

Abstract
Influenza viruses are enveloped, negative stranded, segmented RNA viruses belonging to Orthomyxoviridae family. Each virion consists of three major sub-viral components, namely (i) a viral envelope decorated with three transmembrane proteins hemagglutinin (HA), neuraminidase (NA) and M2, (ii) an intermediate layer of matrix protein (M1), and (iii) an innermost helical viral ribonucleocapsid [vRNP] core formed by nucleoprotein (NP) and negative strand viral RNA (vRNA). Since complete virus particles are not found inside the cell, the processes of assembly, morphogenesis, budding and release of progeny virus particles at the plasma membrane of the infected cells are critically important for the production of infectious virions and pathogenesis of influenza viruses as well. Morphogenesis and budding require that all virus components must be brought to the budding site which is the apical plasma membrane in polarized epithelial cells whether in vitro cultured cells or in vivo infected animals. HA and NA forming the outer spikes on the viral envelope possess apical sorting signals and use exocytic pathways and lipid rafts for cell surface transport and apical sorting. NP also has apical determinant(s) and is probably transported to the apical budding site similarly via lipid rafts and/or through cortical actin microfilaments. M1 binds the NP and the exposed RNAs of vRNPs, as well as to the cytoplasmic tails (CT) and transmembrane (TM) domains of HA, NA and M2, and is likely brought to the budding site on the piggy-back of vRNP and transmembrane proteins.
Budding processes involve bud initiation, bud growth and bud release. The presence of lipid rafts and assembly of viral components at the budding site can cause asymmetry of lipid bilayers and outward membrane bending leading to bud initiation and bud growth. Bud release requires fusion of the apposing viral and cellular membranes and scission of the virus buds from the infected cellular membrane. The processes involved in bud initiation, bud growth and bud scission/release require involvement both viral and host components and can affect bud closing and virus release in both positive and negative ways. Among the viral components, M1, M2 and NA play important roles in bud release and M1, M2 and NA mutations all affect the morphology of buds and released viruses. Disassembly of host cortical actin microfilaments at the pinching-off site appears to facilitate bud fission and release. Bud scission is energy dependent and only a small fraction of virus buds present on the cell surface is released. Discontinuity of M1 layer underneath the lipid bilayer, absence of outer membrane spikes, absence of lipid rafts in the lipid bilayer, as well as possible presence of M2 and disassembly of cortical actin microfilaments at the pinching-off site appear to facilitate bud fission and bud release. We provide our current understanding of these important processes leading to the production of infectious influenza virus particles.
Keywords: Influenza virus, Morphogenesis, Budding, Assembly, Structure, Morphology
1. Introduction
For enveloped viruses, assembly and budding are the penultimate and final steps of virus replication, respectively, prior to release of infectious progeny virus particles. Lytic viruses such as influenza viruses do not form a stable, long-term host–virus relationship within the infected host. The very survival of the viruses thus depends on host to host transmission, which, in turn, depends on the release of progeny viruses from the infected host. The underlying causes of disease syndromes, such as pneumonia and bronchitis, are basically due to the killing of a large number of cells of the infected organ(s)/tissue(s) (e.g. the lungs), leading to the loss of function of the organ/tissue. A number of other factors such as inflammatory cytokines, invasion of inflammatory white blood cells, release of fluids from lymphatic and blood vessels further exacerbate the disease syndrome. Furthermore, humans or animals are normally infected by influenza virus at a very low multiplication of infectivity (moi), except in some recent H5 avian virus infection cases (where it is believed that humans were infected at a very high dose of virus particles while handling infected chickens). Therefore, disease progression and host to host transmission of the influenza virus require efficient release of viral particles from infected host cells.
Since complete influenza virions are not present inside the infected cells and can only be produced by budding from the plasma membrane, both virus assembly and bud release play critical roles in infectious virus production, which in turn affects both the severity of the disease syndrome and transmission of the viruses across humans and animals. Virulence of influenza viruses is usually measured by two parameters: (i) Severity of disease production in the infected host. (ii) Efficiency of transmission from person to person or one host to another. Although these two parameters often go hand in hand, it is possible to have highly transmissible virus with low virulence and, vice versa, a highly pathogenic virus with low transmission.
The goal of this review is to critically analyze the recent advances in our understanding of the processes involved in assembly and budding of influenza viruses. The present review primarily deals with the advances made since the publication of our earlier review (Nayak et al., 2004). In addition, readers should read other reviews on similar subjects published recently (see Chen and Lamb, 2008).
2. Overview of influenza virus composition, assembly and budding
Influenza viruses are members of the Orthomyxoviridae family, which is a family of negative stranded, segmented, enveloped RNA viruses. Each virion contains helical ribonucleocapsids (also called viral ribonucleoprotein [vRNP]) (Palese and Shaw, 2007). Influenza virus virions are pleomorphic and are roughly spheroidal with approximately 100 nm in diameter (Fujiyoshi et al., 1994). The viral envelope consists of a lipid bilayer containing three transmembrane proteins HA (hemagglutinin), NA (neuraminidase), and M2 (ion channel) on the outside and M1 (matrix protein) underneath the membrane. The influenza virus lipid bilayer is a mosaic structure containing both cholesterol-enriched lipid rafts and non-raft lipids derived from the host plasma membrane (Scheiffele et al., 1999, Zhang et al., 2000). HA and NA are anchored in the lipid raft domain of the viral envelope, whereas M2, though a cholesterol-binding protein, is not tightly associated with lipid rafts (Schroeder et al., 2005). HA is the major envelope protein (∼80%) and forms the trimeric spikes with receptor-binding sites and epitopes for neutralizing antibodies. Cleavage of HA into HA1 and HA2 is critical for virus infectivity. NA is the second most abundant (∼17%) envelope protein and forms the tetrameric spikes. NA removes the cell surface receptor (sialic acid) and plays a critical role in the release of progeny virus particles from the cell surface as well as the spread and transmission of virus from host to host. The third envelope protein M2 is a minor component (∼16–20 molecules/virion) and functions as an ion channel (see Lear, 2003, Wu and Voth, 2003). M2 plays a critical role in the early phase of infection leading to the uncoating and release of the vRNPs from M1 matrix. The viral core consists of helical vRNPs containing negative stranded vRNAs and NP along with minor amounts of the nuclear export protein (NEP) and three polymerase (3P) proteins (PB1, PB2, PA) forming the viral RNA polymerase complex (3P complex).
Assembly involves bringing the viral components synthesized in different cellular compartments to the budding site at the plasma membrane of the host cell. Budding is the process leading to the formation, growth and release of virus buds. Although influenza virus assembly and bud release have evolved for the survival of virus species, both infectious virus particles and non-infectious or defective virus particles are produced. The process of assembly and budding is further complicated by the presence of segmented RNA genome. Each infectious influenza virus packages seven to eight different RNA/RNP segments (eight in influenza virus A and B and seven in influenza virus C) and all these individual RNA segments must be present within the individual virus particle for the progeny virus to be infectious and transmissible. However, the majority (≥90%) of released virus particles are non-infectious. Although some particles may lose their infectivity after their release into the external environment, defective or incomplete virus particles can assemble, bud, and are released from the plasma membrane. Production of defective interfering particles (DI, also called “von Magnus particles”) attests to the lack of check point(s) against releasing non-infectious particles (see Nayak et al., 1985). Similarly, recent studies on the production of virus-like particles (VLPs) also support this notion. On the other hand, specific nucleotide sequences of different RNA segments have been shown to favor packaging into virions (Liang et al., 2008, Marsh et al., 2007, Marsh et al., 2008), but not for assembly and bud release. Indeed, virus replication can be abortive at many stages of the viral life cycle. For example, in HeLa cells infected with influenza virus, bud formation occurred on the cell surface, but failed to be released (Gujuluva et al., 1994).
Morphogenesis and budding require four steps—assembly of viral components, bud initiation, bud growth and pinching off from the plasma membrane. These steps in the budding process are sequential and do not have specific start and stop signals. Budding is a complex process and involves physical and structural as well as functional requirements of multiple biological components of both virus and host and is among the least understood processes in viral biology. One of the main reasons for the lack of understanding of the budding process is that the biological processes involving the release of enveloped vesicles from the plasma membrane of cells into an outside environment is not a common phenomenon exhibited by living cells. Release of neurotransmitters from the nerve endings in synapses and exocytic intracellular transport pathway involving vesicle formation, release and fusion are among the close proximate processes mimicking virus budding. However, release of neurotransmitters occurs only in specialized nerve endings in synaptic junctions. Processes of exocytosis and budding into multivesicular body (MVB) have helped to explain some of the steps involved in budding of some viruses such as HIV (Freed and Mouland, 2006). Exocytosis involving release and fusion of vesicles occurs in the intracellular organelles i.e. endoplasmic reticulum (ER) and Golgi complexes and ends in fusion of the vesicles with the plasma membrane leading to either releasing their content (e.g. exocrine proteins such as insulin) or transporting and anchoring the membrane proteins to its destination e.g. plasma membrane.
In analyzing the role of viral components in budding, two methods are most frequently used: (i) producing VLPs (virus-like particles) and (ii) creating mutant viruses by reverse genetics either eliminating or mutating specific viral components. Both of these processes although useful have limitations in understanding virus budding. For example, in VLP analysis, transfections of cDNA or vRNPs are primarily used and quantification of released VLPs from the transfected cells is assayed. However, in cells transfected with cDNAs or vRNPs, normal regulation of synthesis of viral components, present in virus-infected cells, is absent. Furthermore, transfection can lead to high or low expression of specific viral components in individual cells. This may cause artificial release of VLP depending on concentration of specific viral components, which may not mimic bud release in virus-infected cells.
In cDNA transfected cells, high concentration of NP in the nucleus may cause aggregation and nuclear exit without the requirement of M1 (Carrasco et al., 2004) whereas M1 plays a critical role in nuclear exit of NP in virus-infected cells. Similarly, high concentration of HA in the plasma membrane cDNA transfected cells may cause bud release (Chen et al., 2007) in the absence of other vial components, which does not occur in virus-infected cells. Furthermore, VLP release with limited viral components may indicate only minimal requirements of bud formation and bud release and may not reflect the normal budding process in virus-infected cells. On the other hand, reverse genetics which can create specific mutant viruses may be the most important and reliable tool currently available for analyzing the role of individual viral components in budding.
Assay used for the analysis of virus budding is also an important consideration. Usually, particle release [either total particle or plaque forming units (PFU)] is used in determining the efficiency of budding. Although total particle release is a quantitatively effective way in analyzing the budding efficiency, other factors including synthesis, transport and assembly of viral components may affect particle release and therefore may not necessarily reflect the efficiency in bud release only. The other method is examining the morphology of released virus particles and virus buds attached on the cell membrane by electron microscopy (EM). EM analysis may provide more direct information about the budding process and morphogenesis of virus particles. Although fresh field virus isolates and some laboratory adapted virus such as Udorn (A/Udorn/307/72, H3N2) may exhibit elongated or even filamentous form, quantitative formation of elongated or filamentous form of mutant WSN or PR8 virus particles which normally exhibit spheroidal morphology would indicate a budding defect. If the virus bud cannot be released (i.e. defective in the process of bud closure), the bud may continue to grow causing more elongated or even filamentous form. Similarly, conversion of a filamentous virus to spheroidal form may indicate the removal of a budding block causing efficient virus release. Although defect in bud release may not always affect virus morphology, alteration in virus morphology is always an indication of budding defect. Therefore, analysis of virus morphology of either released virus particles or the buds present on the cell surface may be considered the gold standard in assessing budding defect and is the most important tool available at present in studying virus budding.
