

Abstract
The membrane proximal region (MPR) of HIV-1 gp41 is a desirable target for development of a vaccine that elicits neutralizing antibodies since the patient-derived monoclonal antibodies, 2F5 and 4E10, bind to the MPR and neutralize primary HIV isolates. The 2F5 and 4E10 antibodies cross-react with lipids and structural studies suggest that MPR immunogens may be presented in a membrane environment. We hypothesized that covalent attachment of lipid anchors would enhance the humoral immune response to MPR-derived peptides presented in liposomal bilayers. In a comparison of eight lipids conjugated to an extended 2F5 epitope peptide, a sterol, cholesterol hemisuccinate (CHEMS), was found to promote the strongest anti-peptide IgG titers (6.4 × 104) in sera of BALB/C mice. Two lipid anchors, palmitic acid and phosphatidylcholine, failed to elicit a detectable serum anti-peptide IgG response. Association with the liposomal vehicle contributed to the ability of a lipopeptide to elicit anti-peptide antibodies, but no other single factor, such as position of the lipid anchor, peptide helical content, lipopeptide partition coefficient, or presence of phosphate on the anchor clearly determined lipopeptide potency. Conjugation to CHEMS also rendered a 4E10 epitope peptide immunogenic (5.6 × 102 IgG titer in serum). Finally, attachment of CHEMS to a peptide spanning both the 2F5 and 4E10 epitopes elicited serum IgG antibodies that bound to each of the individual epitopes as well as to recombinant gp140. Further research into the mechanism of how structure influences the immune response to the MPR may lead to immunogens that could be useful in prime-boost regimens for focusing the immune response in an HIV vaccine.
Keywords: Adjuvant, IgG, liposomes, neutralizing antibodies
Introduction
Despite extensive research, attempts to elicit broadly neutralizing antibodies (bnAb) to HIV have not yet succeeded. Vaccines targeting the envelope glycoprotein must cope with sequence variation, extensive glycosylation and rapid conformational changes that expose target epitopes before a robust and effective vaccine can be available [1]. Rational design of immunogens to overcome these defenses has been guided by a small number of bnAb isolated from HIV-infected patients. Three of these bnAb (2F5, 4E10 and Z13) target the membrane proximal region (MPR) of gp41, a segment comprised of approximately 35 amino acids N terminal to the transmembrane domain (Figure 1). The MPR is a desirable vaccine target because it is well conserved across viral clades and is essential for virus-cell fusion [2, 3]. However, efforts to date have not succeeded in eliciting antibodies with the breadth or potency of patient-derived bnAb [4].
Figure 1. MPR peptides from the ectodomain of gp41.

Peptide immunogens were designed containing the N terminal region (N-MPR) or C terminal region (C-MPR) of the MPR. The sequences were comprised of the nominal epitopes of monoclonal antibodies 2F5 and 4E10 (underlined), respectively, with additional flanking sequences previously reported to improve binding [5, 6]. The C terminus was amended with a two residue linker and a lysine for on-resin lipid conjugation. A peptide spanning both the N and C terminal sequences (NC-MPR) contained both the 2F5 and the 4E10 epitopes with helix-promoting constraints at the C terminus [31]. ‘B’ indicates aminoisobutyric acid. FP – Fusion peptide; NHR – N heptad repeat; CHR – C heptad repeat; MPR – Membrane proximal region; TM – Transmembrane domain.
A lack of consensus regarding secondary structure has impeded MPR immunogen design. NMR, crystallography, and biophysical studies suggest that the MPR is an α-helix [4]. Crystal structures of 4E10 bound to its peptide epitope corroborate this helical conformation; however, structures of 2F5 bound to its peptide epitope show the peptide adopting an extended conformation with a β-turn [5, 6]. Regardless, attempts to present structurally constrained epitopes, either conjugated to carrier proteins or grafted on recombinant constructs, have not elicited neutralizing antibodies [7, 8]. Strategies to graft the antibody interacting surface onto a generic scaffold represent a promising approach, but neutralizing titers have not been published as of yet [9]. In addition to a lack of consensus regarding the epitope structure, the relatively weak antibody responses to the MPR may result in immune responses to recombinant envelope immunogens directed toward immunodominant regions, such as the gp120 variable loops, or toward determinants on gp41 that mask the MPR from antibody recognition [10, 11]. Law and coworkers grafted a helical 4E10 epitope peptide into the highly immunogenic V3 loop of gp120 but the construct did not elicit 4E10-specific antibodies [7]. Moreover, although MPR antibodies have been associated with neutralizing activity of patient sera in some cohort studies, they are relatively uncommon, further supporting the assertion that poor antibody responses to the epitope may be a barrier to the success of MPR-targeted vaccines [12]. Thus, the structure and immunogenicity of the MPR remain poorly defined, hampering vaccine efforts.
The lipid reactivities of bnAb 2F5 and 4E10 have been a topic of intense study. Both antibodies have unusually long, hydrophobic CDRH3 regions and cross-react with phospholipids and other autoantigens [13-15]. Moreover, biophysical models suggest that the MPR intercalates into the membrane in native virions [16]. These observations have led to suggestions that MPR immunogens may be presented optimally in a lipid bilayer environment. The majority of strategies to insert the epitopes in a lipid environment have involved chimeric viruses or liposomal formulations of recombinant constructs with transmembrane peptide domains [6, 17, 18]. Additionally, variations in lipid membrane composition appear to alter MPR peptide accessibility [19], and modulation of the peptide anchoring mechanism may exert similar effects.
We hypothesized that covalent attachment of lipid anchors would enhance the humoral immune response to MPR-derived peptides presented in liposomal bilayers. Three peptides were selected, corresponding to the 2F5 epitope (N-MPR), the 4E10 epitope (C-MPR) and a helically constrained peptide spanning both epitopes (NC-MPR; summarized in Figure 1). We systematically examined the effects of the lipid anchors on the humoral response in mice immunized with the lipopeptides in liposomes. Of the lipid anchors tested, cholesterol hemisuccinate (CHEMS) attached to the C terminus of the peptide induced the highest titer antibodies against the 2F5 epitope and also elicited antibodies against the 4E10 epitope. These CHEMS-peptide conjugates may be useful in prime-boost regimens for focusing the immune response or as analytical tools for probing antibody responses to the MPR.
Materials and Methods
Materials
Amino acid building blocks, resins and coupling agents were obtained from Novabiochem (Darmstadt, Germany), Anaspec (San Jose, CA) or ChemPep (Miami, FL). Cholesterol, dimyristoylphosphatidylcholine (DMPC), dimyristoylphosphatidylglycerol (DMPG), oxidized phosphatidylcholine (PC; #870601), brain sphingomyelin (SM; #860082), tetramyristoylcardiolipin (CL; #710332), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (#810158), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(carboxyfluorescein) (#810332) were obtained from Avanti Polar Lipids (Alabaster, AL). Dipalmitoylphosphatidylethanolamine (PE; #LP-R4-019) and dipalmitoylglycerol (DPG; #LP-R4-028) were obtained from Genzyme Pharmaceuticals (Cambridge, MA). Palmitic acid (PA; #P5585) and 5-cholenic acid-3β-ol (CHOL; #C2650) were obtained from Sigma-Aldrich (St. Louis, MO). Anhydrous solvents of 99.8% or greater purity were obtained from Acros Organics (Geel, Belgium). Monophosphoryl lipid A derived from Escherichia coli (MPL; #L6638) was obtained from Sigma-Aldrich. 2F5 and 4E10 monoclonal antibodies were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH from Dr. Hermann Katinger. Unless otherwise specified, all other reagents were obtained from Sigma-Aldrich.
Lipopeptide synthesis
Peptides were synthesized on NovaPEG resin in an automated solid phase synthesizer (ABI 433A, Applied Biosystems, Foster City, CA) with standard fluorenylmethyloxycarbonyl/o-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluorophosphate/n-hydroxybenzotriazole (FMOC/HBTU/HOBT) protocols. When appropriate, an orthogonally protected lysine (Fmoc-Lys(1-(4,4-Dimethyl-2,6-dioxo-cyclohexylidene)-3-methylbutyl)-OH; Fmoc-Lys(ivDde)-OH) was incorporated at the C terminus for on-resin conjugation of lipids or biotin. The N terminus was generally Boc-protected unless the peptide was intended for N terminal modification, in which case Fmoc protection was utilized. Removal of the ivDde group was accomplished by 3 × 15 minute treatments of the peptidyl resin with 2% hydrazine hydrate in dimethylformamide (DMF; 10 mL per g resin). The resin was washed in DMF (3 × 10 mL) and dichloromethane (DCM; 3 × 10 mL) and dried under vacuum.
