

Abstract
Recently, the anti-CD3 antibody has been shown to be a promising candidate for the efficient treatment of overt autoimmunity. However, the mechanisms underlying this effect remain unclear. Our previous studies demonstrated that natural killer (NK)T cells and transforming growth factor (TGF)-β were key elements in anti-CD3 F(ab′)2-mediated re-establishment of glucose homeostasis and restoration of self tolerance to islets in type 1 diabetes. In this report, we further investigate the regulatory pathways involved, especially the cellular source of TGF-β production. The treatment of new-onset nonobese diabetic mice with anti-CD3 F(ab′)2 resulted in a significant increase in the numbers of NK cells in spleen and pancreatic lymph nodes that secrete TGF-β. Depletion of this cell population with a specific anti-AsGM1 antibody abrogated anti-CD3 F(ab′)2 therapeutic effects and splenic TGF-β production. When fractionated from recovered mice after CD3 antibody therapy, these NK cells actively suppressed diabetogenic cell proliferation and prevented the cotransfer of diabetes into nonobese diabetic-severe combined immunodeficient mice in a TGF-β-dependent manner. In addition, the regulatory NKT cells from remitting mice were capable of causing NK cells to exhibit a TGF-β-producing phenotype by the secretion of the T helper 2 cytokines interleukins 4 and 10. Overall, these data indicate that NK cells are the main source of TGF-β production after anti-CD3 F(ab′)2 treatment, which are controlled by a population of T helper 2-like NKT cells.
Type 1 diabetes in human and nonobese diabetic (NOD) mice is an autoimmune disease in which pancreatic islet β cells are destroyed by the cellular immune system.1 Based on our understanding of the pathogenesis of β cell destruction in type 1 diabetes, many strategies targeted to immune cells have been developed, including antibodies recognizing antigens expressed on the surface of T cells. CD3-specific antibodies have been believed to be promising candidates to treat overt diabetes.2,3,4 Short-term administration of an anti-CD3 antibody resulted in acquisition of immune tolerance to islets and long-lasting normoglycemia. In surveying the underlying mechanisms, our previous study has identified that natural killer (NK)T cells are key players in the immunoregulation of autoimmunity after anti-CD3 F(ab′)2 therapy.5 Furthermore, anti-CD3 F(ab′)2 treatment heightened the level of production of transforming growth factor (TGF)-β, which is widely accepted as a critical immunoregulatory cytokine in controlling pathogenic cells and maintaining immune homeostasis.6 Interestingly, up-regulated TGF-β appears not to derive from NKT cells or CD4+CD25+ regulatory T cells, as depletion of this regulatory subset does not affect TGF-β secretion in tolerized NOD mice.6 Thus, it is necessary to clarify the identity of lymphocyte population responsible for producing TGF-β after CD3 antibody treatment.
NK cells have been shown to be important components in bridging innate and adaptive immunity. Although this kind of cell plays an effector role in cleaning virally infected cells and rejection of allogenic grafts through cytotoxic capacity and producing pro-inflammatory cytokines,7,8 in some settings, their role is regulatory, as they can also produce multiple immunomodulatory cytokines, eg, interferon-γ, TGF-β, and interleukin (IL)-10.9 Recently, the deficiency of NK cell function in NOD mice has been reported, which contributes to diabetes development.10,11 Accordingly, it is conceivable to prevent the onset of diabetes by modulating NK cells. In fact, a recent study demonstrated that administration of the NK cell activator poly (I:C) in young NOD mice potentially reduced diabetes incidence and insulitis by secreting TGF-β.12 Based on the regulatory function of NK cells in autoimmune disorders, this study examined the role of NK cells in anti-CD3 F(ab′)2-mediated therapeutic effects. We found that anti-CD3 F(ab′)2 antibody treatment increased the frequency and number of NK cells with a hallmark of producing TGF-β and depletion of NK cells abolished anti-CD3 F(ab′)2 effects. Furthermore, NK cells from treated mice inhibited diabetogenic cell response to autoantigen stimulation in vitro and prevented the transfer of diabetes in vivo in a TGF-β-dependent manner.
