is modulated individually. The former supports the idea that ezrin and AP2 binding may compete for L1 unless the localization of L1 (e.g., axon, growth cone, or endocytic vesicle) determines the binding partner. If the latter is true, then the phosphorylation status of Y1176 may determine which molecule binds to L1 through unphosphorylated Y1151. Another possibility is that the requirement for the activation of ERM proteins by phosphorylation and PIP2 binding may independently regulate the composition of the L1 complex by modulating the relative affinities of the components. Nevertheless, the activity of the L1 adhesion complex can be modulated by a balance between AP2 binding and ezrin binding, the former inducing internalization of L1 and the latter supporting traction-force generation by L1. As we proposed previously, the balance between ankyrin binding and ezrin binding could also modulate the activity of the L1 adhesion complex; the former sustains static adhesion and the latter supports retrograde movement (and therefore migration). All these interactions with L1 can be dynamically and locally regulated by signaling pathways such as Src-family kinases and MAP kinase. Therefore, L1 adhesive activity is modulated in an inside-out manner (Hortsch et al., 1998; Gil et al., 2003; Whittard et al., 2006), similar to that of other adhesion proteins.
Acknowledgments
We thank Dr. Monique Arpin, Dr. Atsushi Miyawaki, and Dr. Jan Sap for cDNA. We acknowledge DSHB for the 9E10 antibody and Microscope Shared Research Facility at MSSM for the confocal microscopy.
Contract grant sponsor: NIH; contract grant numbers: GM63192 (to D.P.F.), AA14898, NS050634 (to D.L.B.), DK071308, NS045305 (to S.R.S.); Contract grant sponsor: Damon Runyon Cancer Research Foundation; Contract grant sponsor: Irma T. Hirshl scholar (to D.P.F.); Contract grant sponsor: NARSAD (to S.R.S.). T.S. was supported by NIH grant NS20417 (to David R. Colman) and NYS SCI contract C020935 and is a Seaver fellow.
References
- Buchstaller A, Kunz S, Berger P, Kunz B, Ziegler U, Rader C, Sonderegger P. Cell adhesion molecules NgCAM and axonin-1 form heterodimers in the neuronal membrane and cooperate in neurite outgrowth promotion. J Cell Biol. 1996;135:1593–1607. doi: 10.1083/jcb.135.6.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Itoh K, Lemmon V. L1-mediated branching is regulated by two ezrin-radixin-moesin (ERM)-binding sites, the RSLE region and a novel juxtamembrane ERM-binding region. J Neurosci. 2005;25:395–403. doi: 10.1523/JNEUROSCI.4097-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- Dahlin-Huppe K, Berglund EO, Ranscht B, Stallcup WB. Mutational analysis of the L1 neuronal cell adhesion molecule identifies membrane-proximal amino acids of the cytoplasmic domain that are required for cytoskeletal anchorage. Mol Cell Neurosci. 1997;9:144–156. doi: 10.1006/mcne.1997.0608. [DOI] [PubMed] [Google Scholar]
- Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269:27163–27166. [PubMed] [Google Scholar]
- Derossi D, Chassaing G, Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8:84–87. [PubMed] [Google Scholar]
- Dickson TC, Mintz CD, Benson DL, Salton SR. Functional binding interaction identified between the axonal CAM L1 and members of the ERM family. J Cell Biol. 2002;157:1105–1112. doi: 10.1083/jcb.200111076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn PM, Coote PR, Wood JN, Burgess GM, Rang HP. Bradykinin evoked depolarization of a novel neuroblastoma x DRG neurone hybrid cell line (ND7/23) Brain Res. 1991;545:80–86. doi: 10.1016/0006-8993(91)91272-3. [DOI] [PubMed] [Google Scholar]
- Felsenfeld DP, Choquet D, Sheetz MP. Ligand binding regulates the directed movement of beta1 integrins on fibroblasts. Nature. 1996;383:438–440. doi: 10.1038/383438a0. [DOI] [PubMed] [Google Scholar]
- Felsenfeld DP, Hynes MA, Skoler KM, Furley AJ, Jessell TM. TAG-1 can mediate homophilic binding, but neurite outgrowth on TAG-1 requires an L1-like molecule and beta 1 integrins. Neuron. 1994;12:675–690. doi: 10.1016/0896-6273(94)90222-4. [DOI] [PubMed] [Google Scholar]
- Felsenfeld DP, Schwartzberg PL, Venegas A, Tse R, Sheetz MP. Selective regulation of integrin–cytoskeleton interactions by the tyrosine kinase Src. Nat Cell Biol. 1999;1:200–206. doi: 10.1038/12021. [DOI] [PubMed] [Google Scholar]
- Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, Arpin M. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol. 2004;164:653–659. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen E, Van Camp G, Vits L, Willems PJ. L1-associated diseases: clinical geneticists divide, molecular geneticists unite. Hum Mol Genet. 1997;6:1625–1632. doi: 10.1093/hmg/6.10.1625. [DOI] [PubMed] [Google Scholar]
- Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–714. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil OD, Sakurai T, Bradley AE, Fink MY, Cassella MR, Kuo JA, Felsenfeld DP. Ankyrin binding mediates L1CAM interactions with static components of the cytoskeleton and inhibits retrograde movement of L1CAM on the cell surface. J Cell Biol. 2003;162:719–730. doi: 10.1083/jcb.200211011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada K, Shimizu T, Yonemura S, Tsukita S, Hakoshima T. Structural basis of adhesion-molecule recognition by ERM proteins revealed by the crystal structure of the radixin-ICAM-2 complex. Embo J. 2003;22:502–514. doi: 10.1093/emboj/cdg039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortsch M, Homer D, Malhotra JD, Chang S, Frankel J, Jefford G, Dubreuil RR. Structural requirements for outside-in and inside-out signaling by Drosophila neuroglian, a member of the L1 family of cell adhesion molecules. J Cell Biol. 1998;142:251–261. doi: 10.1083/jcb.142.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignelzi MA, Jr, Miller DR, Soriano P, Maness PF. Impaired neurite outgrowth of src-minus cerebellar neurons on the cell adhesion molecule L1. Neuron. 1994;12:873–884. doi: 10.1016/0896-6273(94)90339-5. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Kizhatil K, Kramarcy NR, Sen A, Sealock R, Bennett V. FIGQY phosphorylation defines discrete populations of L1 cell adhesion molecules at sites of cell-cell contact and in migrating neurons. J Cell Sci. 2001;114:3823–3835. doi: 10.1242/jcs.114.21.3823. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Long KE, Pendergast M, Schaefer AW, Rapoport I, Kirchhausen T, Lemmon V. The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J Neurosci. 1998;18:5311–5321. doi: 10.1523/JNEUROSCI.18-14-05311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. c-Src enhances the spreading of src−/− fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- Lustig M, Erskine L, Mason CA, Grumet M, Sakurai T. Nr-CAM expression in the developing mouse nervous system: ventral midline structures, specific fiber tracts, and neuropilar regions. J Comp Neurol. 2001;434:13–28. doi: 10.1002/cne.1161. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Nakata A, Kamiguchi H. Serine phosphorylation by casein kinase II controls endocytic L1 trafficking and axon growth. J Neurosci Res. 2007;85:723–734. doi: 10.1002/jnr.21185. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Yoshihara F, Tojima T, Ooashi N, Yoon W, Mikoshiba K, Bennett V, Kamiguchi H. L1-dependent neuritogenesis involves ankyrinB that mediates L1-CAM coupling with retrograde actin flow. J Cell Biol. 2003;163:1077–1088. doi: 10.1083/jcb.200303060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Lustig M, Nativ M, Hemperly JJ, Schlessinger J, Peles E, Grumet M. Induction of neurite outgrowth through contactin and Nr-CAM by extracellular regions of glial receptor tyrosine phosphatase beta. J Cell Biol. 1997;136:907–918. doi: 10.1083/jcb.136.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AW, Kamei Y, Kamiguchi H, Wong EV, Rapoport I, Kirchhausen T, Beach CM, Landreth G, Lemmon SK, Lemmon V. L1 endocytosis is controlled by a phosphorylation-dephosphorylation cycle stimulated by outside-in signaling by L1. J Cell Biol. 2002;157:1223–1232. doi: 10.1083/jcb.200203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AW, Kamiguchi H, Wong EV, Beach CM, Landreth G, Lemmon V. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37965–37973. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Pruitt WM, Maness PF. A MAP kinase-signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. J Neurosci. 2000;20:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz MP, Felsenfeld DP, Galbraith CG. Cell migration: regulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol. 1998;8:51–54. doi: 10.1016/s0962-8924(98)80005-6. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Whittard JD, Sakurai T, Cassella MR, Gazdoiu M, Felsenfeld DP. MAP kinase pathway-dependent phosphorylation of the L1-CAM ankyrin binding site regulates neuronal growth. Mol Biol Cell. 2006;17:2696–2706. doi: 10.1091/mbc.E06-01-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EV, Schaefer AW, Landreth G, Lemmon V. Casein kinase II phosphorylates the neural cell adhesion molecule L1. J Neurochem. 1996a;66:779–786. doi: 10.1046/j.1471-4159.1996.66020779.x. [DOI] [PubMed] [Google Scholar]
- Wong EV, Schaefer AW, Landreth G, Lemmon V. Involvement of p90rsk in neurite outgrowth mediated by the cell adhesion molecule L1. J Biol Chem. 1996b;271:18217–18223. doi: 10.1074/jbc.271.30.18217. [DOI] [PubMed] [Google Scholar]