3. Virus morphology
Bud closing and bud release may directly affect the size and shape of released virus particles. Therefore, analysis of the architecture and morphology of virus particles may provide important information about the processes of morphogenesis and budding. Until recently, the morphology of influenza viruses was based on transmission electron microscopy (TEM) images of negatively stained viral particles or thin sections of virus-infected cells. In addition, electron tomography (ET) became available recently to reconstruct the three-dimensional (3D) structure of viral particles and thin sections by combining different tilt views of the same sample (Baumeister, 2002). However, staining and sectioning procedures often introduce artifacts in the shape, size and morphology of virus particles during sample processing due to the use of heavy metal stain at non-physiological pH and sample drying. Influenza viruses are particularly sensitive to these procedures due to the flexible, pH-sensitive viral envelope. In addition, the size and shape of virus particles vary with different virus isolates and laboratory strains. Recently, cryo-electron microscopy (cryoEM) and cryo-electron tomography (cryoET) have been used to examine the structure of these viruses in their natural state without fixing and staining. Furthermore, cryoET can be used to determine the 3D structure of each viral particles by combining different tilt views of the same viral particles (Baumeister, 2002). The 3D structures can then be computationally sliced to reveal the structural arrangement of proteins, nucleic acid and lipids and their possible interactions in their native state within the virus particles.
Noda et al. (2006) recently examined influenza WSN (A/WSN/33, H1N1) virions by ET of both longitudinal and transverse sectioning of individual virus particles embedded in Epon resin mixture. These authors found that WSN virions released from MDCK cells were predominantly spherical in shape. vRNPs inside the virus particle were suspended from the distal end, and were ∼12 nm in width with varying lengths up to 130 nm. In transversely sectioned virions, they found eight electron dense dots arranged as seven in the periphery with one in the center in many virus particles. They concluded that each of these dots represents a vRNP and this observation favored selective over random incorporation of vRNPs in the virus particles.
Using cryoET reconstruction, Harris et al. (2006) analyzed the detailed structure of ice-embedded X31 [A/Aichi/2/68(X31), H3N2] virus particles, which were grown in embryonated chicken eggs. They identified five classes of virions with respect to their shape and size and arrangement of M1 and vRNP within the virion. The most abundant particles (∼80%) were spheroidal with a mean diameter of 120 nm (range 84–170 nm and an average axial ratio 1:2). The next predominant class (∼14%) had an elongated morphology with an average diameter of 100 nm and an axial ratio of 1:4. The other three classes of virions represented only a minor fraction of virions. They noted that the average diameter of elongated particles was shorter than that of the spheroidal particles. These cryoET studies also revealed some interesting insight about the structure and arrangement of major envelope glycoproteins HA and NA. As expected, there were approximately 300 HA (triangular in shape) and 40 NA (square in shape) spikes on the surface of each spheroidal virion. Furthermore, the distribution of HA and NA on the viral surface were not entirely random, but rather with some local clustering of NA surrounded by more abundant HA (Fig. 1 ). Their analysis also revealed an M1 layer, beneath the lipid bilayer. They observed some gaps in the M1 layer, which coincides with the absence of overlying spikes on the outer surface of the membrane, and the authors speculated that these gaps may be the point of pinching-off sites for virus release (see later).
Fig. 1.
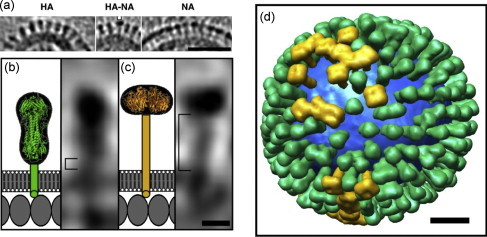
Model virus with HA and NA spikes by cryoET analysis of X31 virus. (a) HA cluster (left); single NA (marked) in a cluster of HA (middle); cluster of mainly NA (right). (b and c) The stem length of HA and NA (square brackets in b and c, respectively. The structures of the stem, transmembrane domain and ectodomain are shown schematically. Molecules in the matrix layer are inferred to be packed in a monolayer (scale bar 5 nm). (d) Model of distribution of HA (green), NA (gold), and lipid bilayers (blue) in a single virion (scale 20 nm). Reproduced from Harris et al. (2006) with permission. The figure was provided by Drs A. Harris and A.C. Steven. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
The cryoET analysis by Harris et al. (2006) also showed that RNPs in the interior of the virus particles is densely packed with similar dimensions as described by Noda et al. (2006). In elongated particles, RNPs formed nearly parallel bundles. However, Harris et al. (2006) could distinguish eight distinct RNP complexes only in a few particles and observed fewer RNPs in smaller particles. They also concluded that the dense inner layer underneath the membrane (presumably M1) had contact with HA and NA tails on the outer side and at one or both ends of vRNPs at the inner side. However, no M1 was found to be associated with vRNP inside the virion (Harris et al., 2006) although studies reported binding of M1 to NP (Noton et al., 2007) and vRNA exposed on the surface of vRNP (Ye et al., 1999) and M1–vRNP interaction was critical for transport of vRNP from the nucleus into cytoplasm and from cytoplasm to the plasma membrane (the budding site).
We have also analyzed PR8 (A/PR/8/34, H1N1) and WSN (A/WSN/33, H1N1) grown in MDCK cells by cryoET. Our analysis yielded similar results as that of Harris et al. (2006) except that PR8 virus particles were substantially more pleomorphic and exhibited particles of various shapes and sizes (Fig. 2 ), whereas WSN, as reported earlier, were relatively uniform and mostly spheroidal in size (Fig. 3 ). PR8 particles ranged from spheroidal to highly elongated, even filamentous representing irregular shape and sizes (Fig. 2). PR8 spheroidal particles were also of various sizes with spike to spike dimension ranging from 73 nm × 57 nm to 288 nm × 267 nm. All particles, including the very small particles (73 nm × 55 nm), exhibit prominent glycoproteins spikes outside, and had some electron dense materials corresponding to the vRNP core inside. However, eight distinct RNP complexes were observed only in a few particles and the majority contained fewer RNP complexes as was noted by Harris et al. (2006). In the elongated particles, vRNPs were parallel to the surface extending essentially end to end (Fig. 2k). Both triangular (HA) and rectangular (NA) spikes were observed on the surface as noted previously (Harris et al., 2006). Reason for extensive heterogeneity observed with PR8 virus is as yet unclear and the role of host (i.e. MDCK vs. chicken eggs) in virus morphology needs to be examined. Furthermore, it remains unclear whether the physical shape and morphology of different classes of virions as well as their RNP content also affected the infectivity of virus particles. It is possible that very small particles contain less vRNP and therefore may not be infectious. Further analysis is also needed to determine the relationship of vRNP content with virus morphology, which in turn may affect infectivity. Unlike PR8, the majority of WSN particles were spheroidal with spike to spike dimension ranging from 133 nm × 92 nm to 94 nm × 86 nm (Fig. 3).
Fig. 2.
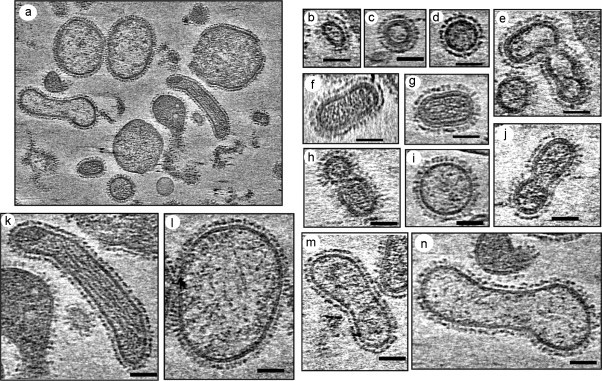
Cryo-electron tomography studies of influenza virus A/PR8 strain showing highly pleomorphic virion architecture. (a) A density slice from a 3D cryo-electron tomography reconstruction of influenza A virus strain PR8. PR8 virus was grown in MDCK cells at 0.001 moi. The tilt series spanning −70° to 70° sample tilt were recorded in a TF20 cryo-electron microscope using the Batch Tomography program (FEI Company), and reconstructed using the Inspect3D (FEI Company) and refined by Protomo program (Winkler and Taylor, 2006). (b–n) Comparison of central slices of viral particles extracted from different cryo-electron tomograms. Different virus particles were picked at random. No attempt was made to determine the percentage of each virus form in the population. Each virus particle contained electron dense spots (RNP) inside, and spikes outside. Both HA and NA spikes, as identified based on morphology as described in Harris et al. (2006), were visible on the outer membrane. Scale bar 50 nm.
Fig. 3.
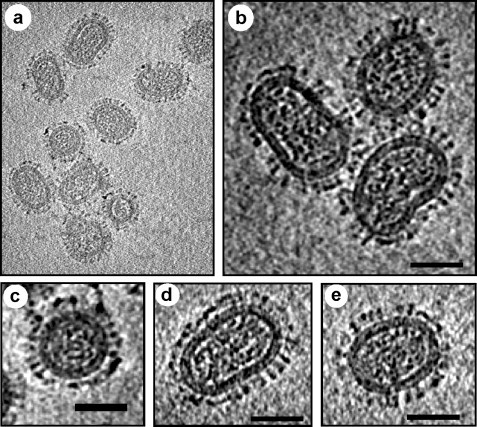
CryoET reconstruction of influenza virus A/WSN strain showing more homogeneous architecture as compared to PR8. The WSN strain virus particles were grown in MDCK cells at 0.05 moi. (a) A density slice from a 3D cryo-electron tomography reconstruction of influenza A strain WSN. Virus morphology was relatively less pleomorphic as compared to PR8 virus. (b–e) Comparison of central slices of viral particles extracted from the tomogram. Different virus particles were picked at random. No attempt was made to determine the percentage of each virus form in the population. Each virus particle contained electron dense spots (RNP) inside, and spikes outside. Both HA and NA spikes, as identified based on morphology as described in Harris et al. (2006), were visible on the outer membrane. Bar scale 50 nm.
CryoET results by us and Harris et al. (2006) agree with the overall vRNP arrangement inside virus particles observed by Noda et al. (2006) using ET of serially thin-sectioned virions, except that the majority of particles contained fewer than eight RNP complexes. Noda et al. (2006) further noted that individual vRNPs were oriented perpendicular to the budding tip and remained suspended from the distal end of budding virions. According to these authors, this orientation and arrangement of clusters of vRNP inside virions supports the model of selected incorporation of vRNPs, which are recruited, packaged and incorporated as a complete vRNP set into the virus buds. However, whether the recruitment and packaging of the complete vRNP set occurred after, during, or before the incorporation of vRNPs into the bud is still an open question since such intracellular specific vRNP complexes have yet to be demonstrated. Noda et al. (2006) noted that they did not observe more than eight vRNP segments inside virions and therefore concluded that a complex of eight specific vRNP segments were incorporated in the bud, thus favoring specific over random incorporation of vRNPs. However, because of the heterogeneity and pleomorphism in both shape and size of virus particles, RNP content of particles varies greatly and each virus particle may not contain a full set of eight vRNPs. Therefore, incorporation of a complete set of vRNP complex may not occur in each virus particle and incorporation of specific vRNP complex may not significantly affect budding and bud release although vRNP incorporation may facilitate bud formation and bud release. Also, further analysis is required to determine whether larger and elongated particles contain more than eight vRNP segments.