Materials and Methods
Mice and Glycemia Screening
NOD and NOD-severe combined immunodeficient (NOD. scid) mice were obtained originally from the Jackson Laboratory and bred in our facilities under specific pathogen-free conditions. Care, use and treatment of mice in this study were in strict agreement with the guidelines in the care and use of laboratory animals set forth by Institute of Basic Medical Sciences. The incidence of diabetes in these mice is 80% to 90% by 30 weeks of age. At 10 week of age, NOD mice were monitored for fasting blood glucose weekly. Diabetes was defined ≥11.3mmol/L on two consecutive measurements.
Reagents
Polyclonal rabbit anti-AsGM1 antibody (Ab) and rabbit IgG were purchased from Wako Pure Chemical. Glutamic acid decarboxylase was prepared in our lab. Ovalbumin (OVA) and porcine insulin were purchased from Sigma-Aldrich. α-Galactosylceramide was purchased from Toronto Research Chemicals Inc. α-Galactosylceramide-loaded dimer was prepared as previous description.5 Fluorescein isothiocyanate-labeled anti-murine CD3 (145-2C11) and T cell receptor-β (H57-597), phycoerythrin-labeled anti-murine DX5 (DX5), and NKG2D (CX5) were purchased from eBioscience. Biotinylated polyclonal Anti-TGF-β1 antibody (1D11) and neutralizing anti-TGF-β Ab were purchased from R&D Systems. The anti-mouse IL-4 (11B11) and IL-10 (JES2A5) neutralizing Ab was purchased from Peprotech.
Anti-CD3 F(ab′)2 Antibody Preparation and in Vivo Treatment
As described previously,5 anti-CD3 F(ab′)2 fragments were obtained by pepsin digestion. The diabetic NOD mice within 7 days of the onset of overt diabetes were treated intravenously with anti-CD3 F(ab′)2 (40 μg/mouse) for 5 consecutive days. The untreated diabetic littermates were regarded as controls.
Flow Cytometry
Lymphocytes were stained with fluorescence-labeled antibody in PBS with 2% heat-inactivated fetal calf serum and 0.2% sodium azide on ice for 25 minutes, and fixed using PBS with 1% paraformaldehyde. Data collection and analysis were performed on a FACS Calibur flow cytometer using CellQuest software (Becton Dickinson).
NK Cell Depletion
NOD mice were injected i.v. with 20 μl of anti-AsGM1 Abs or control rabbit IgG once every 5 days. The elimination of NK cells in mice was confirmed by flow cytometry, and the majority (>90%) was depleted.
Purification of NK and NKT Cells
Single cell suspension was prepared from spleen by passing them through nylon mesh. NK cells (CD3−DX5+) were enriched by magnetic-labeled microbeads (Miltenyi Biotec, Germany) according to manufacturer’s instructions. For purifying NKT cells, hepatic lymphocytes were pooled by 40%/80% percoll isolation and further obtained by gated on TCR-β+α-galactosylceramide-loading dimer+ in flow cytometry. The purity (>90%) of NK and NKT cells was confirmed by flow cytometry. In some experiments, CD3−DX5+ cells were further divided into CD3−CD11c−DX5+ and CD3−CD11c+DX5+ population by CD11c-positive beads (Dynal).
Measurement of T Cell Responses
For measurement of islet Antigen-specific T cell responses, spleen cells (5 × 105/ml) from acutely diabetic NOD mice were co-cultured with glutamic acid decarboxylase (10 μg/ml) or insulin protein (40 μg/ml) for 72 hours at 37°C. As a control, the same number of responder cells was stimulated with OVA (20 μg/ml) for 72 hours. For evaluation of the inhibitory effects of NK or DX5+ dendritic cells (DC) on the islet Ag-specific T cell responses of spleen cells, NK or DX5+ DCs (105/ml) were purified from NOD mice 5 weeks after anti-CD3 F(ab′)2 treatment or control mice and then added in the cultures of spleen cells. In some experiments, anti-TGF-β Ab (20 μg/ml) was added to the culture. As positive controls, OVA-specific T cells were stimulated with anti-CD3 mAb (0.5 μg/ml). On day 3 cultures were pulsed with 0.5μCi/well of [3H] thymidine for the last 16 hours, and the cells were harvested and counted by standard liquid scintillation.