Nomenclature and structures of lipids used in this study are summarized in Table 1. Lipid conjugation was accomplished via amidation of a carboxylated lipid and a deprotected lysine ε-amine at the C terminus. For N terminal conjugation, lipids were attached directly to the deprotected N terminus. Several of the lipids contained carboxyl groups. In the case of DPG, PE, SM, and CL, a carboxyl group was introduced via reaction of an available alcohol (DPG, SM, CL) or amine (PE) with succinic anhydride. For DPG-Suc, 1.8 mmol DPG was dissolved in 5 mL anhydrous DCM and combined with 3.6 mmol succinic anhydride in 10 mL anhydrous pyridine. The mixture was refluxed at 60 oC overnight. For PE-Suc, 1.5 mmol PE was combined with 3 mmol succinic anhydride and 6 mmol triethylamine in 50 mL anhydrous chloroform (CHCl3). The mixture was stirred at room temperature overnight. For CL-Suc, 80 μmol CL was combined with 400 μmol succinic anhydride and 400 μmol triethylamine in 5 mL anhydrous CHCl3. The mixture was refluxed at 60 °C overnight. For SM-Suc, 136 μmol SM was combined with 684 μmol succinic anhydride and 684 μmol triethylamine in 5 mL anhydrous CHCl3. The mixture was refluxed at 60 °C overnight. Reactions were continued to completion as monitored by thin layer chromatography (TLC) and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS; Voyager DE, Applied Biosystems, Foster City, CA) in para-nitroaniline matrix. Products were washed twice with 1M hydrochloric acid (HCl), dried over sodium sulfate and stored dry until use. Carboxylated lipids were obtained in greater than 90% yield. Molecular weights and TLC RF values were as follows: DPG-Suc, 668.19 Da, RF 0.29 in 20:1 DCM:acetone; PE-Suc, 790.02 Da, RF 0.19 in 65:25:4 CHCl3:MeOH:NH4OH; CL-Suc, 1335.90 Da, RF 0.76 in 65:25:4 CHCl3:MeOH:NH4OH; SM-Suc, 767.83 Da, 826.62 Da, 853.02 Da, 910.78 Da, RF 0.09-0.22 in 65:25:4 CHCl3:MeOH:NH4OH. SM-Suc gave a series of peaks because the starting material was a natural product with a distribution of aliphatic chain lengths.
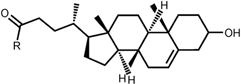
Table 1. Structures and nomenclature of lipids.
Abbreviations: PA – palmitic acid; DPG – dipalmitoylglycerol; PC – phosphatidylcholine; PE – phosphatidylethanolamine; SM – sphingomyelin; CL – cardiolipin; CHOL – cholenic acid; CHEMS – cholesteryl hemisuccinate.
| Lipid structure | Nomenclature |
|---|---|
| PA | |
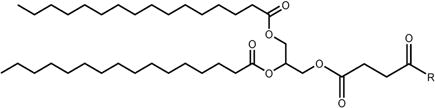 |
DPG |
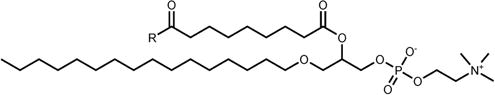 |
PC |
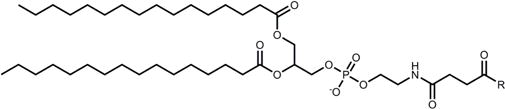 |
PE |
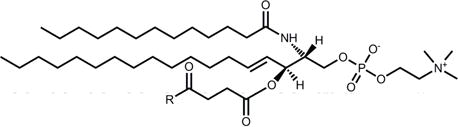 |
SM |
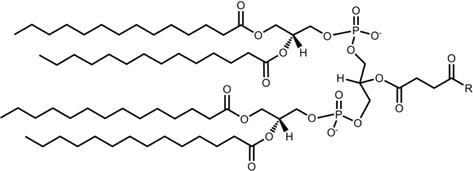 |
CL |
 |
CHOL |
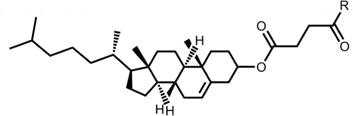 |
CHEMS |
Lipidation was accomplished by activation of 270 μmol carboxylated lipid with 270 μmol each of HBTU, HOBT and diisopropylethylamine (DIEA) in anhydrous DMF/DCM (DCM as needed for lipid solubilization) for 30 min at room temperature followed by addition of 67.5 μmol resin and continued reaction under argon for 24h at room temperature. Following the reaction, the resin was washed with DMF (4 × 10 mL) and DCM (4 × 10 mL) to remove unreacted lipids and dried under vacuum. Peptides were cleaved from the resin by treatment with trifluoroacetic acid containing 2.5% water, 2.5% ethanedithiol and 1% triisopropylsilane for 4 hours under argon. Cleaved peptides were precipitated into cold ethyl ether. The precipitate was pelleted by centrifugation at 3000 rpm (RT6000, Sorvall, Waltham, MA) and washed once with cold ethyl ether. The ether was poured off and the pellet was re-dissolved in methanol (MeOH), transferred to a round bottom flask, dried by rotary evaporation under reduced pressure and further dried under high vacuum. Lipopeptides were further separated from unconjugated peptides by reverse phase high pressure liquid chromatography (RP-HPLC; DX 500, Dionex, Sunnyvale, CA) on a semi-preparative C4 column (214TP510, Grace Vydac, Deerfield, IL) until unconjugated peptide was no longer detectable by MALDI-MS. Lipopeptide fractions were identified by MALDI-MS in 2,5-dihydroxybenzoic acid matrix, pooled and lyophilized. Stock lipopeptide solutions were prepared in MeOH or MeOH/CHCl3 and stored at -20 °C. Final yields were approximately 5-10%.
Biotinylated peptides were prepared for use in ELISA by an analogous method. Biotin was attached to the deprotected C terminal lysyl amine by activation of 500 μmol D-biotin with 500 μmol HBTU/HOBT/DIEA in 1.65 mL anhydrous 1:1 DMF/dimethylsulfoxide (DMSO) for 30 min followed by addition of resin and continued reaction under argon for 24h at room temperature. Following the reaction, the resin was washed with 1:1 DMF/DMSO (3 × 10 mL), DMF (3 × 10 mL) and DCM (3 × 10 mL) and dried under vacuum. Biotinylated peptides were cleaved and purified as described above. Biotin content was quantified by 4′-hydroxyazobenzene-2-carboxylic acid dye exclusion (Sigma #H2153) according to the manufacturer's instructions.
Liposome preparation
Lipopeptides were formulated in liposomes composed of 15:2:3:0.3 DMPC:DMPG:Cholesterol:MPL [20, 21]. Prior to use, glassware was rinsed with MeOH and CHCl3 and dried for at least 90 min at 150 °C to destroy pyrogens. Lipid solutions were combined in borosilicate glass tubes and dried to a thin film by rotary evaporation under reduced pressure. Films were further dried under high vacuum overnight. Lipids were hydrated in endotoxin-free PBS (UCSF Cell Culture Facility) by intermittent vortexing and bath sonication under argon for a brief period (approximately 15 seconds) to disperse the lipids into the buffer. Defined diameter vesicles were formed by extrusion 11 times through 400 nm polycarbonate membranes using a hand-held extruder (Avestin, Ottowa, Canada). To prevent contamination, the extruder was disassembled and thoroughly cleaned with MeOH and sterile PBS between samples. The final formulation contained 1 mg/mL lipopeptide and 0.5 mg/mL monophosphoryl lipid A in 20 mM carrier lipid. To determine the role of N-MPR-CHEMS dose in the anti-N-MPR IgG response, formulations with lower lipopeptide concentrations were prepared where indicated. Liposome preparations contained endotoxin levels less than 0.15 EU/mL when disrupted with 1.5% (v/v) C12E10 detergent and assayed for endotoxin activity by an LAL chromogenic endpoint assay [22] (QCL-1000, Lonza, Walkersville, MD).
Liposomal lipopeptide doses were confirmed after disruption of liposomes by the addition of 1.5% (v/v) C12E10 detergent followed by measurement of lipopeptide concentration by absorbance of aromatic side chains at 280 nm (Nanodrop, Thermo Scientific, Wilmington, DE). Molar extinction coefficients were calculated from the relative contributions of tryptophan, tyrosine, and cysteine residues according to the method of Pace [23], and lipopeptide doses were calculated from the molar extinction coefficient and the molecular weight. Doses were consistently found to be within 5% (2.5 μg) of the expected dose (50 μg). Representative dose measurements are reported in Table 4. Vesicle size was characterized by dynamic light scattering (Zetasizer 3000, Malvern, New Bedford, MA). Liposomes were stored at 4 °C under argon until use.