4. Transport and assembly of viral components and selection of budding site
Bud formation and bud release require assembly of viral components, which can occur either during transport or during budding at the budding site. During the assembly process, viral components, either individually or in the form of complexes, are brought to the budding site to form a higher order of complex which may facilitate bud initiation and/or bud completion. With the majority of viruses either enveloped or non-enveloped, assembly implies the formation of complete capsid, either helical or icosahedral including incorporation of the genome in the capsid. Furthermore, with the majority of enveloped viruses, capsid formation is a requirement for bud formation and bud release as is shown for retrovirus like human immunodeficiency virus (HIV) (Ganser-Pornillos et al., 2008), and alpha viruses like Semliki forest viruses (SFV) (Garoff et al., 1994). Even for enveloped viruses possessing helical nucleocapsids like VSV, formation of nucleocapsid is critical for bud formation and bud release. In fact, the size of the nucleocapsid which is determined by the size of vRNA, determines the size and shape of the released VSV particles. For example, smaller DI (defective interfering) virus particles contain shorter vRNA/RNP (nucleocapsid) compared to the bullet shaped elongated virus particles of wild-type viruses containing the complete and longer vRNA/RNP (Pattnaik and Wertz, 1991). Therefore, with these viruses, capsid assembly is a critical requirement for virus budding. However, requirements of capsid assembly and budding are much more complex for influenza viruses for a number of reasons. Firstly, budding may occur in the absence of vRNPs and/or with incomplete vRNPs. Furthermore, the viral genome consists of multiple segments of vRNAs/vRNPs. Therefore, budding of infectious virus particles requires that each segment of vRNPs must be incorporated into the bud. Secondly, all the components of the virus namely, envelope containing the transmembrane protein (HA, NA, and M2) as well as M1 and vRNPs must be brought either individually or as a complex to the budding site for bud initiation, bud growth, and finally, release of infectious virus buds. Although much is known about the mechanism and the process involved in transporting individual viral components to the budding site, less is known about where such sub-viral complexes are formed and how they are brought to the budding site. Finally, little is known about the requirements of these complexes or the components for bud initiation, bud growth and bud release.
Although assembly and budding are discussed separately, it is in fact a continuous process, one leading to the other as mentioned earlier. Furthermore, it is unclear if there are check points or switches in this continuous process i.e. whether the completion of a previous step is required for initiation of the succeeding step. Normally, one would expect some regulation for the progression of a process involving different steps but such specific stop and start switches are as yet unknown in influenza assembly and budding. Moreover, it is unknown if there are different requirements for the budding process of infectious vs. non-infectious particles leading to favoring one over the other. Also, since influenza viruses bud from the plasma membrane into outside environment, and since complete virus particles are not found inside the cell, assembly of virus components into virus particle must occur at the plasma membrane, the budding site.
As noted earlier, influenza virus particles contain three sub-viral components namely, (a) viral envelope which consists of a lipid bilayer and viral transmembrane proteins (HA, NA, and M2). Lipids are selectively derived from host. (b) M1 protein underneath the lipid bilayer, which forms the bridge between the envelope, and viral core. (c) Virus core (viral nucleocapsid) which consists of vRNP (minus stranded vRNA and NP) and minor amounts of NEP, and 3P (polymerase) protein complex. Each nucleocapsid (vRNP) is a supercoiled ribbon structure with a terminal loop where the vRNA is coiled around NP monomer to form a hairpin structure and vRNA is exposed on the outer surface of NP (Elton et al., 2006). Therefore, assembly involves the formation of these sub-viral complexes and their transport to the budding site, the apical domain of the plasma membrane in polarized epithelial cells whether in cultured cells in laboratory or respiratory epithelium of infected animals.
4.1. Transport and assembly of viral components
Transport of the transmembrane envelope proteins (HA, NA, and M2) has been studied extensively (see Nayak et al., 2004). As mentioned earlier, the viral membrane is a mosaic containing both raft- and non-raft-associated lipids where both HA and NA are inserted in the raft domains and M2 in the non-raft lipid domains. These transmembrane proteins use cellular exocytic transport pathway for apical transport and possess the determinants for both lipid raft association and apical transport in their transmembrane domain (TMD). Lipid raft association of TMD is responsible for apical transport of both HA and NA (Lin et al., 1998, Barman and Nayak, 2000). On the other hand, how M1 and RNP are transported to the budding site (i.e. apical plasma membrane) is not fully understood. M1 is not known to possess any apical determinant but possesses determinants for lipid binding, RNA/RNP/NP binding (Baudin et al., 2001, Noton et al., 2007, Watanabe et al., 1996, Ye et al., 1999), and associating with HA and NA tails (Ali et al., 2000). Therefore, it is likely that some M1 can be transported to the budding site of apical plasma membrane on the piggy-back of HA and NA.
Since RNP is synthesized in the nucleus it must be exported from the nucleus into cytoplasm before being transported to the apical plasma membrane. M1, a small protein possessing nuclear localization signal (NLS) can enter the nucleus, interact with vRNP as well as with NEP forming the daisy-chain complex of (Crm1 and RanGTP)–NEP–M1–RNP, and mediate nuclear export of v-RNP (Akarsu et al., 2003, Whittaker and Digard, 2006). M1–RNP complex has been demonstrated both in infected cells and in virions (Zhirnov, 1992, Ye et al., 1999). Interaction of M1 with RNP preventing transcription is critically required for exit of vRNPs into cytoplasm and incorporation into virions since only transcriptionally inactive vRNP with the polymerase complex present only at the end of vRNP are found in virus particles (Murti et al., 1988). Recent studies suggest that NP/RNP may possess an as yet undefined determinant for apical transport (Carrasco et al., 2004). NP/RNP exits nucleus from its apical side and is transported to the apical plasma membrane of polarized MDCK cells (Fig. 4 ). NP/RNP was also shown to interact with actin microfilaments (Avalos et al., 1997) and associate with lipid rafts (Carrasco et al., 2004). Therefore, it is likely that RNP along with the associated M1 can be directed to the apical budding site via its association with cortical actin microfilaments and lipid rafts. However, neither the apical determinant(s) of NP/RNP nor the cellular machinery involved in its apical transport has been identified.
Fig. 4.
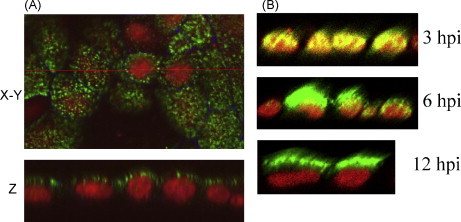
Viral glycoprotein-independent apical transport of NP in polarized MDCK cells. VLP (delHA, del-NA, and del-M2-CT-TMD) infected (A) or wt virus-infected (B) MDCK cells were fixed (at 12 hpi. for panel A) with paraformaldehyde, permeabilized with saponin and triple-stained for NP (green), tight junction protein ZO-1 (blue) and DNA (red, with propidium iodide) and examined by confocal microscopy. Single optical section in the xy plane (A, upper, top/apical surface) and xz planes (A, lower and B) are shown for merged fluorescence. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Finally, since the genome of influenza virus is segmented, multiple vRNA/vRNP segments (eight separate segments for influenza type A and B, seven segments for influenza type C viruses) must be incorporated into each infectious virus particle (see later). However, as noted earlier, although packaging of different RNP segments in the virus bud is critically important for infectivity of virus particle, assembly or incorporation of genomic segments do not appear to play an important role in the budding of virions. On the other hand, M1 plays a critical role in the assembly process, as it interacts with multiple components and brings viral components together. M2 interacts with M1 via cytoplasmic tail and thereby plays an important role in virus assembly, genome packaging and budding (Iwatsuki-Horimoto et al., 2006, McCown and Pekosz, 2005, McCown and Pekosz, 2006). The following complexes of viral components have been identified either in the infected cells or in virions and are likely to play major role in virus assembly and some also in budding process: HA–HA (trimer), HA–M1, NA–NA (tetramer), NA–M1, M1–M1 (multimer), M1–M2, M1–NP, M1–RNA, M1–RNP, M1–RNP–NEP, NP–RNA, PB1–PB2–PA (3P), 3P–RNP, 3P–RNP–M1, PB1/PB2–NP. However, complexes involving vRNP–vRNP interaction have not been physically demonstrated, although inferred from biological, mutational and morphological analysis.
4.2. Selection of the budding site
Influenza viruses bud specifically from the apical domain of the plasma membrane of infected polarized cells, regardless in cultured cells or in respiratory epithelium of infected animals. However, what determines the selection of the apical budding site is not clear. For majority of the viruses, viral glycoproteins are thought to be important in the selection of the budding site since virus glycoproteins, even when expressed alone in the absence of other viral components, predominantly accumulate at the site of virus budding. Viruses such as hepatitis B virus, bunyaviruses, coronaviruses, and others that bud from the internal sub-cellular organelles, possess intrinsic determinants for the same sub-cellular localization as the site of virus budding (for review, see Hobman, 1993). On the other hand, for viruses budding from the plasma membrane, the viral glycoproteins possess either apical or basolateral sorting signals and are directed to the specific site where virus assembly and budding occur in polarized epithelial cells. The surface glycoproteins of viruses such as influenza virus, parainfluenza virus, or respiratory syncitial viruses (RSV) budding from the apical plasma membrane, possess apical sorting signal(s) and predominantly accumulate at the apical plasma membrane in polarized epithelial cells. Conversely, for viruses that are released from the basolateral membrane, their surface glycoproteins, possessing basolateral sorting signal, are transported basolaterally in polarized epithelial cells even when these proteins are expressed alone. Vesicular stomatitis virus (VSV), Semliki forest virus (SFV), vaccinia virus, and certain retroviruses including human immunodeficiency virus type 1 (HIV-1) exhibit basolateral budding. Furthermore, in different cells and tissues where some viruses bud from the opposite domains of the plasma membrane, their glycoproteins are distributed accordingly. For example, Semliki forest viruses (SFV) buds apically from FRT cells but basolaterally from CaCo-2 cells; similarly, in the absence of any other viral protein, p62/E2, the envelope glycoproteins of SFV, are targeted apically in FRT cells but basolaterally in CaCo-2 cells (Zurzolo et al., 1992). However, there are examples of polarized virus budding occurring independently of the polarized envelope viral glycoprotein sorting. For example, although measles virus glycoproteins H and F are transported in a random fashion or to basolateral membrane, respectively, virus budding occurred predominantly from the apical surface of polarized MDCK cells (Maisner et al., 1998). Similarly, the spike protein of coronavirus is not involved in the polarized budding of this virus (Rossen et al., 1998). Moreover, Marburg virus buds predominantly from the basolateral surface, while its glycoprotein is transported to the apical surface (Sanger et al., 2001).
Influenza virus, which assembles and buds from the apical plasma membrane in polarized epithelial cells, has been used extensively as a model for studying protein targeting. The three envelope glycoproteins, HA, NA and M2 proteins are transported to the apical plasma membrane in virus-infected cell, as well as in cDNA transfected polarized epithelial cells when expressed alone. Since HA is the major glycoprotein on influenza virus envelope, its role on the apical budding of virus was determined. Using a transfectant influenza virus (HATyr) containing basolaterally targeted HA (Cys543 → Tyr543), it was shown that the basolateral targeting of HA did not significantly alter the apical budding of influenza virus (Barman et al., 2003, Mora et al., 2002). Over 99% of the virus particles containing the HAtyr were released from the apical side even though the majority of HAtyr was directed to the basolateral side. However, when virus budding was examined by thin section transmission electron microcopy one significant difference was observed in HAtyr-infected cells. In the lateral intercellular space of HAtyr-infected cells, the presence of empty virus-like structures (Barman et al., 2003) with the same size diameter as the virus particles at apical surface was often observed. These virus-like structures were not seen in wt WSN-infected cells and found only in HAtyr-infected cells (Barman et al., 2003). Likely, these particles represent abortive virus buds containing HA and M1 but not vRNP, suggesting that vRNP may play a role in polarized budding of influenza virus. Later on Carrasco et al. (2004) demonstrated that in later stages of influenza virus-infected cells, NP/vRNP localized at the apical but not basolateral membranes in both polarized (MDCK) and non-polarized (BHK and 293T) cells although HA was present on apical membrane of MDCK cells and on both apical and lateral membranes of 293T cells. These findings suggested that apical targeting of NP/vRNP was independent of HA. Furthermore, apical targeting of NP was also shown to be independent of M1 and NEP which did not accumulate to the apical membrane (Carrasco et al., 2004). Also, when expressed alone in BHK cells (non-polarized fibroblast) using cDNA transfection, NP was also localized specifically to the apical side. It was further shown that apical distribution of NP correlated with its association to detergent resistant lipid rafts by flotation assay of membranes from virus-infected and plasmid-transfected cells. However, in these experiments, apical localization of NP in transfected polarized epithelial (such as MDCK) cells was not demonstrated.We have examined the localization of NP/vRNP in VLP-infected polarized MDCK cells which lack the expression of viral envelope proteins. Accordingly, infectious VLPs were produced using methods similar to the virus rescue protocol using cDNA transfection of 293T cells. Stop codons were introduced after ATG of HA and NA and after 26 aa position of M2 cDNAs (to allow expression of M1 but not M2) in pol I and II cDNA plasmids and HA, NA, and M2 were expressed in cDNA transfected cells using protein expression plasmids. VLPs obtained from this system were used to infect polarized MDCK cells. In VLP-infected polarized MDCK cells lacking the expression of HA and NA and M2 proteins except the N-terminus 26 aa of M2 (without the cytoplasmic tail and transmembrane domain), NP was expressed efficiently and found to accumulate at the apical plasma membrane, similar to that observed in wt virus-infected cells (Fig. 4). As expected from previous observation (Elton et al., 2005), exit of NP was observed to occur through apical side of the nucleus in wild-type (wt) virus-infected (Fig. 4, panel B) as well as in VLP-infected (not shown) polarized MDCK cells. Taken together these results demonstrate that NP/vRNP can be transported independently to the apical plasma membrane of polarized epithelial cells in the absence of transmembrane viral proteins. Therefore, transmembrane proteins alone do not determine the site of virus budding and NP also plays an important role in apical budding of influenza A viruses. Determinants of NP and cellular machinery involved in its apical transport are yet to be determined. Cortical actin microfilaments as well as lipid rafts may aid in apical transport since NP binds to both of these host components.