Regulatory NK Cell Differentiation
NK (CD3−CD11c−DX5+) cells were isolated from acutely diabetic NOD mice and cultured alone, with syngenic hepatic NKT cells from anti-CD3 F(ab′)2-treated remitting mice or autologous NKT cells (at 2:1 ratio) for 3 days. In some setting, neutralizing antibodies to IL-4 and/or IL-10 (5 μg/ml) or isotype IgG were added into the culture. At the end of co-culture, the supernatants were collected for determination of TGF-β production.
Enzyme-Linked Immunosorbent and Spot Assays for TGF-β Production
Splenocytes (5 × 105), pancreas lymph node cells (2 × 105), or purified NK cells (1 × 105) were incubated in 96-well flat-bottom microtiter plates in the presence of 40 μg/ml insulin protein or not. Supernatants were harvested after 48 hours. The levels of TGF-β and IL-12 were determined in triplicate in 0.1 ml of supernatant by sandwich enzyme-linked immunosorbent assay. The enzyme-linked immunosorbent assay kits used in this study were purchased from R&D Systems.
TGF-β enzyme-linked immunospot assay was conducted according to the instructions of the manufacturer. In brief, plates were coated with purified anti-TGF-β overnight and blocked. NK cells (2 × 105) derived from pancreatic lymph nodes of treated or untreated mice were suspended in medium and co-cultured with stimulus. After 72 hours in 5% CO2 at 37C, cells were removed and the reaction was visualized by addition of the individual biotinylated anticytokine antibody and subsequent alkaline phosphatase–conjugated streptavidin. Spots were counted using an immunospot image analyzer (Bioreader 4000 PRO-X).
Histopathology
NOD mice were sacrificed by cervical dislocation, and the pancreas was immediately removed. Each pancreas was fixed with 10% buffered formalin, embedded in paraffin, sectioned at 4.5 μm, and stained with H&E. Insulitis grade was determined as follows: 0, normal islet; 1, mononuclear infiltration, largely in the periphery, in <25% of the islet; 2, 25% to 50% of the islets showing mononuclear infiltration; 3, >50% of the islet showing mononuclear infiltration; 4, small, retracted islet with few mononuclear cells.
Adoptive Transfer of Diabetes
Purified NK or DX5+ DCs from the spleen of anti-CD3 F(ab′)2-treated or control mice were mixed with splenocytes (1 × 107 cells) from untreated, acutely diabetic NOD mice and given i.v. into the tail veins of 4- to 8-week-old NOD.scid mice. Age-matched NOD.scid mice receiving only 1 × 107 diabetic splenocytes were used as a positive control. In combination with adoptive transfer, some mice were injected intraperitoneally with anti-TGF-β Abs at a dose of 1 mg on day 0, 3, 5, 8, and thereafter every 5 days. Recipients were tested every week for diabetes and diagnosed as described above.
Statistical Analysis
Kaplan-Meier method was used to compare diabetes remission. Student’s t-test was used to compare mean values. Values of P < 0.05 were considered significant.
Results
Anti-CD3 F(ab′)2 Treatment Reversed New-Onset Diabetes and Diminished Insulitis in NOD mice
First, as described previously,5,6 the efficacy of anti-CD3 F(ab′)2 antibody on treating new-onset diabetes was tested. Freshly diabetic NOD mice were randomized into two groups and injected intravenously with a low dose of anti-CD3 F(ab′)2 antibodies on five consecutive days or left untreated. The results showed that over 80% of anti-CD3 F(ab′)2-treated mice returned to normal glucose level and exhibited diabetes-free survival by 9 weeks post-treatment (Figure 1A and B). In contrast, all untreated control mice showed a progressively heightened glycemia accompanied by typical symptoms of type 1 diabetes.