Table 4. Doses of selected formulations measured by A280.
| Formulation | Dose (μg) |
|---|---|
| N-MPR | 47.79 |
| N-MPR-DPG | 51.33 |
| N-MPR-PC | 49.18 |
| N-MPR-CL | 52.18 |
| N-MPR-CHOL | 49.81 |
| N-MPR-CHEMS | 50.74 |
Liposome association of lipopeptides
The extent of retention of MPR lipopeptides in liposomes was measured following sedimentation of liposomes by an ultracentrifugation method [24]. Essentially, liposomes were formulated with lipopeptides exactly as described above and centrifuged at 65,000 rpm (150,000g) for 45 min at 4 °C (TLA 100.2 rotor, Beckman TL-100, Beckman Coulter, Fullerton, CA). The liposome pellet was resuspended in PBS by pipetting and an aliquot was disrupted by mixing with 1.5% (v/v) C12E10 detergent. Lipopeptide concentration in the pellet and supernatant was quantified by the absorbance of aromatic side chains at 280 nm as described above. The percent of liposome association was defined as (Pellet)/(Pellet + Supernatant) × 100. When control liposomes containing rhodamine-labeled or fluorescein-labeled phosphatidylethanolamine were sedimented in this manner, greater than 90% of the fluorescence was consistently recovered in the liposome pellet (data not shown).
Circular dichroism
Liposomal lipopeptide samples were prepared as described above with the following modifications. Stock liposome solutions containing 5 mM carrier lipid and 500 μM lipopeptide were prepared in 10 mM phosphate, pH 7.4. To minimize light scattering, liposomes were prepared by bath sonication under argon until a size of less than 100 nm was obtained. For analysis, samples were diluted to 5 μM lipopeptide in 10 mM phosphate buffer containing 1 mM carrier lipid. Spectra were obtained with a J-715 spectrapolarimeter (Jasco, Easton, MD) and data were processed using Jasco software. Data were acquired in continuous scanning mode with a pathlength of 1 cm, 0.1 nm interval and scan speed of 1 nm/s. Each spectrum represents an average of two scans. A background spectrum of “empty” liposomes in buffer was subtracted from each sample spectrum. Percent helicity was estimated from θ222 according to the method of Taylor and Kaiser [25].
Tryptophan fluorescence
Lipopeptide membrane partitioning was characterized by measurement of tryptophan fluorescence intensity as described with modifications [26]. Briefly, DMPC:DMPG:Cholesterol liposomes were prepared in phosphate-buffered saline as described above. Lipopeptide stock solutions were prepared in MeOH and 12 nmol lipopeptide was injected via glass syringe (Hamilton, Reno, NV) into 1.2 mL buffer containing diluted liposomes (10-150 μM lipid). The samples were mixed by inversion and allowed to equilibrate in the dark at room temperature overnight. Fluorescence emission spectra were obtained on a SPEX Fluorolog spectrophotometer (Horiba Jobin Yvon, Edison, NJ) with 1 cm pathlength, 2.5 mm excitation slit, 5.0 mm emission slit, 1 s integration time and 1 nm interval. For each liposome concentration, a background spectrum of “empty” liposomes in buffer was subtracted from the sample spectrum. Fluorescence intensity was determined by integration of the tryptophan fluorescence peak and data were normalized to the highest intensity in each sample series. Partition coefficients were calculated from the double reciprocal plot of normalized fluorescence intensity versus lipid concentration, according to the equation F = (F0*L*Kp)/(55.6 + Kp*L) [27].
Animal immunizations
All animal procedures were conducted in accordance with the policies and approval of the UCSF Institutional Animal Care and Use Committee. 8 week-old female BALB/C mice (Jackson Laboratories, Bar Harbor, ME) were housed in a UCSF specific pathogen-free barrier facility. Animals received subcutaneous immunizations in alternating hind hocks on Days 0 and 14 as described [28]. Each injection contained 50 μg lipopeptide, 25 μg MPL and 1 μmol lipid vehicle in 50 μL sterile phosphate-buffered saline. All formulations contained MPL and 50 μg lipopeptide unless otherwise specified. On Day 28 blood was collected from the submandibular vein for characterization of antibody responses. Cells were removed by centrifugation at 14,000 rpm for 15 min (5415C, Eppendorf, Westbury, NY) and sera were stored at -80 °C until use.
ELISA
ELISAs were developed to quantify binding of immune sera to peptides, lipids, and recombinant gp140. Peptide ELISAs were conducted using MPR peptides biotinylated as described above and captured on 96 well streptavidin-coated plates (#15120, Pierce, Rockford, IL). Assays were performed according to the manufacturer's instructions with modifications. Biotinylated peptides were added to wells in PBS containing 0.1% Tween-20 (PBS-T) and incubated for 2 hr at 37 °C. Following a wash step, sera were serially diluted in PBS containing 0.1% casein (C7078, Sigma-Aldrich) (PBS-C), added to wells and incubated for 30 min at 37 °C. After reconstitution, horseradish peroxidase-conjugated secondary antibodies (IgG, IgG1, IgG2a; Jackson Immunoresearch, West Grove, PA) were diluted 1:1 in glycerol for long-term storage at -20 °C and further diluted 1:1000 in PBS-C immediately prior to use. Following a wash step, secondary antibodies were added to wells and incubated for 30 min at 37 °C. Following a final wash step, a tetramethylbenzidine substrate solution (#T0440, Sigma-Aldrich) was added to wells and incubated for 30 min at room temperature. The reaction was stopped with 0.5M H2SO4 and the yellow product was monitored at 450 nm (Optimax, Molecular Devices, Sunnyvale, CA). All incubations were done in 100 μL volumes and wells were washed 6 times with PBS-T between each step. Titer was defined as the reciprocal dilution of immune sera yielding an optical density twice that of 1:200 preimmune sera after subtraction of background wells lacking serum. IgG1/IgG2a ratios were calculated as an average of optical density quotients measured at 3 dilutions after subtraction of background values. All samples were assayed in duplicate.
Lipid ELISAs were performed as described with modifications [29]. Lipids were diluted to 0.2 mg/mL in EtOH and 50 μL per well were added to flat-bottomed untreated polystyrene plates (Fisher) and allowed to dry overnight. Plates were blocked with 0.5% casein for 2 hr. After a wash step, immune sera were diluted 1:200 in 10% fetal bovine serum in PBS and incubated in wells for 1 hr. Wells were washed and peroxidase-conjugated anti-mouse IgG was diluted 1:1000 in PBS-C and added to wells for 1 hr. Following a final wash step, a tetramethylbenzidine substrate solution was added to wells and incubated for 30 min at room temperature. The reaction was stopped with 0.5M H2SO4 and the yellow product was monitored at 450 nm (Optimax). All incubations were done in 100 μL volumes at room temperature and wells were washed 6 times with PBS between each step.
Recombinant gp140 ELISAs were performed follows: Ba-1 gp140 (Immune Technology Corp, New York, NY) was diluted to 5 μg/mL in 50 mM sodium carbonate, pH 9.6 and 100 μL per well were added to flat-bottomed high capacity immunoassay plates (Costar). Plates were sealed with parafilm and incubated at 4 °C overnight. Plates were blocked with 0.5% casein for 2 hr. After a wash step, immune sera were diluted 1:50 in PBS-C and incubated in wells for 1 hr. Wells were washed and peroxidase-conjugated anti-mouse IgG was diluted 1:1000 in PBS-C and added to wells for 30 min. Following a final wash step, a tetramethylbenzidine substrate solution was added to wells and incubated for 30 min at room temperature. The reaction was stopped with 0.5M H2SO4 and the yellow product was monitored at 450 nm (Optimax). All incubations were done in 100 μL volumes at 37 °C and wells were washed 6 times with PBS-T between each step.
Statistical analysis
Statistical significance was assessed by analysis of variance and two-tailed Student's t test. Differences were considered significant if they exhibited p values < 0.05 in the Student's t test. Data analyses were performed using Microsoft Excel and SigmaPlot.
Results
Preparation of lipopeptides and liposomes
This study sought to address the role of lipid structure in the humoral immune response to MPR lipopeptides formulated in liposomes. Three peptides were selected for lipid modification, corresponding to the N terminal sequence (N-MPR; 2F5 epitope), the C terminal sequence (C-MPR; 4E10 epitope) and an extended peptide spanning the N and C termini (NC-MPR; summarized in Figure 1). The sequences of N-MPR and C-MPR included flanking residues that were found to maximize binding affinities for their respective antibodies in vitro [6, 30]. Two helix-promoting isobutyric acid residues were incorporated into NC-MPR, as previously implemented in the design of a helically constrained 4E10 epitope peptide [31]. The N terminus of NC-MPR was extended to include the full 2F5 epitope. An orthogonally protected lysine was included for lipid conjugation at the C terminus to mimic the native structure, in which the C terminus is anchored to the membrane.
Lipid anchors were selected to represent several general lipid types: fatty acids, diacylglycerols, phospholipids and sterols (Table 1). Additionally, some are implicated in cross-reactivity with 4E10 and 2F5 (cardiolipin) or in virus-cell fusion (virion lipid phosphatidylethanolamine; raft lipids sphingomyelin and cholesterol) [14, 32]. Consideration was also given to lipid anchors that may facilitate elicitation of antibodies binding to both peptide and lipid moieties. Specifically, lipids lacking a phosphate (palmitic acid and diacylglycerol) were selected for comparison to phosphate-containing lipids because the phosphate and head group moieties are important in recognition by anti-phospholipid antibodies [33]. Cholenic acid (CHOL) was chosen in addition to cholesterol hemisuccinate (CHEMS) due to work indicating that the 3β-hydroxyl is a primary moiety responsible for recognition of cholesterol by anti-cholesterol antibodies [34]. Additional lipids, such as galatosyl ceramide, which may serve as a receptor for MPR binding, would be of interest in future studies but were not included here [35].