5. Role of virus components in budding
5.1. Role of structural proteins
A number of viral components are involved in the assembly and budding processes. However, some may be involved in different stages of bud initiation, bud growth or bud release while others may be involved in multiple steps of the budding process including bud release. Again, the assessment of their involvement has been analyzed quantitatively by particle or pfu release and/or examining the morphology of the virus buds on the cell surface or released particles. It appears that all three transmembrane proteins (HA, NA, and M2) are involved in the assembly and budding processes. However, direct involvement of HA alone in the budding process appears to be controversial. VLP assays by transfection in baculovirus expression system in Sf9 insect cells (Latham and Galarza, 2001) and T7 expression system in COS-1 cells (Gomez-Puertas et al., 2000) indicated that HA alone in the absence of M1 cannot cause VLP production. On the other hand, using VLP assay in mammalian cells (293T and COS-1 cells) Chen et al. (2007) concluded that HA but not M1 was critical for budding and bud release. However, using mutant viruses produced by reverse genetics, it was reported that HA tail mutation alone did not affect virus budding including the shape and size of released particles whereas specific NA mutants in the CT affected virus morphology, generating elongated virus buds and highly irregular particle shape. Furthermore, mutant viruses lacking tails of both HA and NA (HAt-/NAt-) caused the formation of bizarre virus morphology (Jin et al., 1997). Barman et al. (2004) showed that some NA CT substitution and NA TMD replacement mutants produced elongated particles clearly indicating a defect in the pinching-off process of virus buds caused by NA mutants. Similarly, some M1 mutants produced elongated virus particles indicating the involvement of M1 in the last step of bud release (Burleigh et al., 2005, Nayak et al., 2004).
Complete or partial deletion of WSN M2 tail were shown to cause attenuation of virus growth, produce elongated, even filamentous particles in some mutants, indicating an important role of M2 tail in viral assembly and morphogenesis (Iwatsuki-Horimoto et al., 2006). Mutations in Udorn M2 tail were also shown to affect particle release in VLP assay by cDNA transfection suggesting their role in budding (Chen et al., 2008). These M2 mutants were extremely defective in multi-cycle virus growth, and unlike the wt M2, they were defective in M1–M2 interaction as determined by co-immunoprecipitation. However, since the morphology of virus buds on the cell surface and released VLPs were not examined quantitatively by EM, it is unclear if these M2 mutants affected only bud release and bud morphology but also the virus assembly and bud initiation processes.
Furthermore as indicated earlier, budding and release of VLPs using cDNA transfection may only indicate the minimum requirements for budding and bud release and may not mimic the budding process in virus-infected cells since the regulation and control involved in synthesis and assembly of different viral components in virus-infected cells may not be present in cells transfected by cDNAs. Also, VLP production may be affected by over-expression or under-expression of specific viral components in individual cells depending on the permissiveness of specific cells for cDNA transfection and entry of the copy number of individual cDNAs. For example, although equimolar concentrations of different cDNAs are used in transfection, some cells may be transfected with higher copy number of a specific cDNA. Increased expression of specific protein and under expression of other proteins may cause an artificial concentration effect of a specific protein on bud formation and bud release and therefore, may not reflect the role of a specific protein in bud formation and bud release from virus-infected cells.
M1 is clearly an important protein involved not only in virus assembly but also in virus budding and bud release. As noted earlier, M1 affects virus assembly by interacting with transmembrane proteins (HA, NA, and M2) on the outer side and core vRNP on the inner side as well as by bringing and concentrating these viral components to the budding site. Furthermore, M1 is present underneath the lipid bilayer and interacts with each other causing asymmetry in the lipid bilayer and thereby, facilitating membrane bending required for bud initiation. In addition, M1 appears to be a critical component in the final step in bud closing, causing bud release. Bourmakina and García-Sastre (2005) suggested that a threshold level of M1 was required for bud release since low levels of M1 and M2 reduced virus release. However, the virus morphology or viral protein composition was not affected by the low levels of M1 and M2. M1 was found to be critical for maintaining the filamentous morphology of A/Udorn/72 virus particles (Bourmakina and García-Sastre, 2003) and a single mutation in M1 (1A variant) caused transformation from filamentous to spherical particle (Roberts et al., 1998). The role of M1 in virus morphology and virus release is clearly evident also from the budding defect causing formation of elongated buds and released particles by some M1 mutants (Burleigh et al., 2005, Nayak et al., 2004). In addition, an influenza C M1 mutant rMG96A (M1A24T) appears to affect virus morphology by modulating its membrane affinity (Muraki et al., 2007). These studies indicate that M1 plays an important role in many aspects of virus budding including bud closing. Other structural components of influenza virus particles including NEP and polymerase proteins although are critical in many aspects of influenza virus replication appear not be involved in the assembly or the budding process.
5.2. Role of the eight vRNP segments in virus budding
It appears that incorporation of vRNP segments in the virus particles are not absolutely required for budding and bud release since expression of structural proteins (HA, NA, M1, and M2) can release VLP (Gomez-Puertas et al., 2000, Latham and Galarza, 2001, Chen et al., 2007). However, incorporation of all eight (seven for influenza C) vRNA segments are required for the formation of infectious virus particles and M1 and NP play important roles for the incorporation of vRNPs (Neumann et al., 2000, Chen et al., 2007, Li et al., 2009) in virus particles. How these multiple vRNA/vRNP segments are incorporated into virus particles still remains unclear.
Two models have been proposed for the incorporation of eight vRNA/vRNP segments into virions: “random packaging” and “specific packaging”. The “random packaging” model predicts the presence of common structural elements in all vRNPs causing them to be incorporated randomly into virions and therefore incorporation of vRNPs will be concentration dependent. Support for this model comes from the observation that influenza A virions can possess more than eight vRNPs (9–11 vRNAs per virion) (Bancroft and Parslow, 2002, Enami et al., 1991) and at most 1 in 10 or more virus particles are infectious. On the other hand, the “specific packaging” model assumes specific structural features are present in each vRNA/vRNP segment, enabling them to be selectively incorporated into virions. Evidence for this model is deduced mainly from the finding that the various vRNAs are equimolar within viral particles even though their concentrations in infected cells may vary (Smith and Hay, 1982). The selective packaging model has been favored by the earlier studies demonstrating that the small DI vRNAs can competitively inhibit the packaging of their normal counterparts but not that of other vRNAs (Duhaut and McCauley, 1996, Nakajima et al., 1979, Nayak et al., 1985, Nayak et al., 1989, Odagiri and Tobita, 1990). Recent studies have demonstrated the presence of segment-specific packaging signal(s) in 3′ and 5′ UTR as well as adjacent coding regions (varying with specific RNA segment as well as with 3′ or 5′ends). Specific packaging signals have been found for all eight RNA segments (Watanabe et al., 2003, Fujii et al., 2003, Fujii et al., 2005, Liang et al., 2005, Muramoto et al., 2006, Ozawa et al., 2007, Li et al., 2009) and incorporation of some specific RNA segments is critical for the incorporation of other RNA segments (Muramoto et al., 2006, Marsh et al., 2008).
ET studies of serially sectioned influenza A virus particles showing that the RNPs of influenza A virus are organized in a distinct pattern (seven segments of different lengths surrounding a central segment) also argues against random incorporation of RNPs into virions, and supports the “specific packaging” model (Noda et al., 2006). Such a model would require that specific vRNA–vRNA interaction among the vRNP segments in trans would form multi-segmental vRNP macromolecules for incorporation into virus particles and that large vRNP complexes containing eight unique vRNPs in trans are stable. However, such intracytoplasmic multi RNA/RNP complexes have not yet been demonstrated. More importantly, bud closure and virus release should not occur until such vRNP complexes containing eight specific vRNP segments are formed and incorporated in the bud. In support of this model, Fujii et al., 2003 demonstrated that the efficiency of infectious virion production correlated with the number of different vRNA segments. They observed that the higher the number of different vRNA segments the higher was the efficiency of virion production. Recently, specific nucleotide residues in 3′ and 5′ end (coding and non-coding) of PB1, PB2, and PA (Liang et al., 2008, Marsh et al., 2008) as well as in HA (Marsh et al., 2007), have been further shown to play critical role for packaging of specific vRNA segment into progeny virions. The major weakness of this model is that bud closure and virus release do not appear to depend on the incorporation of eight specific RNA segments and particles with fewer RNP segments are found (see Section 3). However, it is possible that segment-specific complex formation and incorporation of viral RNA may occur but may not affect bud closing and bud release.
6. Role of host components in budding
It is well established that budding of enveloped viruses requires the function of both viral and host components. As mentioned earlier, budding and bud release require assembly of viral components either prior to or during the budding process which includes bud initiation in the form of membrane bending, bud growth, i.e. development of fully mature buds and finally, bud scission leading to bud release from the plasma membrane into the outer environment. It is unlikely that accumulation and aggregation of viral components alone in the lipid raft microdomains of the plasma membrane will be sufficient to complete these complex multiple steps from bud initiation to bud release (Reynwar et al., 2007). The role of host components in the assembly processes including transport of viral components to the specific membrane sites, interaction leading to accumulation of different viral components at the budding site, and initiation of membrane bending is well established (Nayak et al., 2004). However, the requirements of specific host components in bud maturation and bud release of influenza viruses are poorly understood.