Figure 1.
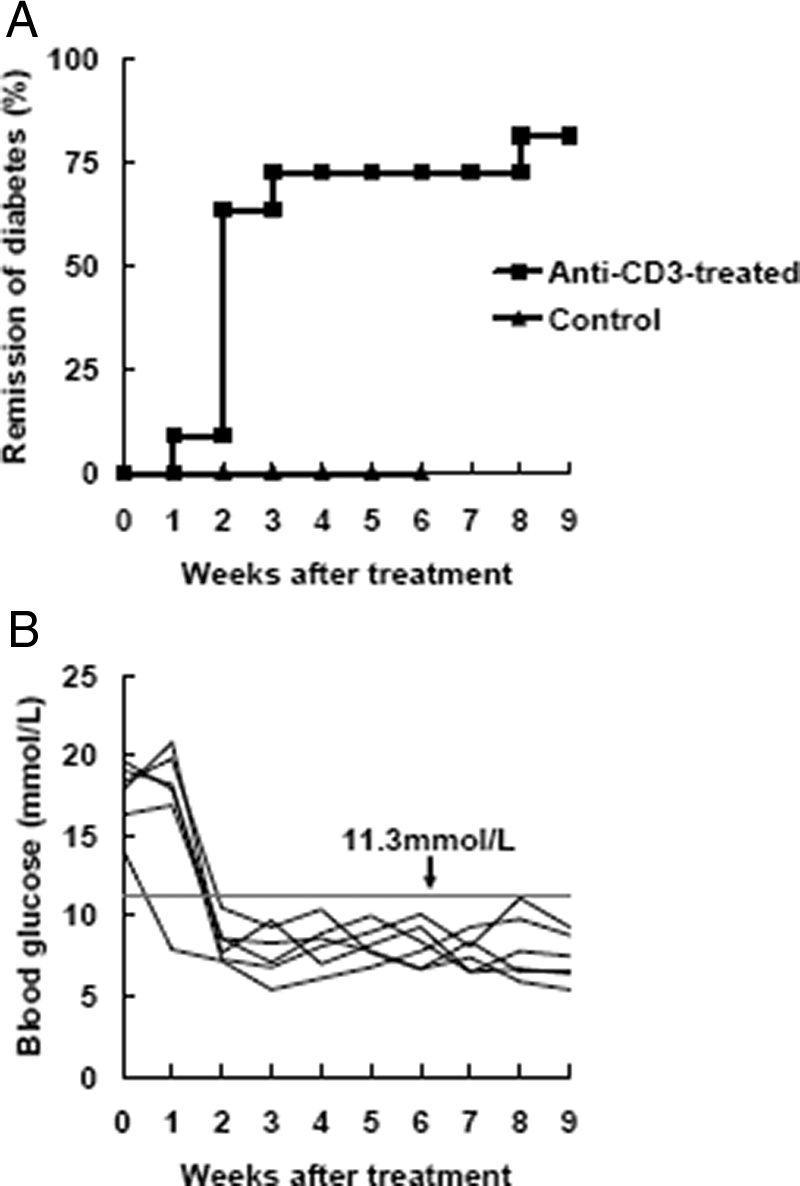
Anti-CD3 F(ab′)2 antibody efficiently reverses new-onset diabetes in NOD mice. Antibody preparation and injection is described in Materials and Methods. A: New-onset diabetic NOD mice were injected with anti-CD3 F(ab′)2. The diabetic untreated littermates were regarded as controls. Mice were screened for glucose levels every week after antibody treatment and complete remission was defined as a return to normal glycemia. Results were analyzed statistically between the control group (triangle, n = 15) and the antibody-treated group (square, n = 20). B: Representative data of glucose control from six anti-CD3 F(ab′)2-treated NOD mice. Blood glucose was detected weekly after antibody treatment. A value less than 11.3mmol/L was regarded as normal glycemic.