For those lipids lacking a carboxyl group, one was introduced by reaction with succinic anhydride (TLC and MW data in Methods). For peptide modification, the on-resin lipidation strategy allowed complete removal of unreacted lipid via extensive washing of the resin prior to cleavage. The remaining contaminant, unreacted peptide, was removed by RP-HPLC. Molecular weights of lipid- and biotin-modified peptides are reported in Table 2. Modified peptides were obtained in approximately 5-10% yield; steric hinderance in modification of the C terminal lysyl ε-amine and loss upon RP-HPLC purification may have contributed to the relatively poor yield. Human monoclonal antibodies 2F5 and 4E10 bound strongly to biotinylated MPR peptides containing their epitopes (N-MPR and C-MPR, respectively) by ELISA (Figure 2). The cause for weak binding of 2F5 to C-MPR is uncertain but may be attributed to partial overlap in the peptide sequences (Figure 1). Regardless, sera of mice immunized with N-MPR lipopeptides did not bind to C-MPR by ELISA and vice versa (data not shown). Liposomal formulation of MPR lipopeptides resulted in vesicles approximately 175-250 nm in diameter (Table 3). Addition of peptide or lipopeptide did not appreciably affect vesicle size, with the exception of N-MPR-PE liposomes, which were slightly smaller than the others.
Table 2. Molecular weights of lipid- and biotin-modified peptides determined by MALDI.
| Name | MW (exp) | MW (obs) |
|---|---|---|
| N-MPR-PA | 2467.9 | 2468.0 |
| N-MPR-DPG | 2880.0 | 2903.7 (Na+) |
| N-MPR-PC | 2863.4 | 2865.1 |
| N-MPR-PE | 3003.5 | 3002.7 |
| N-MPR-SM | 2979.2, 3038.0, 3064.4* | 3049.3, 3146.0, 3186.33* |
| N-MPR-CL | 3587.2 | 3587.2 |
| N-MPR-CHOL | 2586.1 | 2588.2 |
| N-MPR-CHEMS | 2698.3 | 2718.2 (Na+) |
| C-MPR-PA | 2306.8 | 2326.3 (Na+) |
| C-MPR-DPG | 2749.4 | 2748.0 |
| C-MPR-PC | 2702.3 | 2703.4 |
| C-MPR-CHOL | 2425.0 | 2427.5 |
| C-MPR-CHEMS | 2537.1 | 2539.6 |
| NC-MPR-DPG | 4859.4 | 4861.7 |
| NC-MPR-CHEMS | 4677.6 | 4696.8 (Na+) |
| N-MPR-biotin | 2455.6 | 2455.0 |
| C-MPR-biotin | 2294.7 | 2296.8 |
| NC-MPR-biotin | 4435.2 | 4433.3 |
Figure 2. Binding of human monoclonal antibodies 2F5 and 4E10 to MPR peptides attached to the surface of ELISA plates.

Panel A: 4E10 mAb bound to biotinylated peptides containing the ‘NFWDIT’ epitope (C-MPR and NC-MPR) but not a peptide containing only the ‘ELDKWA’ epitope (N-MPR). Data representing 4E10 binding to ‘N-MPR’ and ‘None’ overlap in the figure. Panel B: 2F5 mAb bound to biotinylated peptides containing the ‘ELDKWA’ epitope, with very weak binding to the ‘NWFDIT’ peptide. Data are representative of two independent experiments.
Table 3. Vesicle size and lipopeptide association of lipopeptide liposomes.
Vesicle size was determined by dynamic light scattering and the fraction of lipopeptide in the liposome pellet following ultracentrifugation was measured by A280 as described in the Methods. ND – not determined.
| Formulation | Vesicle Diameter (nm) | Standard Deviation (nm) | %Association |
|---|---|---|---|
| Empty | 214.9 | 2.2 | 0.00 |
| N-MPR | 185.6 | 3.7 | 24.18 |
| N-MPR-PA | 175.5 | 9.1 | ND |
| N-MPR-DPG | 218.5 | 4.4 | 100.00 |
| N-MPR-PC | 212.6 | 17.1 | 81.89 |
| N-MPR-PE | 118.5 | 9.6 | ND |
| N-MPR-SM | 193.7 | 2.5 | ND |
| N-MPR-CL | 241.1 | 6.5 | 92.07 |
| N-MPR-CHOL | 153.2 | 8.9 | 94.90 |
| N-MPR-CHEMS | 192.9 | 6.8 | 99.71 |
| N-MPR-CHEMS (no liposome) | ND | ND | 0.00 |
| C-MPR | 222.4 | 2.5 | 98.97 |
| C-MPR-PA | 213.4 | 1.3 | ND |
| C-MPR-DPG | 216.6 | 2.1 | ND |
| C-MPR-PC | 246.2 | 1.8 | ND |
| C-MPR-CHOL | 211.7 | 0.2 | ND |
| C-MPR-CHEMS | 220.7 | 9.0 | 100.00 |
| NC-MPR | 240.0 | 2.1 | 100.00 |
| NC-MPR-DPG | 235.7 | 1.1 | ND |
| NC-MPR-CHEMS | 249.5 | 5.7 | 100.00 |
| CHEMS-NC-MPR | 288.7 | 13.5 | 97.42 |
| CHEMS-NC-MPR-CHEMS | 225.9 | 9.6 | 95.24 |
Sedimentation of liposomes formulated with N-MPR lipopeptides resulted in 80-100% retention of lipopeptides in the liposome pellet (Table 3). In contrast, when a formulation containing unconjugated peptide was sedimented, only 24% of the peptide was associated with the pellet; the majority remained in the supernatant. C-MPR and NC-MPR peptides were fully retained in the liposome pellet regardless of lipid conjugation. Importantly, when a suspension of N-MPR-CHEMS without liposomes was sedimented under the same conditions, the lipopeptide absorbance remained entirely in the supernatant. This indicates that lipopeptide detected in the pellet is associated with the liposome and is not in independent micelles.
The attached lipid moiety alters N-MPR lipopeptide structure and behavior in membrane vesicles
When formulated in liposomes, N-MPR secondary structure was greatly altered by the attached lipid moiety (Figure 3a). Whereas attachment of CHEMS to N-MPR resulted in a modest increase in helicity (26.5% versus 20.7%), attachment of DPG substantially increased helical content (47.8% versus 20.7%). In contrast, attachment of CHEMS to NC-MPR only modestly affected its already helical conformation (Figure 3b). For NC-MPR, the data suggest a trend in which C terminal attachment promotes helicity (8% and 5% respective increases when comparing ‘C Terminus’ versus ‘Unconjugated’ and ‘Both Termini’ versus ‘N Terminus’), whereas N terminal attachment decreases helicity (2% and 5% respective decreases when comparing ‘N Terminus versus ‘Unconjugated’ and ‘Both Termini’ versus ‘C Terminus’). The NC-MPR spectra are in agreement with those reported for 4E10 epitope peptides with nearly identical C terminal helix restraints [31]. By comparison, the lower overall helicity of NC-MPR may be attributed to the contribution of the extended N terminal segment not present in the peptide synthesized by Cardoso et al.
Figure 3. Effect of lipid and attachment site on the circular dichroism spectra of MPR lipopeptides.

Panel A: Attachment of DPG to N-MPR promoted helix formation more strongly than did attachment of CHEMS to N-MPR. Panel B: NC-MPR-CHEMS conjugates exhibited helical character regardless of CHEMS attachment site. Data are normalized for concentration and peptide length. Inset: Percent helicity estimated by the method of Taylor and Kaiser [25]. Spectra represent averages of two scans.
Tryptophan fluorescence experiments revealed that the attached lipid moiety also affects partitioning of N-MPR into lipid bilayers (Figure 4). Both PA and CHEMS conjugates exhibited incremental differences in tryptophan fluorescence as a function of liposome concentration. This indicates that as the concentration of liposomes is increased, additional lipopeptides partition into the membrane. However, tryptophan fluorescence of N-MPR-DPG was unaffected by increasing lipid concentration over the range measured. The Kp of N-MPR-DPG was estimated to be at least an order of magnitude greater than that of N-MPR-PA or N-MPR-CHEMS (5.84 × 108 versus 2.01 × 107 and 1.95 × 107, respectively). This observation suggests that N-MPR-DPG partitions more strongly into bilayer membranes than the other conjugates. Alternatively, the possibility that DPG promotes self-aggregation cannot be excluded. As hydrophobic bilayer environments are known to promote helicity of peptides [36], the increased helicity of N-MPR-DPG (Figure 3a) relative to N-MPR-CHEMS may correspond to increased membrane partitioning. Taken together, these data indicate that the attached lipid alters both the peptide's secondary structure and its behavior in bilayer vesicles.