Furthermore, different steps of the budding process may be either positively or negatively regulated i.e. some host components may facilitate specific steps of the budding process while others may regulate or inhibit one or more steps in budding. Therefore, these inhibitory components are to be removed from the budding site whereas others which facilitate budding are to be brought to the budding site. Furthermore, the same host component may aid in some step(s) of budding but may interfere with other steps. For example, lipid rafts are clearly important in transporting and concentrating viral components such as HA and NA to the apical domain of the plasma membrane and are also involved in transporting RNP and M1 to the budding site and facilitating their interaction during virus assembly. Lipid rafts also may cause membrane bending and bud initiation. However, lipid rafts may regulate and interfere with bud closing and bud release, and removal of lipid rafts may facilitate bud release (Barman and Nayak, 2007), the final step in the budding process. Similarly, cortical actin microfilaments may aid in stabilizing multiple vRNPs into complexes and bringing the vRNP/M1 complex to the budding site and also promote bud growth and maturation by pushing the vRNP complex into the bud. But actin microfilaments may interfere in last step of bud fission. Disruption of actin microfilaments helps in bud release in HeLa (Gujuluva et al., 1994) and MDCK (Roberts and Compans, 1998, Simpson-Holley et al., 2002) cells. Furthermore, microfilament-disrupting agents caused selective inhibition of filament formation and resulted in the preferential assembly and release of spherical particles (Roberts and Compans, 1998, Simpson-Holley et al., 2002). Influenza virus budding also is an active energy-dependent process requiring ATP (see Nayak et al., 2004). Recently, proteomic analysis of purified influenza virus particles has shown the presence of 36 host encoded proteins in virions. Among these, at least seven host proteins including B-actin, annexin A5, tubulin, cyclophilin A and A2, colifilin, and GAPDH were present in virus particles after subtilisin protease treatment whereas CD9 and CD59 were absent in protease-treated virions suggesting the incorporation of some host proteins inside virus particles (Shaw et al., 2008). It is possible that some of these host proteins may also have role in bud closing. As mentioned earlier, B-actin interacts with vRNP/NP (Avalos et al., 1997) and affects virus budding and virus release (Roberts and Compans, 1998, Simpson-Holley et al., 2002).
For a number of enveloped viruses, the role of specific host components in bud release has been demonstrated. The role of VPS (vesicular protein sorting) components in bud release of HIV and other enveloped viruses has been investigated extensively. Bud scission of these viruses depends on the interaction of their L domain(s) with the components(s) of VPS pathways involved in giving rise to multivesicular bodies (MVB). Tsg101 and AIP1/Alix, the components of ESCRT (endosomal sorting complex required for transport) interact with L domains and require the function of AAA-ATPase of Vps4 for bud release of HIV (see Fujii et al., 2007, Demirov and Freed, 2004). However, influenza virus proteins do not contain any identifiable L domain(s) and its budding is not affected by dominant negative Vps4 (Chen et al., 2007) or by proteasome inhibitors (Hui and Nayak, 2001, Khor et al., 2003). Recently, budding of RSV was shown to be affected by dominant negative mutant (FIP2deltaC2) of Rab11 family interacting protein2 (FIP2) producing elongated filaments (Utley et al., 2008). RSV does not have an identifiable L domain and is not affected by dominant negative Vps4 or by proteasome inhibitors (Utley et al., 2008). However, specific RSV protein domains interacting with FIP2 protein has not yet been identified.
By mutational analysis using reverse genetics, mutants of three influenza proteins NA, M2 and M1 have been shown to interfere individually in the final step in bud closure producing elongated and tethered virus particles (Nayak et al., 2004, Barman et al., 2004, Burleigh et al., 2005, Bourmakina and García-Sastre, 2003, Bourmakina and García-Sastre, 2005, Roberts et al., 1998, Chen et al., 2008). However, cause of the budding defect by these mutant influenza viruses including possible interaction with host components is yet to be determined. Involvement of transmembrane proteins affecting virus morphogenesis and bud release is rather uncommon. However, NS3 and NS3A which are non-structural glycosylated proteins present at the surface of BTV-infected cells affect virus budding (Hyatt et al., 1993). Also, F13L envelope protein of vaccinia virus (Honeychurch et al., 2007) has been shown to possess L like domain and use Vps pathways of ESCRT complex.
Lipid rafts are lipid microdomains enriched in sphingolipids and cholesterol. As mentioned above, lipid rafts have been shown to play critical roles in many aspects of the virus life cycle, including viral entry and fusion, viral protein transport and targeting, and finally viral assembly and budding process (see Nayak and Barman, 2002, Nayak and Hui, 2004, Nayak et al., 2004, Ono and Freed, 2005, Schmitt and Lamb, 2004, Schmitt and Lamb, 2005). They contain lipids in liquid order (l o) phase and are relatively resistant to nonionic detergent at a low temperature (see Brown and London, 1998, Simons and Toomre, 2000). Among the three influenza viral envelope proteins, HA and NA, but not M2, use lipid rafts as a platform for apical transport and remain associated with lipid raft microdomains present on cellular and viral membranes (Nayak and Hui, 2004, Nayak et al., 2004). However, Schroeder et al. (2005) reported that M2 is a cholesterol-binding protein and proposed that M2 may play a critical role in virus budding. Mutational analysis of HA and NA transmembrane domain (TMD) and cytoplasmic tail (CT) showed that strong raft association is not obligatory for their apical transport and virus assembly (Barman and Nayak, 2000, Barman et al., 2004, Takeda et al., 2003). Lipid rafts have been also proposed to be involved in transporting NP/vRNP to the apical side of polarized epithelial cells (Carrasco et al., 2004). More recently, it was shown that membrane accumulation of hemagglutinin and its tight association with lipid raft domains triggered activation of the MAPK cascade via protein kinase C alpha activation and induced RNP export from nucleus into cytoplasm (Marjuki et al., 2006). Authors hypothesized the presence of an auto-regulative mechanism which coordinates timing of RNP export to a point when all viral components are ready for virus budding at the plasma membrane (Marjuki et al., 2006).
In the envelope of released virions, both HA and NA remain raft associated, whereas M2 does not associate with lipid rafts. Therefore, the influenza viral envelope is not a homogeneous lipid membrane but a mosaic mixture of both raft-associated and non-raft-associated lipid microdomains even though the majority of lipids present in the viral envelope are in the highly ordered l o phase, and the influenza virus envelope is enriched in cholesterol-dependent detergent-insoluble lipids (Zhang et al., 2000, Scheiffele et al., 1999). Furthermore, protein–protein interactions may facilitate bringing non-raft-associated proteins to lipid raft microdomains. For example, interactions of influenza virus M1 with HA and NA bring M1, a non-raft-associated protein, into lipid rafts (Ali et al., 2000). Using recombinant influenza A virus containing mutant HA that does not associate with lipid rafts, Takeda et al. (2003) observed reduced budding in cells infected with mutant virus compared to that seen with wild-type virus. These observations suggest that influenza viruses use lipid rafts as their budding platform. However, little is known about the role of lipid rafts in the budding process, specifically in bud release. Cholesterol is a known critical structural component of lipid rafts and depletion of cholesterol leads to disorganization of lipid raft microdomains and dissociation of proteins bound to the lipid rafts (Hanada et al., 1995, Hannan and Edidin, 1996, Zhang et al., 2000, Scheiffele et al., 1999). Recently, it was shown that depletion of cholesterol from virus-infected cells by MβCD treatment for a short time at the late phase of infection facilitated bud completion and increased virus particle release (Barman and Nayak, 2007). MβCD is a strictly surface-acting agent and can selectively and rapidly remove cholesterol from the plasma membrane in preference to other membrane lipids and has been widely used in studying the effects of cholesterol depletion and lipid raft disassembly (Gosselin-Grenet et al., 2006, Ono and Freed, 2001). Therefore, short MβCD treatment at the late phase of infection was done to avoid the effect of lipid raft disruption on protein transport (Carrasco et al., 2004, Keller and Simons, 1998, Prydz and Simons, 2001) and virus assembly (Leser and Lamb, 2005, Scheiffele et al., 1999, Takeda et al., 2003, Zhang et al., 2000). The results show that depletion of cholesterol from virus-infected cells by MβCD treatment for a short time facilitated bud completion and increased virus particle release. Increased release of virus particles was quantified by three independent assays: (i) higher infectious virus titer as determined by PFU assay, (ii) increased particle release as determined by protein analysis, and (iii) increased number of virus particles as determined by negative-stain EM. Increased release of virus particles correlated with lipid raft disruption as measured by the decreased TX-100 insolubility of cell surface HA and NA. Finally, and most importantly, the presence of exogenous cholesterol was shown to reverse the effect of MβCD. Exogenous cholesterol increased the TX-100 insolubility of HA and NA, reduced the release of virus particles, and produced deformed, elongated particles (Fig. 5 ). Recently, Wang et al. (2007) reported that the interferon-inducible protein viperin inhibited influenza virus release, more specifically the bud pinching-off process (Wang et al., 2007). Authors have further stated that the expression of viperin disrupted lipid rafts and concluded that viperin induced disruption of lipid rafts was responsible for reduced virus release. Their conclusion contradicts the observation stated above that disruption of lipid raft by cholesterol depletion enhances virus particle release and that higher integrity of lipid raft by exogenous cholesterol caused reduction of particle release (Barman and Nayak, 2007). One possible explanation of these conflicting results may be the definition of lipid raft disruption and the assay used in determining lipid raft disruption by Wang et al. (2007). In standard assays, insolubility of the cell surface HA by 1% TX-100 treatment at 4 °C is used for comparing and defining the integrity and disruption of lipid rafts. However, Wang et al. (2007) used very low concentration (0.1%) TX-100 solubility followed by 1% TX-100 treatment of the total HA, not just cell surface HA in viperin expressed and mock treated cells for assaying lipid raft disruption. Furthermore in their analysis, immature (non-lipid-raft-associated) and mature (lipid raft-associated) forms of HA were not separated or quantified. It is therefore likely that viperin expression did not cause a major disruption of lipid rafts and that budding defect caused by viperin expression was due to some other effect of viperin and not due to lipid raft disruption.
Fig. 5.
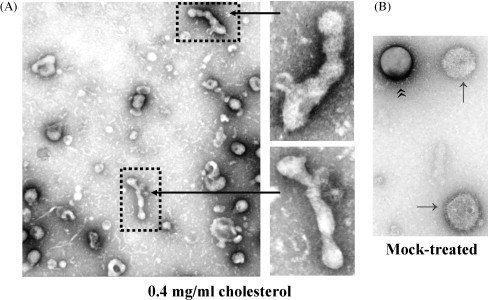
Deformed particles are produced in the presence of exogenous cholesterol. MDCK cells were infected (3.0 moi) with WSN virus and at 5 hpi virus-infected cells were either treated with 0.4 mg/ml water soluble cholesterol containing 6 mM MβCD (A) or mock treated (B). At 12 hpi, released virus particles were purified and examined by negative-stain EM (Barman and Nayak, 2007). Panel A, left picture was taken at 29,000×, and portions were further magnified at 72,000× (panel A, right). Panel B picture was taken at 72,000× magnification. Daisy-chain structures of some virus particles (panel A) released from cells in the presence of exogenous cholesterol indicate defective bud pinching-off process.
Bud completion and the pinching-off process require fusion of apposing plasma membranes and viral membranes, leading to fission of virus bud and bud release. It is possible that lipid rafts may have two opposite effects on the influenza virus budding process. Initially, lipid rafts may facilitate bud formation by bringing the viral components to and concentrating the components at the budding site, as well as causing asymmetry in the membrane bilayers and favoring membrane bending and bud initiation (Nayak et al., 2004). However, at the final stage of bud completion, the lipid raft microdomain may slow down and regulate bud closure because of its increased viscosity and rigidity. This idea is supported by the observation that exogenous cholesterol increased the TX-100 insolubility of raft-associated proteins, inhibited virus release (Barman and Nayak, 2007), and produced deformed, and elongated buds with multiple incomplete buds attached to each other (Fig. 5). These results suggest defective bud scission in the presence of excess exogenous cholesterol. The mosaic nature of the viral membrane containing both raft-associated and non-raft-associated lipid microdomains may have different functions in the virus budding process involving bud initiation and bud closure. Disruption of lipid rafts at the final stage may facilitate bud closure, thereby increasing virus release.
7. Budding process
Influenza viruses selectively bud from the apical domain of polarized epithelial cells, which would require the transport of all viral components to the budding site, and their interaction and assembly either prior to or subsequent to their arrival to the budding site. As mentioned earlier, transporting viral components to the budding site requires the involvement of exocytic pathway and their components. Similarly, in the assembly process, multiple cellular components including actin microfilaments, lipid rafts as well as exocytic pathway play critical role in concentrating the viral components and providing favorable environment for their interaction and formation of sub-viral complexes of higher order in the assembly process. The budding process itself requires three major steps: bud initiation, bud growth, and bud completion releasing the virus from the host cell membrane. Each of these steps involves interaction of multiple host and viral components.