To determine whether the restoration of normoglycemia by CD3-specific antibody is caused by interference with cell infiltration, the mice were subjected to histopathological analysis at various time points after completion of the treatment regimen. Most of the islets in control hyperglycemic and diabetic mice exhibited intra-insulitis (Figure 2A), the majority of islets in treated mice were not inflamed (Figure 2, B–D). Also, the insulitis scores for the islets 10 weeks post-treatment indicated that the treated group had a higher number of islets with peri-insulitis (35% vs. 14%) or no insulitis (31% vs. 6%) relative to the untreated diabetic mice (Figure 2E). On the other hand, the number of islets with severe and mild intra-insulitis were reduced in the treated versus control mice (20% and 14% vs. 53% and 27%, respectively). Overall, the treatment with anti-CD3 F(ab′)2 antibody led to the establishment of benign milieus in pancreatic islets facilitating recovery of endogenous insulin-producing cell function.
Figure 2.

Anti-CD3 F(ab′)2 treatment diminishes insulitis in NOD mice. A: Representative islets from untreated diabetic mice (n = 3) with infiltration of numerous lymphocytes. B: Representative islets from mice 10 weeks after anti-CD3 F(ab′)2 treatment (n = 4). C: Representative islets from mice 20 weeks after anti-CD3 F(ab)′2 treatment (n = 4). D: Representative islets from mice 35 weeks after anti-CD3 F(ab′)2 treatment (n = 5). E: The insulitis scores of pancreatic islets from anti-CD3 F(ab′)2-treated mice (n = 6) or controls (n = 5). 75 islets for control group and 84 islets for treated group were examined, respectively. Magnification: ×200 (A), ×100 (B–D). Arrows denote islets.
Anti-CD3 F(ab′)2 Treatment Up-Regulated NK Cell Number and enhanced Their Production of TGF-β
Previous studies indicated that the improved histology of pancreatic islets and restoration of normoglycemia was not attributed to immunosuppression, but to re-establishing immune tolerance to self islets.2,5 TGF-β, whose production is significantly elevated after treatment, has been proved to be pivotal for maintaining this process by controlling pathogenic cells. Neutralization of TGF-β activity dramatically abrogated anti-CD3 therapeutic effects.13 However, the cellular source of TGF-β is still controversial, although several studies reported that CD4+CD25+ regulatory T cells were able to produce TGF-β.14,15 Our observation demonstrated that depletion of this regulatory subset had minimal influence in TGF-β production and glucose homeostasis after anti-CD3 F(ab′)2 treatment.6 Thus, in this system TGF-β appears not to be mainly produced by CD4+CD25+ regulatory T cells. It is known that all leukocyte types can secrete TGF-β. Of these lymphocyte populations, NK cells are emerging as an important origin of this kind of cytokine.16,17 Hence, to determine whether NK cells are involved in anti-CD3 F(ab′)2-mediated diabetes reversion and TGF-β production, we observed the change of NK cells in quantity and quality after anti-CD3 F(ab′)2 treatment. The data showed that not only the proportion but also the number of NK cells in spleen and pancreatic lymph nodes did increase remarkably from treated mice compared with the control diabetic mice (Figure 3A and B). Interestingly, this effect was more significant on 12th week post-treatment, indicating these NK cells might play a role in maintaining long-lasting tolerance and glucose homeostasis in remitting mice. So, next we addressed the capacity of these NK cells to produce TGF-β. NK cells from mice 3 or 12 weeks after treatment remarkably secreted much more TGF-β than those from control age-matched mice (Figure 3, C and D). This phenomenon could also be seen in mice 20 weeks or much longer after treatment. As a result, anti-CD3 F(ab′)2 administration induces a population of TGF-β-producing NK cells, which may regulate peripheral immune balance and retard the attack to self islets by pathogenic cells. In addition, as IL-12 is a powerful stimulator for NK cell expansion,18 we investigated whether increased number of NK cells after anti-CD3 F(ab′)2 treatment was due to up-regulated expression of IL-12. Intriguingly, IL-12 production by pancreas lymph node lymphocytes of anti-CD3 F(ab′)2-treated mice was much lower than the untreated diabetic controls at different time points post-treatment (see supplemental Figure S1 at http://ajp.amjpathol.org), which indicates the increase of NK cell number is not attributed to enhanced expression of IL-12.