Figure 4. Partitioning of N-MPR lipopeptides into DMPC:DMPG:Cholesterol vesicles.

PA and CHEMS conjugates exhibited comparable partitioning, whereas DPG conjugates appeared maximally partitioned at the lowest lipid concentrations measured. Kp values for PA and CHEMS conjugates are calculated as described in Methods. The invariant fluorescence intensity of N-MPR-DPG yielded a poor curve fit and the calculated Kp was estimated as a lower bound of the true value. Data are representative of two independent experiments.
CHEMS conjugation elicits the greatest anti-peptide immune response to N-MPR and induces an anti-peptide response to C-MPR in BALB/C mice
An immunization dose of 50 μg was selected after a review of the literature concerning peptide and lipopeptide antigens revealed a dose range of 5 μg to 130 μg for mouse immunization studies [20, 37]. Additionally, the relatively high dose of 50 μg was chosen as a result of the widely reported poor antibody response to the MPR following immunization or infection [4]. When formulated in liposomes and administered to BALB/C mice, N-MPR lipid conjugates exhibited considerable differences in their ability to induce anti-peptide antibodies (Figure 5). Sterols and lipids containing two or more acyl chains generally elicited anti-peptide titers in the range of 104 to 105. These lipopeptides elicited balanced IgG1/IgG2a responses, suggesting a balanced T helper response, with a slight preponderance of IgG1. Anti-peptide IgA responses were not detected in serum (data not shown). Unconjugated peptide formulated in liposomes induced a greater anti-peptide response (detected in 2 of 5 mice) than either palmitic acid or PC conjugates, both of which failed to elicit a detectable response despite the presence of MPL in the formulation. N-MPR-PC, in which the peptide was attached to the distal end of an acyl chain, may have functioned more as a single chain due to the distribution of polar groups (peptide and head group) throughout the molecule.
Figure 5. Anti-peptide serum IgG titer, IgG1/IgG2a ratio and lipid reactivity of sera of mice immunized with N-MPR lipopeptides.

Panel A: N-MPR lipopeptides elicited serum anti-peptide IgG titers in the range of 104 to 105. Antibodies were detected in 2 of 5 mice that received liposomes containing peptide not conjugated to lipid (‘Unconj’). In all other responding groups, antibodies were detected in every mouse. Antibodies were not detected in mice receiving PC or PA conjugates. ‘Empty’ denotes animals that received liposomes lacking a peptide immunogen. All formulations contained MPL. ‘Control’ animals received no injection. Titers are expressed as geometric means. Panel B: Serum IgG responses exhibited a balanced IgG1/IgG2a ratio with a slight preponderance of IgG1. Panel C: Immune sera contained detectable antibodies to cholesterol but not cardiolipin or phosphatidylglycerol. Each group consisted of at least n = 5 animals. Error bars represent standard deviations. * p = 0.046 vs. SM, 0.012 vs. PE, 0.008 vs. CL, 0.004 vs. CHOL, 0.00002 vs. DPG, 0.007 vs. Unconj.
To assess the contribution of N-MPR-CHEMS dose in the anti-N-MPR antibody response, a range of doses from 5 μg to 50 μg was administered to BALB/C mice (Figure 6). The results indicated that N-MPR-CHEMS could elicit anti-N-MPR serum IgG titers greater than 3×103 at lipopeptide doses as low as 5 μg. Mice that received liposomes containing 50 μg N-MPR-CHEMS but lacking monophosphoryl lipid a (MPL) did not generate a detectable anti-N-MPR serum IgG response, indicating that MPL is required for the adjuvant activity of MPR lipopeptides. Additionally, conjugation of C-MPR to CHEMS, but not DPG or CHOL, elicited a weak serum IgG response against the C-MPR peptide (GMT 5.6 × 102, not shown).
Figure 6. Dose response of anti-N-MPR serum IgG titer following N-MPR-CHEMS immunization.

Mice exhibited anti-N-MPR IgG in sera following immunization with 5, 15, 25 or 50 μg N-MPR-CHEMS. The formulation was otherwise constant as described in the Methods. A group which received 50 μg of N-MPR-CHEMS in liposomes lacking MPL did not generate a detectable antibody response (not shown). Each group consisted of at least n = 5 animals. Error bar represents the standard deviation. * p = 0.0003 vs. 15 μg, 0.0002 vs. 5 μg.
Lipid reactivity of murine antisera was assayed because cross-reactivity of 2F5 and 4E10 with anionic phospholipids is thought to be important in their ability to neutralize HIV [38]. The lipopeptide formulations did not elicit antibodies against either cardiolipin or phosphatidylglycerol but did evoke a weak response against cholesterol. No difference in anti-cholesterol antibodies was detected between sera of mice that received the CHOL lipopeptide, in which the 3β-hydroxyl is available, and the CHEMS lipopeptide, in which the 3β-hydroxyl is masked. Cholesterol antibodies were likely generated by the unmodified cholesterol in the carrier formulation in addition to the lipopeptide itself. These assays were repeated with Tris-buffered saline to address concerns that the presence of soluble phosphate in the assay buffer may have inhibited anti-phospholipid antibody binding. However, phospholipid reactivity was also not detected in these assays (data not shown).
Alteration of the CHEMS attachment site modulates the anti-peptide humoral response to the N terminus but not the C terminus of NC-MPR
To further probe the utility of CHEMS conjugation for promoting antibody responses to the MPR, NC-MPR lipopeptides were synthesized in which CHEMS was attached to the C terminus, the N terminus, or both (Figure 7a). Sera of mice immunized with these conjugates were assayed for binding to the full sequence (NC-MPR), the N terminus (N-MPR; 2F5 epitope), and the C terminus (C-MPR; 4E10 epitope). All three molecules elicited antibodies that bound to the individual 2F5 and 4E10 epitopes. Notably, the NC-MPR-CHEMS C terminal conjugate elicited a stronger response to N-MPR than to itself. The other two conjugates elicited significantly lower antibodies to N-MPR (p < 0.004), suggesting that attachment of CHEMS to the N terminus diminished the antibody response to the N terminal segment of the peptide. Reduction in antibodies to the N terminus did not appear to arise from reduced interaction with the liposomal formulation (Table 3). However, conjugation to the C terminus exerted no detectable effect on the antibody response to the C terminal segment. Interestingly, a control unconjugated NC-MPR peptide formulated in liposomes elicited high titers to the N terminus but no detectable responses to the C terminus. None of the conjugates elicited detectable antibodies to cardiolipin or phosphatidylglycerol (data not shown).
Figure 7. Anti-peptide IgG titers in sera of mice immunized with NC-MPR-CHEMS lipopeptides with CHEMS conjugated at the N terminus, C terminus, or both termini assayed for binding to N-MPR, C-MPR or NC-MPR by ELISA.

Panel A, CHEMS attachment site exerted a dramatic effect on serum anti-peptide IgG responses to NC-MPR lipopeptides. Attachment to the N terminus dramatically reduced antibodies directed against the N terminal peptide (N-MPR), whereas attachment site did not significantly alter levels of antibody directed against the C terminal peptide (C-MPR). The attachment site of CHEMS to the immunizing NC-MPR lipopeptide is indicated in the figure inset, and the ELISA antigen is indicated on the horizontal axis. Each group consisted of at least n = 5 animals. Error bars represent standard deviations. Panel B, IgG in sera of individual mice immunized with NC-MPR lipopeptides bound weakly to recombinant gp140. Each bar represents an individual animal. * p = 0.003 vs. N Terminus, 0.004 vs. Both Termini. # p = 0.044 vs. N Terminus, 0.010 vs. Both Termini. ** p = 0.011 vs. N terminus, 0.013 vs. Both Termini. ## p = 0.026 vs. Both Termini.
Finally, we sought to determine if these conjugates could elicit antibodies that bind to recombinant gp140 (Figure 7b). The gp140 construct used (Clade B, Strain Ba-1) differed from the MPR consensus sequence by only one residue (N677E). In control experiments, bnAb 2F5 and bnAb 4E10 bound strongly to this gp140 at 1 μg/mL (data not shown). Several immune sera bound weakly to gp140, but only at a very low dilution (1:50), suggesting that the majority of antibodies recognize structures other than that of the native protein. Although NC-MPR-DPG elicited greater reactivity to gp140 than NC-MPR-CHEMS (3/5 responders versus 1/5 responders), the reactivity is low and it is unclear if this difference is meaningful. Since the sequence of interest is positioned at the end of the C terminus of the recombinant construct, there was concern that adsorption on the ELISA plate may alter the structure and interfere with binding. However, binding was not stronger when the recombinant construct was attached to hexahistidine-binding plates via a hexahistidine tag (data not shown).