7.1. Bud initiation
Bud initiation requires outward bending of the plasma membrane and involves transition of a more planar membrane structure to a curved structure at the budding site. Although the structural nature and biochemical properties as well as the physical forces at these sites responsible for membrane bending and bud initiation are unknown, it is likely that both lipid rafts and raft-associated proteins present at the budding site play an important role in causing membrane curvature and bud initiation. Lipid rafts causing asymmetry in lipid bilayers can cause intrinsic curvature of one lipid monolayer relative to the other monolayer leading to membrane bending (see Nayak and Hui, 2004). Membrane deformation can be caused by selective transfer of lipids between the lipid bilayers, interaction of cholesterol into the budding leaflet as well as hydrolytic cleavage of phosphocholine head groups of sphingomyelin by sphingomyelinase generating smaller head groups (Holopainen et al., 2000). Additionally, BAR (Bin/Amphiphysin/Rsv) domain has been shown to cause membrane curvature (Peter et al., 2004) and is known to be present in a number of proteins involved in vesicle formation and recycling. However, role of any of these proteins in influenza virus budding is unknown. In addition to lipid microdomains, the presence of specific viral proteins including HA, NA, M2, and M1 proteins may further facilitate membrane bending (see Nayak and Hui, 2004). Among these, M1 interacting with the inner leaflet of lipid bilayers is likely to play a critical role in bud initiation. Clustering of M1 due to M1/M1 interaction underneath the lipid bilayers can cause outward membrane bending and bud initiation.
7.2. Bud growth
Bud growth leading to bud maturation is the intermediate stage between the bud initiation and bud release. Bud growth determines the size and the morphology of released virus particles. However, what factors or forces determine and regulate bud growth remains unclear. For most viruses regardless containing either icosahedral (e.g. SFV) or helical (e.g. VSV) nucleocapsids, the size of the nucleocapsids determines the size of the virions. However, influenza viruses are highly pleomorphic and the size of the released particles can vary from spheroidal, elongated and even filamentous (Fig. 2) and the content of the nucleocapsids is not the major factor for bud growth. Influenza virus bud growth appears rather to depend on two forces, a pulling and a pushing force. The pulling force is primarily provided by the transmembrane proteins along with M1 which are pulling nucleocapsids into the bud. On the other hand, the cortical actin microfilaments which bind to viral RNPs provide the pushing force for incorporating the nucleocapsids and M1 into the bud. Electron tomography analysis of virus buds attached to the cell surface shows that helical nucleocapsids are perpendicular to the cell membrane while being incorporated into the buds and that buds are essentially complete still remaining attached to the cell membrane and are of similar size (Fig. 6, Fig. 7 ).
Fig. 6.
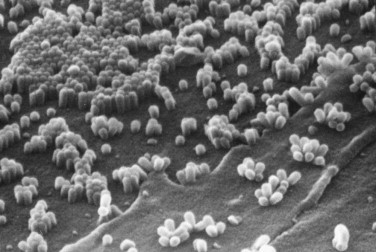
Scanning electron micrographs of spheroidal influenza virus buds attached to infected cells (40,000×). This micrograph was kindly provided by and is printed with the permission of David Hockley of the National Institute of Biological Standard and Control at Hertfordshire, UK.
Fig. 7.

Virus buds at the cell surface by ET. At 12 hpi WSN-infected MDCK cells were processed for thin section and examined by ET. This picture represents one slice through the central region of the virus buds. One can see the parallel arrangement of the vRNPs inside the bud perpendicular to cell surface. The bud neck (⇒) shows gaps indicating possible absence of M1. HA and NA spikes are seen on the bud envelope.
As mentioned earlier, influenza virus particles are highly pleomorphic in shape and size. Basically, there are two types of pleomorphism observed among influenza viruses: (i) strain-specific i.e. strain to strain variation which may also vary depending on the host cell; (ii) variation within the population of plaque purified virus in the same cell. Clearly, the genome of the virus strain is an important factor in determining the particle size and shape of a specific virus strain (e.g. Udorn vs. WSN). Specific viral genes involved in determining filamentous vs. spheroidal forms have been identified (see Section 5). Similarly, role of polarized epithelial cells and intact actin microfilaments were found to be critical in maintaining the filamentous form of Udorn virus. However, the cause of pleomorphism in plaque purified influenza viruses is not well understood. Whatever may be the viral and cellular factors involved in viral pleomorphism, these are likely to affect bud growth and closing and will eventually affect the shape and size of the virus particles. This is not to state that factors affecting bud closing will always affect bud size and bud shape; but factors affecting bud shape and bud size will always affect bud closing. The viral and host factors affecting the size of virus particles will either hinder or facilitate bud closing.
7.3. Bud closing
Bud closing is the final step for the scission of the bud and release of the virus particle into the outer environment. Bud closure would involve fusion of two ends of the apposing viral membranes as well as that of the apposing cell membranes leading to fission of the virus bud from the infected cell membrane (Fig. 8 ). This would require bringing and holding the apposing membrane ends next to each other in close proximity so that each end can find its counterpart causing fusion of corresponding lipid bilayers. Virus buds would then become separated from the membrane of the parent infected cell. This model holds that two lipid bilayers are to be held in very close proximity for fusion to occur. Host and viral factors could have both positive and negative impact on bud release. Some factors could interfere in bringing the apposing ends close to each other and therefore, should be removed. Others could help in bringing and holding the membrane ends close to each other for fusion to occur and therefore should be brought to the pinching-off site. As mentioned above, lipid rafts and the cortical actin microfilaments, though critical in many aspects of the budding process, are inhibitory in the final step of bud closing and therefore should be removed from the pinching-off site. Clearly, there are other host factors, yet to be identified, which are required for bringing and holding the apposing viral and cellular membranes next to each other for facilitating fusion and fission. Among the viral components, M2 may play critical role in the pinching-off process. M2 when present in the neck of the bud may aid in bud release (Schroeder et al., 2005) by bringing non-lipid rafts in this region (Fig. 8). Absence of M1 protein underneath the lipid bilayers and absence of spikes on the outer surface may indicate the absence of lipid rafts (Fig. 8). From CT analysis, such lipid microdomains have been proposed to be the preferred sites for the bud pinching off (Harris et al., 2006).
Fig. 8.
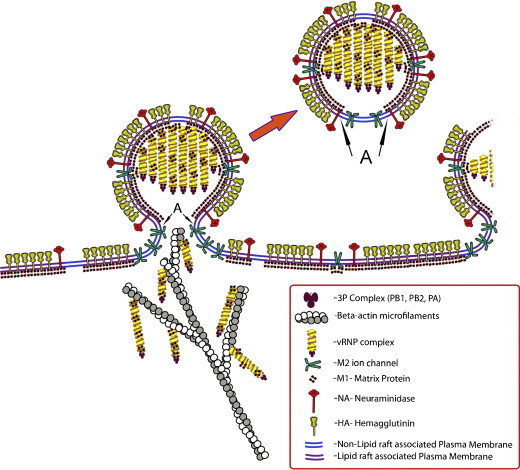
Schematic illustration of the pinching-off process of influenza virus bud. The pinching-off region (neck) is shown to be viral membrane devoid of lipid rafts (Barman and Nayak, 2007), devoid of HA and NA spikes outside and M1 inside the lipid bilayers (Harris et al., 2006) and may contain M2 (Schroeder et al., 2005).
As mentioned earlier, bud closing of influenza virus is very inefficient and only a small fraction of virus buds are released while the majority of virus buds remain attached to the cell membrane even though they appear mature (Fig. 6). Both host and virus factors may be contributing towards the rate limiting step of the pinching-off process. Influenza virus budding also appears to be an active, energy-dependent process and metabolic inhibitors such as antimycin A, CCCP, FCCP, oligomycin as well as ATP analogues such as ATPγS and AMP-PNP inhibited influenza virus budding (Hui and Nayak, 2001). Therefore, limited energy at the end of infectious cycle may be a factor for the inefficient release of virus particles. Among other host factors, actin microfilaments may interfere with bud closing, and conversely, disassembly of cortical actin microfilaments may facilitate it. This notion is supported by several observations, including the release of virus particles in abortively infected HeLa cells (Gujuluva et al., 1994), conversion of filamentous Udorn (H3N2) virus to spherical virus and enhanced release of WSN and PR8 spherical particles in polarized MDCK cells (Roberts and Compans, 1998, Simpson-Holley et al., 2002) by microfilament-disrupting agents.
Similarly, lipid rafts may interfere with bud release, owing to its increased viscosity and rigidity. Indeed, virus release was enhanced by cholesterol depletion and virus budding was reduced by addition of exogenous cholesterol (Barman and Nayak, 2007). Interestingly, members of the ESCART components and proteins containing WW domains, which are critical for bud closing of many viruses (Freed and Mouland, 2006), appeared not to be involved in influenza virus bud closing (Chen and Lamb, 2008). Similarly, proteasome inhibitors including ubiquitination inhibitors did not affect influenza virus budding (Hui and Nayak, 2001).
Scission of influenza virus buds from infected cells is the last step in completion of virus life cycle. This step appears to be rate limiting since morphological analysis by both thin section transmission (Barman and Nayak, data not shown) and scanning EM (Fig. 6) shows that a large number of mature virus particles remain attached to the cell membrane and only a relatively small fraction of virus buds (∼10%) are released. The kinetics of virus release relative to the presence of virus buds awaits further investigation in order to understand the cause(s) and mechanism(s) for such inefficient bud completion.
8. Conclusion
Influenza virus assembly and budding are the last steps in the virus replication process and are critically important to the survival and transmission of the virus and to the pathogenesis of diseases. Budding requires transport and assembly of the virus components at the budding site leading to bud initiation, growth and release. In this review, we have covered the steps involved in the assembly and budding processes and the roles played by both virus and host components in these steps. Although much is known about the different steps of virus replication including entry, uncoating, synthesis of viral RNA and proteins, transport of viral components, our knowledge about the process of bud closing and bud release is very limited. A better understanding of this important process in influenza virus biology awaits answers to the following questions:
-
1.
What are the specific determinants of viral components required for preferential budding of influenza virus from the apical plasma membrane of polarized epithelial cells? In addition to HA and NA, the role of NP in selecting the apical budding site requires further investigation.
-
2.
How are all the eight specific vRNP segments incorporated into a single virus bud? Is there a stable complex of eight RNP segments in the cytoplasm of infected cells outside the virus buds? Why are there no apparent check points in the budding of infectious vs. non-infectious viruses? Is bud closure and release a random, unregulated process? Do the majority of released virus particles contain eight vRNPs or do the genomic content of individual particles vary?
-
3.
What is the cause of such a high degree of pleomorphism among the released virus particles even with the same genomic content?
-
4.
Are there any regulations of bud closing? If not, why are bud closing and release so inefficient? If so, what are the host and viral factors involved in regulating bud closing? A more detailed structural analysis of native and mutant virus particles, as well as the budding and pinching-off sites on the plasma membrane by electron tomography would be critical for understanding the bud release process.
In summary, our knowledge about the roles of host factors in influenza virus bud closing is critically lacking. A better understanding of the influenza virus budding process will not only help us in identifying new targets for therapeutic intervention against pandemic and epidemic influenza, but also will facilitate in the production of more potent and cheaper vaccines. For example, more efficient bud closing will produce more virus particles in the process of vaccine production and may also produce higher yield of VLP antigen and thus may lead to better and cheaper vaccines.
Acknowledgments
Research activities in the authors’ labs (DPN and ZHZ) were supported by USPHS grants from NIH/NIAID (AI16348 DPN, AI41681 DPN, AI069015 ZHZ) and NIGMS (GM071940 ZHZ). We thank the following members in the Zhou group for their assistance in the research reported in Figs. 2, 3 and 7: Ivo Atanasov (Figs. 2 and 3), Wei Dai (Fig. 7), Jennifer Wong (Fig. 3), and Lingpeng Cheng (Fig. 2).