Discussion
The discovery of broadly neutralizing monoclonal antibodies reactive with the MPR region of gp41 from patient-derived cells raised the hope for an HIV vaccine against the epitopes recognized by these antibodies [1, 4, 8, 38]. Numerous studies of MPR-specific neutralizing antibodies suggest that presentation of MPR immunogens in a membrane environment could facilitate elicitation of neutralizing responses [4]. However, recombinant viruses and MPR-transmembrane fusion constructs in lipid vesicles have not elicited high titer neutralizing antibodies [6, 17, 18, 39, 40].
We hypothesized that covalent attachment of lipid anchors to MPR segments would improve upon these approaches by increasing anti-peptide antibody titers, altering epitope structure within the membrane, or eliciting neutralizing antibodies. We compared sterols, fatty acids and phospholipids for promoting humoral responses to covalently attached antigens. The key finding of this study is that the structure of the lipid anchor exerts significant influence on the anti-peptide serum IgG titer. Unexpectedly, cholesterol hemisuccinate (CHEMS) elicited significantly greater anti-peptide serum IgG responses than all other lipid anchors, including cholenic acid (CHOL), a similar molecule (geometric mean titers of 6.4 × 104 and 1.8 × 104, respectively; p = 0.004; Figure 5). Although the differences in anti-N-MPR serum IgG titers were significant, differences were relatively small amongst the more potent anchors. Furthermore, N-MPR-CHEMS elicited anti-N-MPR serum IgG titers greater than 3×103 at lipopeptide doses as low as 5 μg (Figure 6). Conjugation to CHEMS also rendered C-MPR immunogenic, and the two lipid-anchored NC-MPR peptides tested elicited antibodies that bound weakly to gp140 by ELISA. Thus, these lipids augment the toolbox available to HIV-1 vaccine researchers for probing antibody responses to the MPR and designing MPR-targeted vaccines.
Lipid conjugation appears to be most useful for eliciting antibody responses to hydrophilic peptides, which would not otherwise be retained in liposomes (e.g. N-MPR), and weakly immunogenic hydrophobic peptides (e.g. C-MPR), which are retained in liposomes but do not generate an antibody response. Unconjugated NC-MPR peptide, which is both hydrophobic and strongly immunogenic, elicited serum IgG titers against the N terminus of the MPR that were comparable to those generated by the NC-MPR-CHEMS conjugate (Figure 7). Thus, it is clear that improved peptide association with the lipid bilayer contributes to the ability of lipid conjugation to promote antibody responses to liposomal peptides, as reported by others [41]. However, two key observations support the conclusion that the adjuvant effects of CHEMS conjugation does not simply arise from improved retention in the lipid vehicle. First, considerable differences in the anti-N-MPR antibody response were observed amongst N-MPR lipopeptides that were retained in liposomes to an equivalent extent (Figure 5 and Table 3). Amongst N-MPR lipopeptides, no single factor, such as structure of the lipid anchor, peptide helical content, lipopeptide partition coefficient, or presence of phosphate on the anchor determined the ability of a lipopeptide to elicit anti-peptide antibodies. Second, CHEMS conjugation was required to elicit anti-C-MPR antibodies when either C-MPR or NC-MPR was the immunizing antigen (Figure 7 and Results). Since neither of these peptides requires a lipid anchor to be retained in liposomes (Table 3), attachment of CHEMS appears to promote antibody responses in these cases.
It is unclear to us why a sterol-anchored peptide would elicit a greater antibody response than a peptide anchored by aliphatic chains. The mechanism does not appear to arise from induced changes in secondary structure; N-MPR-CHEMS, which differed little from free N-MPR peptide by circular dichroism, elicited nearly an order of magnitude higher geometric mean titer (GMT) than N-MPR-DPG (6.4 × 104 and 6.7 × 103, respectively; Figure 5), which exhibited considerably greater helical content (26.5% and 47.8%, respectively; Figure 3). Membrane partitioning does not explain the disparity in anti-peptide titers either, as N-MPR-DPG partitioned much more strongly into liposomes than N-MPR-CHEMS (Kp > 5.84 × 108 and Kp = 1.95 × 107; Figure 4). Moreover, although N-MPR-CHEMS and N-MPR-PA exhibited very similar partitioning behavior, N-MPR-PA failed to elicit any detectable peptide antibodies (Figure 5). Thus, the adjuvant activity of CHEMS conjugates arises from some other mechanism. In an earlier study, when a peptide derived from the V3 loop of gp120 was acylated and incorporated into a liposomal vaccine, peptide exposure on the surface of liposomes was critical in elicitation of anti-V3 antibodies [20]. In light of this report, CHEMS conjugates may adopt a more highly exposed surface structure than CHOL, DPG, or other less immunogenic lipopeptides. However, efforts to quantitate liposome surface accessibility of lipid-modified MPR peptides are complicated by the ability of the 2F5 and 4E10 antibodies to intercalate into the membrane and “extract” their epitopes [16]. Alternatively, the lipid moiety may alter the processing of associated T helper epitopes or facilitate membrane transfer to cells that provide more efficient presentation to B lymphocytes [42]. The lipid composition of the carrier vesicle may also be important, as several groups have shown that the MPR structure in membranes is modulated by membrane composition [19, 43]. Regardless, further studies with additional peptide antigens will be needed to determine if the strategies presented here are broadly applicable or can only be applied to MPR antigens.
The weak response of immune sera to the native protein (Figure 7b) likely results from the ability of a peptide antigen to adopt a multitude of conformations in vivo and thus elicit antibodies recognizing multiple structures. Of those, only a portion will correspond to the native protein structure, even when structural constraints are imposed (e.g. lipid conjugation or addition of specific amino acids) [4]. This remains a challenge, and further work is needed to achieve the correct structure. Our strategy is analogous to that reported by Giannecchini and colleagues, in which octadecanoic acid was attached to the C terminus of MPR of feline immunodeficiency virus [44]. However, this immunogen elicited only weak anti-peptide antibodies (ELISA OD < 1.0 at 1:100 serum dilution) in cats. Thus, there is a need for immunogens that not only target the appropriate antigenic structure, but also elicit high titer antibodies. Coutant and coworkers also recently derivatized an MPR peptide with phosphatidylethanolamine to probe its physiological structure within membranes [43], but did not report antibody titers. Our findings suggest that lipid-anchored MPR peptides elicit substantial serum IgG titers in mice; the titers are an order of magnitude higher than those reported by Lenz and colleagues in BALB/C mice immunized with liposome-anchored trimeric gp41, although their preparations did not contain the potent adjuvant MPL [18].
The use of liposomes containing monophosphoryl lipid A (MPL) for induction of antibody and cytotoxic T lymphocyte responses against liposome-associated peptides and proteins has been pioneered by Alving and colleagues [20, 33, 41, 45, 46]. Indeed, in the present study, MPL was required to generate a detectable anti-N-MPR serum IgG response following immunization with liposomal N-MPR-CHEMS. Adjuvant mechanisms attributed to liposomes containing MPL include enhanced uptake, processing and presentation by antigen presenting cells [45, 47], prolonged persistence at the injection site and activation of innate immunity through binding to Toll-like receptor 4 [41, 45, 47]. Moreover, several studies have demonstrated that covalent attachment of peptides to liposomes enhances humoral immune responses to liposome-associated peptides and proteins [20, 48, 49]. As compared to non-covalent encapsulation, White and colleagues demonstrated increased antibody responses to a peptide derived from the V3 loop of gp120 when the peptide was acylated at the N terminus prior to liposome formulation or attached via a reversible disulfide bond to liposomes containing a thiolated cholesterol derivative [20].
Liposomal MPL is generally regarded as a T helper type 1 (Th1) adjuvant in mice, although exceptions have been reported [50]. In this case, the serum anti-N-MPR IgG1/IgG2a ratio indicated a balanced T helper response (Figure 5b). The slight excess of IgG1 suggests a Th2 component to the response [51]. Most broadly neutralizing human antibodies identified to date have been of the IgG1 and IgG3 subclasses, which in humans arise from a Th1 response [1]. Thus, additional adjuvants that direct Th1 responses may improve the ability of MPR lipopeptides to elicit neutralizing antibodies.
Liposomes adjuvanted with MPL have also been used to elicit anti-lipid antibodies of diverse specificities [33]. A murine monoclonal antibody to phosphatidylinositol phosphate with no known HIV-1 binding specificity has also been shown to neutralize primary isolates, suggesting that membrane binding alone may be sufficient for neutralization [52]. The failure to elicit anti-phospholipid antibodies in the present study is at odds with a recent report in which immunization of BALB/C mice with a liposome-associated peptide adjuvanted by MPL elicited dual specificity, low titer (O.D. ∼1.0 at 1:00 serum dilution) antibodies that recognized both peptide and lipid determinants [46, 53]. In these studies the MPR sequence was modified with a universal T helper epitope from tetanus toxin but did not contain a covalent lipid. As induction of anti-lipid antibodies by liposomes is affected by a number of factors, including formulation and injection route, modulation of these parameters in future studies may enable MPR lipopeptides to elicit lipid cross-reactive antibodies [54].