References
- Akarsu H., Burmeister W.P., Petosa C., Petit I., Muller C.W., Ruigrok R.W., Baudin F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2) EMBO J. 2003;22(18):4646–4655. doi: 10.1093/emboj/cdg449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A., Avalos R.T., Ponimaskin E., Nayak D.P. Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J. Virol. 2000;74(18):8709–8719. doi: 10.1128/jvi.74.18.8709-8719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos R.T., Yu Z., Nayak D.P. Association of influenza virus NP and M1 proteins with cellular cytoskeletal elements in influenza virus-infected cells. J. Virol. 1997;71(4):2947–2958. doi: 10.1128/jvi.71.4.2947-2958.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft C.T., Parslow T.G. Evidence for segment-nonspecific packaging of the influenza a virus genome. J. Virol. 2002;76(14):7133–7139. doi: 10.1128/JVI.76.14.7133-7139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman S., Adhikary L., Chakraborti A.K., Bernas C., Kawaoka Y., Nayak D.P. Role of transmembrane domain and cytoplasmic tail amino acid sequences of influenza A virus neuraminidase in raft-association and virus budding. J. Virol. 2004;78(10):5258–5269. doi: 10.1128/JVI.78.10.5258-5269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman S., Adhikary L., Kawaoka Y., Nayak D.P. Influenza A virus hemagglutinin containing basolateral localization signal does not alter the apical budding of a recombinant influenza A virus in polarized MDCK cells. Virology. 2003;305(1):138–152. doi: 10.1006/viro.2002.1731. [DOI] [PubMed] [Google Scholar]
- Barman S., Nayak D.P. Analysis of the transmembrane domain of influenza virus neuraminidase, a type II transmembrane glycoprotein, for apical sorting and raft association. J. Virol. 2000;74:6538–6545. doi: 10.1128/jvi.74.14.6538-6545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman S., Nayak D.P. Lipid raft disruption by cholesterol depletion enhances influenza A virus budding from MDCK cells. J. Virol. 2007;81(22):12169–12178. doi: 10.1128/JVI.00835-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin F., Petit I., Weissenhorn W., Ruigrok R.W.H. In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology. 2001;281:102–108. doi: 10.1006/viro.2000.0804. [DOI] [PubMed] [Google Scholar]
- Baumeister W. Electron tomography: towards visualizing the molecular organization of the cytoplasm. Curr. Opin. Struct. Biol. 2002;12:679–684. doi: 10.1016/s0959-440x(02)00378-0. [DOI] [PubMed] [Google Scholar]
- Bourmakina S.V., García-Sastre A. The morphology and composition of influenza A virus particles are not affected by low levels of M1 and M2 proteins in infected cells. J. Virol. 2005;79(12):7926–7932. doi: 10.1128/JVI.79.12.7926-7932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourmakina S.V., García-Sastre A. Reverse genetics studies on the filamentous morphology of influenza A virus. J. Gen. Virol. 2003;84:517–527. doi: 10.1099/vir.0.18803-0. [DOI] [PubMed] [Google Scholar]
- Brown D.A., London E. Functions of lipid rafts in biological membrane. Annu. Rev. Cell Dev. Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Burleigh L.M., Calder L.J., Skehel J.J., Steinhauer D.A. Influenza A viruses with mutations in the m1 helix six domain display a wide variety of morphological phenotypes. J. Virol. 2005;79(2):1262–1270. doi: 10.1128/JVI.79.2.1262-1270.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco M., Amorom M.J., Digard P. Lipid raft-dependent targeting of the influenza A virus nucleoprotein to the apical plasma membrane. Traffic. 2004;5(12):979–992. doi: 10.1111/j.1600-0854.2004.00237.x. [DOI] [PubMed] [Google Scholar]
- Chen B.J., Lamb R.A. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology. 2008;372(2):221–232. doi: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.J., Leser G.P., Jackson D., Lamb R.A. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 2008;82(20):10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.J., Leser G.P., Morita E., Lamb R.A. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 2007;81(13):7111–7123. doi: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirov D.G., Freed E.O. Retrovirus budding. Virus Res. 2004;106(2):87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Duhaut S.D., McCauley J.W. Defective RNAs inhibit the assembly of influenza virus genome segments in a segment-specific manner. Virology. 1996;216(2):326–337. doi: 10.1006/viro.1996.0068. [DOI] [PubMed] [Google Scholar]
- Elton D., Amorim M.J., Medcalf L., Digard P. ‘Genome gating’; polarized intranuclear trafficking of influenza virus RNPs. Biol. Lett. 2005;1(2):113–117. doi: 10.1098/rsbl.2004.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elton D., Digard P., Tiley L., Ortin J. Structure and function of the influenza virus RNP. In: Kawaoka Y., editor. Influenza Virology; Current Topics. Caister Academic Press; Wymondham: 2006. pp. 1–36. [Google Scholar]
- Enami M., Sharma G., Benham C., Palese P. An influenza virus containing nine different RNA segments. Virology. 1991;185(1):291–298. doi: 10.1016/0042-6822(91)90776-8. [DOI] [PubMed] [Google Scholar]
- Freed E.O., Mouland A.J. The cell biology of HIV-1 and other retroviruses. Retrovirology. 2006;3:77. doi: 10.1186/1742-4690-3-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K., Fujii Y., Noda T., Muramoto Y., Watanabe T., Takada A., Goto H., Horimoto T., Kawaoka Y. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J. Virol. 2005;79(6):3766–3774. doi: 10.1128/JVI.79.6.3766-3774.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K., Hurley J.H., Freed E.O. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat. Rev. Microbiol. 2007;5(12):912–916. doi: 10.1038/nrmicro1790. [DOI] [PubMed] [Google Scholar]
- Fujii Y., Goto H., Watanabe T., Yoshida T., Kawaoka Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. U.S.A. 2003;100(4):2002–2007. doi: 10.1073/pnas.0437772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyoshi Y., Kume N.P., Sakata K., Sato S.B. Fine structure of influenza A virus observed by electron cryo-microscopy. EMBO J. 1994;13(2):318–326. doi: 10.1002/j.1460-2075.1994.tb06264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos B.K., Yeager M., Sundquist W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008;18(2):203–217. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garoff H., Wilschut J., Liljestrom P., Wahlberg J.M., Bron R., Suomalainen M., Smyth J., Salminen A., Barth B.U., Zhao H. Assembly and entry mechanisms of Semliki Forest virus. Arch. Virol. 1994;(Suppl. 9):329–338. doi: 10.1007/978-3-7091-9326-6_33. [DOI] [PubMed] [Google Scholar]
- Gomez-Puertas P., Albo C., Perez-Pastrana E., Vivo A., Portela A. Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 2000;74(24):11538–11547. doi: 10.1128/jvi.74.24.11538-11547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin-Grenet A.S., Mottet-Osman G., Roux L. From assembly to virus particle budding: pertinence of the detergent resistant membranes. Virology. 2006;344(2):296–303. doi: 10.1016/j.virol.2005.09.035. [DOI] [PubMed] [Google Scholar]
- Gujuluva C.N., Kundu A., Murti K.G., Nayak D.P. Abortive replication of influenza virus A/WSN/33 in HeLa229 cells: defective viral entry and budding processes. Virology. 1994;204(2):491–505. doi: 10.1006/viro.1994.1563. [DOI] [PubMed] [Google Scholar]
- Hanada K., Nishijima M., Akamatsu Y., Pagano R.E. Both sphingolipids and cholesterol participate in the detergent insolubility of alkaline phosphatase, a glycosylphosphatidylinositol-anchored protein, in mammalian membranes. J. Biol. Chem. 1995;270:6254–6260. doi: 10.1074/jbc.270.11.6254. [DOI] [PubMed] [Google Scholar]
- Hannan L.A., Edidin M. Traffic, polarity, and detergent solubility of a glycosylphosphatidylinositol-anchored protein after LDL-deprivation of MDCK cells. J. Cell Biol. 1996;133:1265–1276. doi: 10.1083/jcb.133.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A., Cardone G., Winkler D.C., Heymann J.B., Brecher M., White J.M., Steven A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. U.S.A. 2006;103(50):19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobman T.C. Targeting of viral glycoproteins to the Golgi complex. Trends Microbiol. 1993;1(4):124–130. doi: 10.1016/0966-842X(93)90126-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holopainen J.M., Angelova M.I., Kinnunen P.K. Vectorial budding of vesicles by asymmetrical enzymatic formation of ceramide in giant liposomes. Biophys. J. 2000;78(2):830–838. doi: 10.1016/S0006-3495(00)76640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeychurch K.M., Yang G., Jordan R., Hruby D.E. The vaccinia virus F13L YPPL motif is required for efficient release of extracellular enveloped virus. J. Virol. 2007;81(13):7310–7315. doi: 10.1128/JVI.00034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E.K., Nayak D.P. Role of ATP in influenza virus budding. Virology. 2001;290(2):329–341. doi: 10.1006/viro.2001.1181. [DOI] [PubMed] [Google Scholar]
- Hyatt A.D., Zhao Y., Roy P. Release of bluetongue virus-like particles from insect cells is mediated by BTV nonstructural protein NS3/NS3A. Virology. 1993;193(2):592–603. doi: 10.1006/viro.1993.1167. [DOI] [PubMed] [Google Scholar]
- Iwatsuki-Horimoto K., Horimoto T., Noda T., Kiso M., Maeda J., Watanabe S., Muramoto Y., Fujii K., Kawaoka Y. The cytoplasmic tail of the influenza A virus M2 protein plays a role in viral assembly. J. Virol. 2006;80(11):5233–5240. doi: 10.1128/JVI.00049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., Leser G.P., Lamb R.A., Zhang J. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997;16(6):1236–1247. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller P., Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J. Cell Biol. 1998;140:1357–1367. doi: 10.1083/jcb.140.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor R., McElroy L.J., Whittaker G.R. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic. 2003;4(12):857–868. doi: 10.1046/j.1398-9219.2003.0140.x. [DOI] [PubMed] [Google Scholar]
- Latham T., Galarza J.M. Formation of wild-type and chimeric influenza virus-like particles following simultaneous expression of only four structural proteins. J. Virol. 2001;75(13):6154–6165. doi: 10.1128/JVI.75.13.6154-6165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear J.D. Proton conduction through the M2 protein of the influenza A virus; a quantitative, mechanistic analysis of experimental data. FEBS Lett. 2003;552(1):17–22. doi: 10.1016/s0014-5793(03)00778-6. [DOI] [PubMed] [Google Scholar]
- Leser G.P., Lamb R.A. Influenza virus assembly and budding in raft-derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology. 2005;342(2):215–227. doi: 10.1016/j.virol.2005.09.049. [DOI] [PubMed] [Google Scholar]
- Li Z., Watanabe T., Hatta M., Watanabe S., Nanbo A., Ozawa M., Kakugawa S., Shimojima M., Yamada S., Neumann G., Kawaoka Y. Mutational analysis of conserved amino acids in the influenza A virus nucleoprotein. J. Virol. 2009;83(9):4153–4162. doi: 10.1128/JVI.02642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Hong Y., Parslow T.G. cis-Acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J. Virol. 2005;79(16):10348–10355. doi: 10.1128/JVI.79.16.10348-10355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Huang T., Ly H., Parslow T.G., Liang Y. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J. Virol. 2008;82(1):229–236. doi: 10.1128/JVI.01541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Naim H.Y., Rodriguez A.C., Roth M.G. Mutations in the middle of the transmembrane domain reverse the polarity of transport of the influenza virus hemagglutinin in MDCK epithelial cells. J. Cell Biol. 1998;142(1):51–57. doi: 10.1083/jcb.142.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisner A., Klenk H., Herrler G. Polarized budding of measles virus is not determined by viral surface glycoproteins. J. Virol. 1998;72(6):5276–5278. doi: 10.1128/jvi.72.6.5276-5278.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjuki H., Alam M.I., Ehrhardt C., Wagner R., Planz O., Klenk H.D., Ludwig S., Pleschka S. Membrane accumulation of influenza A virus hemagglutinin triggers nuclear export of the viral genome via protein kinase Calpha-mediated activation of ERK signaling. J. Biol. Chem. 2006;281(24):16707–16715. doi: 10.1074/jbc.M510233200. [DOI] [PubMed] [Google Scholar]