The findings reported here may prove useful in studies of the MPR as a target for design of immunogens that elicit neutralizing antibodies. Importantly, the data bolster the assertion that the immunogenicity of the MPR arises predominantly from the N terminal portion. This fact was borne out through immunization studies with peptides containing only a single bnAb epitope (N-MPR and C-MPR) or both epitopes (NC-MPR). N-MPR-CHEMS elicited an anti-N-MPR GMT of 6.4 × 104 whereas C-MPR-CHEMS elicited anti-C-MPR titers of less than 6 × 102. Additionally, mice immunized with NC-MPR derivatized with CHEMS at the C terminus generated extremely high titers (GMT 2.5 × 105) against the N terminal region of the peptide but only low titers against the C terminal segment (GMT 9 × 102). The poor immunogenicity of the 4E10 epitope may arise from masking of the epitope within the membrane, as is predicted to occur in native envelope spikes [16]. However, other studies indicate that the peptide sequence itself is poorly immunogenic [7]. If this is due to autoreactivity of antibodies that target the 4E10 epitope, more potent adjuvants or alternative immunization schedules may be needed to circumvent a peripheral tolerance barrier [38].
The lipopeptide immunogens described here may be useful in a prime-boost immunization regimen for focusing the immune response to the MPR, as proposed by others [4, 6]. First, the immune system would be primed with highly immunogenic, membrane-bound peptides that induce antibody responses targeted to MPR peptides in the context of membrane, minimizing antibodies directed against other immunodominant, non-neutralizing epitopes. Second, the immune system would be boosted with a recombinant construct in which the MPR is constrained in the appropriate structural confirmation [9, 55]. Only MPR-reactive antibodies of the appropriate confirmation would be boosted, minimizing antibodies directed against irrelevant MPR structures. Thus, CHEMS may be a simple but effective lipid anchor for creating lipopeptide immunogens as part of an MPR-targeted HIV vaccine.
Acknowledgments
We are grateful to Nichole Macaraeg and Katherine Jerger for assistance with animal studies. We thank Dr. Jay Levy, Dr. Anthony DeFranco and Dr. Zhaohua Huang for helpful discussions, Dr. Gary Fujii for suggestions regarding the liposome formulation and Veena Thomas for assistance with circular dichroism. This work was supported by NIH R01 GM061851 and by a grant from the National Institutes of Health, University of California, San Francisco – Gladstone Institute of Virology & Immunology Center for AIDS Research, P30-AI027763. 2F5 and 4E10 monoclonal antibodies were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH from Dr. Hermann Katinger. D. Watson was supported by a U.S. Department of Homeland Security Graduate Fellowship, administered by the Oak Ridge Institute for Science and Education under U.S. Department of Energy contract number DE-AC05-00OR22750. The opinions expressed herein do not necessarily reflect the policies and views of DHS, DOE, or ORISE.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Karlsson Hedestam G, Fouchier R, Phogat S, Burton D, Sodroski J, Wyatt R. The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nature Reviews Microbiology. 2008;6(2):143–55. doi: 10.1038/nrmicro1819. [DOI] [PubMed] [Google Scholar]
- 2.Salzwedel K, Johnston P, Roberts S, Dubay J, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. Journal of Virology. 1999;73(3):2469–80. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zwick MB, Labrijn AF, Wang M, Spenlehauer C, Saphire EO, Binley JM, et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. Journal of Virology. 2001;75(22):10892–905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montero M, van Houten N, Wang X, Scott J. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiology and Molecular Biology. 2008;72(1):54–85. doi: 10.1128/MMBR.00020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardoso RMF, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, et al. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22(2):163–73. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, et al. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. Journal of Virology. 2004;78(19):10724–37. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Law M, Cardoso RM, Wilson IA, Burton DR. Antigenic and immunogenic study of membrane-proximal external region-grafted gp120 antigens by a DNA prime-protein boost immunization strategy. Journal of Virology. 2007;81(8):4272–85. doi: 10.1128/JVI.02536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGaughey G, Barbato G, Bianchi E, Freidinger R, Garsky V, Hurni W, et al. Progress towards the development of a HIV-1 gp41-directed vaccine. Current HIV Research. 2004;2:193–204. doi: 10.2174/1570162043484933. [DOI] [PubMed] [Google Scholar]
- 9.Davis K, Bibollet-Ruche F, Li H, Decker J, Kutsch O, Morris L, et al. HIV-2/HIV-1 envelope chimeras detect high titers of broadly reative HIV-1 V3-specific antibodies in human plasma. Virology. 2008 doi: 10.1128/JVI.01743-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrity R, Rimmelzwaan G, Minassian A, Tsai W, Lin G, de Jong J, et al. Refocusing neutralizing antibody response by targeted dampening of an immunodominant epitope. Journal of Immunology. 1997;159(1):279–89. [PubMed] [Google Scholar]
- 11.Alam SM, Scearce RM, Parks RJ, Plonk K, Plonk SG, Sutherland LL, et al. Human immunodeficiency virus type 1 gp41 antibodies that mask the membrane proximal region epitopes: antibody binding kinetics, induction, and potential for regulation in acute infection. Journal of Virology. 2008;82(1):115–25. doi: 10.1128/JVI.00927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penn-Nicholson A, Han D, Kim S, Park H, Ansari R, Montefiori DC, et al. Assessment of antibody responses against gp41 in HIV-1 infected patients using soluble gp41 fusion proteins and peptides derived from M group consensus envelope. Virology. 2008;372(2):442–56. doi: 10.1016/j.virol.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matyas G, Beck Z, Karasavvas N, Alving CR. Lipid binding properties of 4E10, 2F5, and WR304 monoclonal antibodies that neutralize HIV-1. Biochimica et Biophysica Acta - Biomembranes. 2008;1788(3):660–5. doi: 10.1016/j.bbamem.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 14.Haynes BF, Fleming J, St Clair E, Katinger H, Stiegler G, Kunert R, et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. 2005;208(5730):1906–8. doi: 10.1126/science.1111781. [DOI] [PubMed] [Google Scholar]
- 15.Alam SM, McAdams M, Boren D, Rak M, Scearce RM, Gao F, et al. The role of antibody polyspecificity and lipid reactivity in binding of broadly neutralizing anti-HIV-1-envelope human monoclonal antibodies 2F5 and 4E10 to glycoprotein 41 membrane proximal envelope epitopes. Journal of Immunology. 2007;178(7):4424–35. doi: 10.4049/jimmunol.178.7.4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, et al. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28(1):52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Luo M, Yuan F, Liu Y, Jiang S, Song X, Jiang P, et al. Induction of neutralizing antibody against human immunodeficiency virus type 1 (HIV-1) by immunization with gp41 membrane-proximal external region (MPER) fused with porcine endogenous retrovirus (PERV) p15E fragment. Vaccine. 2006;24(4):435–42. doi: 10.1016/j.vaccine.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Lenz O, Dittmar M, Wagner A, Ferko B, Vorauer-Uhl K, Stiegler G, et al. Trimeric membrane-anchored gp41 inhibits HIV membrane fusion. Journal of Biological Chemistry. 2005;280(6):4095–101. doi: 10.1074/jbc.M411088200. [DOI] [PubMed] [Google Scholar]
- 19.Huarte N, Lorizate M, Kunert R, Nieva JL. Lipid modulation of membrane-bound epitope recognition and blocking by HIV-1 neutralizing antibodies. FEBS Letters. 2008;582(27):3798–804. doi: 10.1016/j.febslet.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 20.White WI, Cassatt DR, Madsen J, Burke SJ, Woods RM, Wassef NM, et al. Antibody and cytotoxic T-lymphocyte responses to a single liposome-associated peptide antigen. Vaccine. 1995;13(12):1111–22. doi: 10.1016/0264-410x(94)00058-u. [DOI] [PubMed] [Google Scholar]
- 21.Ernst W, Kim H, Tumpey T, Jansen A, Tai W, Cramer D, et al. Protection against H1, H5, H6 and H9 influenza A infection with liposomal matrix 2 epitope vaccines. Vaccine. 2006;24(24):5158–68. doi: 10.1016/j.vaccine.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 22.Harmon P, Cabral-Lilly D, Reed R, Maurio F, Franklin J, Janoff A. The release and detection of endotoxin from liposomes. Analytical Biochemistry. 1997;250:139–46. doi: 10.1006/abio.1997.2216. [DOI] [PubMed] [Google Scholar]