- Marsh G.A., Hatami R., Palese P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J. Virol. 2007;81(18):9727–9736. doi: 10.1128/JVI.01144-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh G.A., Rabadan R., Levine A.J., Palese P. Highly conserved regions of influenza A virus polymerase gene segments are critical for efficient viral RNA packaging. J. Virol. 2008;82(5):2295–2304. doi: 10.1128/JVI.02267-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCown M.F., Pekosz A. The influenza A virus M2 cytoplasmic tail is required for infectious virus production and efficient genome packaging. J. Virol. 2005;79(6):3595–3605. doi: 10.1128/JVI.79.6.3595-3605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCown M.F., Pekosz A. Distinct domains of the influenza a virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J. Virol. 2006;80(16):8178–8189. doi: 10.1128/JVI.00627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora R., Rodriguez-Boulan E., Palese P., Garcia-Sastre A. Apical budding of a recombinant influenza A virus expressing a hemagglutinin protein with a basolateral localization signal. J. Virol. 2002;76(7):3544–3553. doi: 10.1128/JVI.76.7.3544-3553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraki Y., Murata T., Takashita E., Matsuzaki Y., Sugawara K., Hongo S. A mutation on influenza C virus M1 protein affects virion morphology by altering the membrane affinity of the protein. J. Virol. 2007;81(16):8766–8773. doi: 10.1128/JVI.00075-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramoto Y., Takada A., Fujii K., Noda T., Iwatsuki-Horimoto K., Watanabe S., Horimoto T., Kida H., Kawaoka Y. Hierarchy among viral RNA (vRNA) segments in their role in vRNA incorporation into influenza A virions. J. Virol. 2006;80(5):2318–2325. doi: 10.1128/JVI.80.5.2318-2325.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murti K.G., Webster R.G., Jones I.M. Localization of RNA polymerases on influenza viral ribonucleoproteins by immunogold labeling. Virology. 1988;164(2):562–566. doi: 10.1016/0042-6822(88)90574-0. [DOI] [PubMed] [Google Scholar]
- Nakajima K., Ueda M., Sugiura A. Origin of small RNA in von Magnus particles of influenza virus. J. Virol. 1979;29(3):1142–1148. doi: 10.1128/jvi.29.3.1142-1148.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak D.P., Barman S. Role of lipid rafts in virus assembly and budding. Adv. Virus Res. 2002;77:1977–1983. doi: 10.1016/s0065-3527(02)58001-5. [DOI] [PubMed] [Google Scholar]
- Nayak D.P., Chambers T.M., Akkina R.K. Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. Curr. Top. Microbiol. Immunol. 1985;114:103–151. doi: 10.1007/978-3-642-70227-3_3. [DOI] [PubMed] [Google Scholar]
- Nayak D.P., Chambers T.M., Akkina R.M. Structure of defective-interfering RNAs of influenza viruses and their role in interference. In: Krug R.M., editor. The Influenza Viruses. Plenum Press; New York: 1989. pp. 269–317. [Google Scholar]
- Nayak D.P., Hui E.-K.W. The role of lipid microdomains in virus biology. In: Quinn P.J., editor. vol. 37. Kluwer Academic/Plenum Publishers; New York: 2004. pp. 443–491. (Subcellular Biochemistry). [DOI] [PubMed] [Google Scholar]
- Nayak D.P., Hui E.-K.W., Barman S. Assembly and budding of influenza virus. Virus Res. 2004;106:147–165. doi: 10.1016/j.virusres.2004.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G., Watanabe T., Kawaoka Y. Plasmid-driven formation of influenza virus-like particles. J. Virol. 2000;74(1):547–551. doi: 10.1128/jvi.74.1.547-551.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T., Sagara H., Yen A., Takada A., Kida H., Cheng R.H., Kawaoka Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature. 2006;439(7075):490–492. doi: 10.1038/nature04378. [DOI] [PubMed] [Google Scholar]
- Noton S.L., Medcalf E., Fisher D., Mullin A.E., Elton D., Digard P. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J. Gen. Virol. 2007;88(8):2280–2290. doi: 10.1099/vir.0.82809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odagiri T., Tobita K. Mutation in NS2, a nonstructural protein of influenza A virus, extragenically causes aberrant replication and expression of the PA gene and leads to generation of defective interfering particles. Proc. Natl. Acad. Sci. U.S.A. 1990;87(15):5988–5992. doi: 10.1073/pnas.87.15.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A., Freed E.O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. U.S.A. 2001;98(24):13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A., Freed E.O. Role of lipid rafts in virus replication. Adv. Virus Res. 2005;64:311–358. doi: 10.1016/S0065-3527(05)64010-9. [DOI] [PubMed] [Google Scholar]
- Ozawa M., Fujii K., Muramoto Y., Yamada S., Yamayoshi S., Takada A., Goto H., Horimoto T., Kawaoka Y. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 2007;81(1):30–41. doi: 10.1128/JVI.01434-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P., Shaw M.L. Orthomyxoviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E., editors. Fields Virology. 5th ed. Lippincott Williams and Wilkins; Philadelphia: 2007. pp. 1647–1689. [Google Scholar]
- Pattnaik A.K., Wertz G.W. Cells that express all five proteins of vesicular stomatitis virus from cloned cDNAs support replication, assembly, and budding of defective interfering particles. Proc. Natl. Acad. Sci. U.S.A. 1991;88(4):1379–1383. doi: 10.1073/pnas.88.4.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter B.J., Kent H.M., Mills I.G., Vallis Y., Butler P.J., Evans P.R., McMahon H.T. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303(5657):495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Prydz K., Simons K. Cholesterol depletion reduces apical transport capacity in epithelial Madin–Darby canine kidney cells. Biochem. J. 2001;357(Pt 1):11–15. doi: 10.1042/0264-6021:3570011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynwar B.J., Illya G., Harmandaris V.A., Muller M.M., Kremer K., Deserno M. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 2007;447(7143):461–464. doi: 10.1038/nature05840. [DOI] [PubMed] [Google Scholar]
- Roberts P.C., Compans R.W. Host cell dependence of viral morphology. Proc. Natl. Acad. Sci. U.S.A. 1998;95(10):5746–5751. doi: 10.1073/pnas.95.10.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts P.C., Lamb R.A., Compans R.W. The M1 and M2 proteins of influenza A virus are important determinants in filamentous particle formation. Virology. 1998;240(1):127–137. doi: 10.1006/viro.1997.8916. [DOI] [PubMed] [Google Scholar]
- Rossen J.W., de Beer R., Godeke G.J., Raamsman M.J., Horzinek M.C., Vennema H., Rottier P.J. The viral spike protein is not involved in the polarized sorting of coronaviruses in epithelial cells. J. Virol. 1998;72(1):497–503. doi: 10.1128/jvi.72.1.497-503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger C., Muhlberger E., Ryabchikova E., Kolesnikova L., Klenk H.D., Becker S. Sorting of Marburg virus surface protein and virus release take place at opposite surfaces of infected polarized epithelial cells. J. Virol. 2001;75(3):1274–1283. doi: 10.1128/JVI.75.3.1274-1283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P., Rietveld A., Wilk T., Simons K. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 1999;274(4):2038–2044. doi: 10.1074/jbc.274.4.2038. [DOI] [PubMed] [Google Scholar]
- Schmitt A.P., Lamb R.A. Escaping from the cell: assembly and budding of negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004;283:145–196. doi: 10.1007/978-3-662-06099-5_5. [DOI] [PubMed] [Google Scholar]
- Schmitt A.P., Lamb R.A. Influenza virus assembly and budding at the viral budozone. Adv. Virus Res. 2005;64:383–416. doi: 10.1016/S0065-3527(05)64012-2. [DOI] [PubMed] [Google Scholar]
- Schroeder C., Heider H., Moncke-Buchner E., Lin T.I. The influenza virus ion channel and maturation cofactor M2 is a cholesterol-binding protein. Eur. Biophys. J. 2005;34(1):52–66. doi: 10.1007/s00249-004-0424-1. [DOI] [PubMed] [Google Scholar]
- Shaw M.L., Stone K.L., Colangelo C.M., Gulcicek E.E., Palese P. Cellular proteins in influenza virus particles. PLoS Pathog. 2008;4(6):e1000085. doi: 10.1371/journal.ppat.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K., Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1(1):31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Simpson-Holley M., Ellis D., Fisher D., Elton D., McCauley J., Digard P. A functional link between the actin cytoskeleton and lipid rafts during budding of filamentous influenza virions. Virology. 2002;301(2):212–225. doi: 10.1006/viro.2002.1595. [DOI] [PubMed] [Google Scholar]
- Smith G.L., Hay A.J. Replication of the influenza virus genome. Virology. 1982;118(1):96–108. doi: 10.1016/0042-6822(82)90323-3. [DOI] [PubMed] [Google Scholar]
- Takeda M., Leser G.P., Russell C.J., Lamb R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. U.S.A. 2003;100(25):14610–14617. doi: 10.1073/pnas.2235620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley T.J., Ducharme N.A., Varthakavi V., Shepherd B.E., Santangelo P.J., Lindquist M.E., Goldenring J.R., Crowe J.E., Jr. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc. Natl. Acad. Sci. U.S.A. 2008;105(29):10209–10214. doi: 10.1073/pnas.0712144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Hinson E.R., Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe. 2007;2(2):96–105. doi: 10.1016/j.chom.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Watanabe K., Handa H., Mizumoto K., Nagata K. Mechanism for inhibition of influenza virus RNA polymerase activity by matrix protein. J. Virol. 1996;70(1):241–247. doi: 10.1128/jvi.70.1.241-247.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Watanabe S., Noda T., Fujii Y., Kawaoka Y. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J. Virol. 2003;77(19):10575–10583. doi: 10.1128/JVI.77.19.10575-10583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker G.R., Digard P. Entry and intracellular transport of influenza virus. In: Kawaoka Y., editor. Influenza Virology; Current Topics. Caister Academic Press; Wymondham: 2006. pp. 37–64. [Google Scholar]
- Winkler H., Taylor K.A. Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy. 2006;106:240–254. doi: 10.1016/j.ultramic.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Wu Y., Voth G.A. Computational studies of proton transport through the M2 channel. FEBS Lett. 2003;552(1):23–27. doi: 10.1016/s0014-5793(03)00779-8. [DOI] [PubMed] [Google Scholar]
- Ye Z., Liu T., Offringa D.P., McInnis J., Levandowski R.A. Association of influenza virus matrix protein with ribonucleoproteins. J. Virol. 1999;73(9):7467–7473. doi: 10.1128/jvi.73.9.7467-7473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Pekosz A., Lamb R.A. Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins. J. Virol. 2000;74(10):4634–4644. doi: 10.1128/jvi.74.10.4634-4644.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhirnov O.P. Isolation of matrix protein M1 from influenza viruses by acid-dependent extraction with nonionic detergent. Virology. 1992;186(1):324–330. doi: 10.1016/0042-6822(92)90090-c. [DOI] [PubMed] [Google Scholar]
- Zurzolo C., Polistina C., Saini M., Gentile R., Aloj L., Migliaccio G., Bonatti S., Nitsch L. Opposite polarity of virus budding and of viral envelope glycoprotein distribution in epithelial cells derived from different tissues. J. Cell Biol. 1992;117(3):551–564. doi: 10.1083/jcb.117.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]