- 23.Pace C, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Science. 1995;4(11):2411–23. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun J, Vernier G, Wigelsworth D, Collier R. Insertion of anthrax protective antigen into liposomal membranes: effects of a receptor. Journal of Biological Chemistry. 2007;282(2):1059–65. doi: 10.1074/jbc.M609869200. [DOI] [PubMed] [Google Scholar]
- 25.Taylor J, Kaiser E. Structure-function analysis of proteins through the design, synthesis, and study of peptide models. Methods of Enzymology. 1987;154:473–98. doi: 10.1016/0076-6879(87)54091-5. [DOI] [PubMed] [Google Scholar]
- 26.Chattopadhyay A, Mukherjee S, Rukmini R, Rawat S, Sudha S. Ionization, partitioning, and dynamics of tryptophan octyl ester: Implications for membrane-bound tryptophan residues. Biophysical Journal. 1997;73(2):839–49. doi: 10.1016/S0006-3495(97)78116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Z, Haugland R. Partition coefficients of fluorescent probes with phospholipid membranes. Biochemical and Biophysical Research Communications. 1991;181(1):166–71. doi: 10.1016/s0006-291x(05)81396-8. [DOI] [PubMed] [Google Scholar]
- 28.Kamala T. Hock immunization: A humane alternative to mouse footpad injections. Journal of Immunological Methods. 2007;328(12):204–14. doi: 10.1016/j.jim.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diaz C, Balasubramanian K, Schroit AJ. Synthesis of disulfide-containing phospholipid analogs for the preparation of head group-specific lipid antigens: Generation of phosphatidylserine antibodies. Bioconjugate Chemistry. 1998;9(2):250–4. doi: 10.1021/bc970156x. [DOI] [PubMed] [Google Scholar]
- 30.Brunel FM, Zwick MB, Cardoso RM, Nelson JD, Wilson IA, Burton DR, et al. Structure-function analysis of the epitope for 4E10, a broadly neutralizing human immunodeficiency virus type 1 antibody. Journal of Virology. 2006;80(4):1680–7. doi: 10.1128/JVI.80.4.1680-1687.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cardoso RMF, Brunel FM, Ferguson S, Zwick M, Burton DR, Dawson PE, et al. Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10. Journal of Molecular Biology. 2007;365(5):1533–44. doi: 10.1016/j.jmb.2006.10.088. [DOI] [PubMed] [Google Scholar]
- 32.Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland F, Krausslich H. The HIV lipidome: A raft with an unusual composition. Proceedings of the National Academy of Sciences. 2006;103(8):2641–6. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alving C, Rao M. Lipid A and liposomes containing lipid A as antigens and adjuvants. Vaccine. 2008;26(24):3036–45. doi: 10.1016/j.vaccine.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Dijkstra J, Swartz GM, Raney JJ, Aniagolu J, Toro L, Nacy CA, et al. Interaction of anti-cholesterol antibodies with human lipoproteins. Journal of Immunology. 1996;157(5):2006–13. [PubMed] [Google Scholar]
- 35.Alfsen A, Bomsel M. HIV-1 gp41 envelope residues 650-685 exposed on native virus act as a lectin to bind epithelial cell galatosyl ceramide. Journal of Biological Chemistry. 2002;277(28):25649–59. doi: 10.1074/jbc.M200554200. [DOI] [PubMed] [Google Scholar]
- 36.Li S, Deber C. Peptide environment specifies conformation. Helicity of hydrophobic segments compared in aqueous, organic, and membrane environments. Journal of Biological Chemistry. 1993;268(31):22975–8. [PubMed] [Google Scholar]
- 37.Engler O, Schwendener R, Dai W, Wolk B, Pichler W, Moradpour D, et al. A liposomal peptide vaccine inducing CD8+ T cells in HLA-A2.1 transgenic mice, which recognise human cells encoding hepatitis C virus (HCV) proteins. Vaccine. 2004;23(1):58–68. doi: 10.1016/j.vaccine.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Haynes BF, Alam SM. HIV-1 hides an Achilles' heel in virion lipids. Immunity. 2008;28(1):10–2. doi: 10.1016/j.immuni.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Huang Y, Fayad R, Spear G, Qiao L. Induction of mucosal and systemic neutralizing antibodies against human immunodeficiency virus type 1 (HIV-1) by oral immunization with bovine papillomavirus-HIV-1 gp41 chimeric virus-like particles. Journal of Virology. 2004;78(15):8342–8. doi: 10.1128/JVI.78.15.8342-8348.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sathaliyawala T, Rao M, Maclean DM, Birx DL, Alving CR, Rao VB. Assembly of human immunodeficiency virus (HIV) antigens on bacteriphage T4: a novel in vitro approach to construct multicomponent HIV vaccines. Journal of Virology. 2006;80(15):7688–98. doi: 10.1128/JVI.00235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alving CR, Koulchin V, Glenn G, Rao M. Liposomes as carriers of peptide antigens: Induction of antibodies and cytotoxic T lymphocytes to conjugated and unconjugated peptides. Immunological Reviews. 1995;145:5–31. doi: 10.1111/j.1600-065x.1995.tb00075.x. [DOI] [PubMed] [Google Scholar]
- 42.Robinson J, Case M, Brooks C. Palmitic acid conjugation of a protein antigen enhances major histocompatibility complex class II-restricted presentation to T cells. Immunology. 1992;76(4):593–8. [PMC free article] [PubMed] [Google Scholar]
- 43.Coutant J, Yu H, Clement M, Alfsen A, Toma F, Curmi P, et al. Both lipid environment and pH are critical for determining physiological solution structure of 3-D-conserved epitopes of the HIV-1 gp41-MPER peptide P1. FASEB Journal. 2008;22(12):4338–51. doi: 10.1096/fj.08-113142. [DOI] [PubMed] [Google Scholar]
- 44.Giannecchini S, D'Ursi A, Esposito C, Scrima M, Zabogli E, Freer G, et al. Antibodies generated in cats by a lipopeptide reproducing the membrane-proximal external region of the feline immunodeficiency virus transmembrane enhance virus infectivity. Clinical and Vaccine Immunology. 2007;14(8):944–51. doi: 10.1128/CVI.00140-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verma J, Rao M, Amselem S, Krzych U, Alving C, Green S, et al. Adjuvant effects of liposomes containing lipid A: Enhancement of liposomal antigen presentation and recruitment of macrophages. Infection and Immunity. 1992;60(6):2438–44. doi: 10.1128/iai.60.6.2438-2444.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karasavvas N, Beck Z, Tong J, Matyas GR, Rao M, McCutchan FE, et al. Antibodies induced by liposomal protein exhibit dual binding to protein and lipid epitopes. Biochemical and Biophysical Research Communications. 2008;366(4):982–7. doi: 10.1016/j.bbrc.2007.12.057. [DOI] [PubMed] [Google Scholar]
- 47.Dal Monte P, Szoka F., Jr Antigen presentation by B cells and macrophages of cytochrome c and its antigenic fragment when conjugated to the surface of liposomes. Vaccine. 1989;7(5):401–8. doi: 10.1016/0264-410x(89)90153-9. [DOI] [PubMed] [Google Scholar]
- 48.Friede M, Muller S, Briand J, Plaue S, Fernandes I, Frisch B, et al. Selective induction of protection against influenza virus infection in mice by a lipid-peptide conjugate delivered in liposomes. Vaccine. 1994;12(9):791–7. doi: 10.1016/0264-410x(94)90287-9. [DOI] [PubMed] [Google Scholar]
- 49.Fernandes I, Frisch B, Muller S, Schuber F. Synthetic lipopeptides incorporated in liposomes: in vitro stimulation of the proliferation of murine splenocytes and in vivo induction of an immune response against a peptide antigen. Molecular Immunology. 1997;34(89):569–76. doi: 10.1016/s0161-5890(97)00090-4. [DOI] [PubMed] [Google Scholar]
- 50.McAleer J, Vella A. Understanding how lipopolysaccharide impacts CD4 T-cell immunity. Critical Reviews in Immunology. 2008;28(4):281–99. doi: 10.1615/critrevimmunol.v28.i4.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hussain R, Dawood G, Abrar N, Toossi Z, Minai A, Dojki M, et al. Selective increases in antibody isotypes and immunoglobulin G subclass responses to secreted antigens in tuberculosis patients and healthy household contacts of the patients. Clinical and Diagnostic Laboratory Immunology. 1995;2(6):726–32. doi: 10.1128/cdli.2.6.726-732.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown BK, Karasavvas N, Beck Z, Matyas G, Birx DL, Polonis VR, et al. Monoclonal antibodies to phosphatidylinositol phosphate neutralize human immunodeficiency virus type 1: role of phosphate-binding subsites. Journal of Virology. 2007;81(4):2087–91. doi: 10.1128/JVI.02011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beck Z, Karasavvas N, Matyas G, Alving CR. Membrane-specific antibodies induced by liposomes can simultaneously bind to HIV-1 protein, peptide, and membrane lipid epitopes. Journal of Drug Targeting. 2008;16(78):535–42. doi: 10.1080/10611860802228517. [DOI] [PubMed] [Google Scholar]
- 54.Banerji B, Kenny J, Scher I, Alving C. Antibodies against liposomes in normal and immune-defective mice. Journal of Immunology. 1982;128(4):1603–7. [PubMed] [Google Scholar]
- 55.Frey G, Peng H, Rits-Volloch S, Morelli M, Cheng Y, Chen B. A fusion-intermediate state of HIV-1 gp41 is targeted by broadly neutralizing antibodies. Proceedings of the National Academy of Sciences. 2008;105(10):3739–44. doi: 10.1073/pnas.0800255105. [DOI] [PMC free article] [PubMed] [Google Scholar]