Figure 3.

Anti-CD3 F(ab′)2 administration expands the NK cell population and enhances their anti-inflammatory response. The proportion (A) and absolute number (B) of NK cells in spleen or pancreatic lymph nodes of NOD mice after anti-CD3 F(ab′)2 treatment were determined by fluorescence-activated cell sorting analysis. Representative data of three experiments were shown. Pancreas lymph node NK cells were isolated from control (open square) or anti-CD3 F(ab′)2-treated mice (closed square), respectively, and cultured for 48 hours without antigen stimulation. The production of TGF-β was determined by enzyme-linked immunosorbent assay (C) and enzyme-linked immunospot (D). E: Cell-bound TGF-β was detected on NK cells from anti-CD3 F(ab′)2-treated mice or controls. Plots representative of four experiments are shown. Each group consisted of 4 to 5 mice. *P < 0.05 as compared with control mice.
On the other hand, recent reports showed a crucial role of membrane-bound TGF-β for the function of CD4+CD25+ regulatory T cells.19,20,21 To determine whether NK cells also express cell-bound TGF-β, we detected TGF-β expression on the surface of NK cells by fluorescence-activated cell sorting. Consequently, we did not find TGF-β expression on the NK cell surface (Figure 3E), even those producing large amounts of soluble TGF-β after anti-CD3 F(ab′)2 treatment. These data indicated that NK cells primarily produced soluble TGF-β, which mediated NK cell regulatory function.
Another question to be clarified is whether anti-CD3 F(ab′)2 treatment led to NK cell activation. To address this, active status of NK cells was examined. Because NK cells express a diverse array of activating receptors, here we focused on NKG2D expression in NK cells by fluorescence-activated cell sorting, which is critical for function of mouse NK cells22 and whose expression is impaired in NOD NK cells.23 The result showed that NKG2D expression in NK cells from anti-CD3 F(ab′)2-treated mice was low and comparable with control counterparts (see supplemental Figure S2 at http://ajp.amjpathol.org). These data indicate that these NK cells with increased production of TGF-β are likely not activated.
It is worthy to note that a previous report showed the presence of a population of cells co-expressing NK and DC markers (DX5 and CD11c) in naive mice. These cells exhibit regulatory function in some settings such as CD40L blockade to prevent autoimmune disease in the RIP-LCMV mouse model for virally induced type 1 diabetes.24 To uncover the virtual players, CD3−DX5+ population was further divided into CD11c+ and CD11c− subsets by magnetic sorting then the abilities of these two subsets (CD3−CD11c−DX5+ and CD3−CD11c+DX5+, respectively) to secrete TGF-β were tested. The results showed that soluble TGF-β after anti-CD3 F(ab′)2 treatment was mainly produced by CD3−CD11c−DX5+ population, that was NK cells, instead of CD3−CD11c+DX5+ population (see supplemental Figure S3A at http://ajp.amjpathol.org), indicating that key players in anti-CD3 F(ab′)2-mediated diabetes reversion and tolerance restoration were NK cells rather than DX5+ DC population.
AsGM1-Positive Cell Depletion Abrogated Anti-CD3 F(ab′)2 Therapeutic Effects and Decreased TGF-β Production of Splenocytes
To further address the role of NK cells in anti-CD3 F(ab′)2 therapeutic effects, we depleted AsGM1-positive cells with specific antibodies and investigated the influence on anti-CD3 F(ab′)2 effects as well as TGF-β production. The AsGM1-positive cell elimination significantly abrogated diabetes remission following anti-CD3 F(ab′)2 therapy while treatment with isotype antibodies had no such effect (Figure 4A). Moreover, AsGM1-positive cell depletion also decreased TGF-β production of splenocytes from treated mice (Figure 4B), indicating in anti-CD3 F(ab′)2-treated remitting NOD mice a subset of NK cells produces immunoregulatory TGF-β thereby modulates overt autoimmunity to homeostasis.
Figure 4.
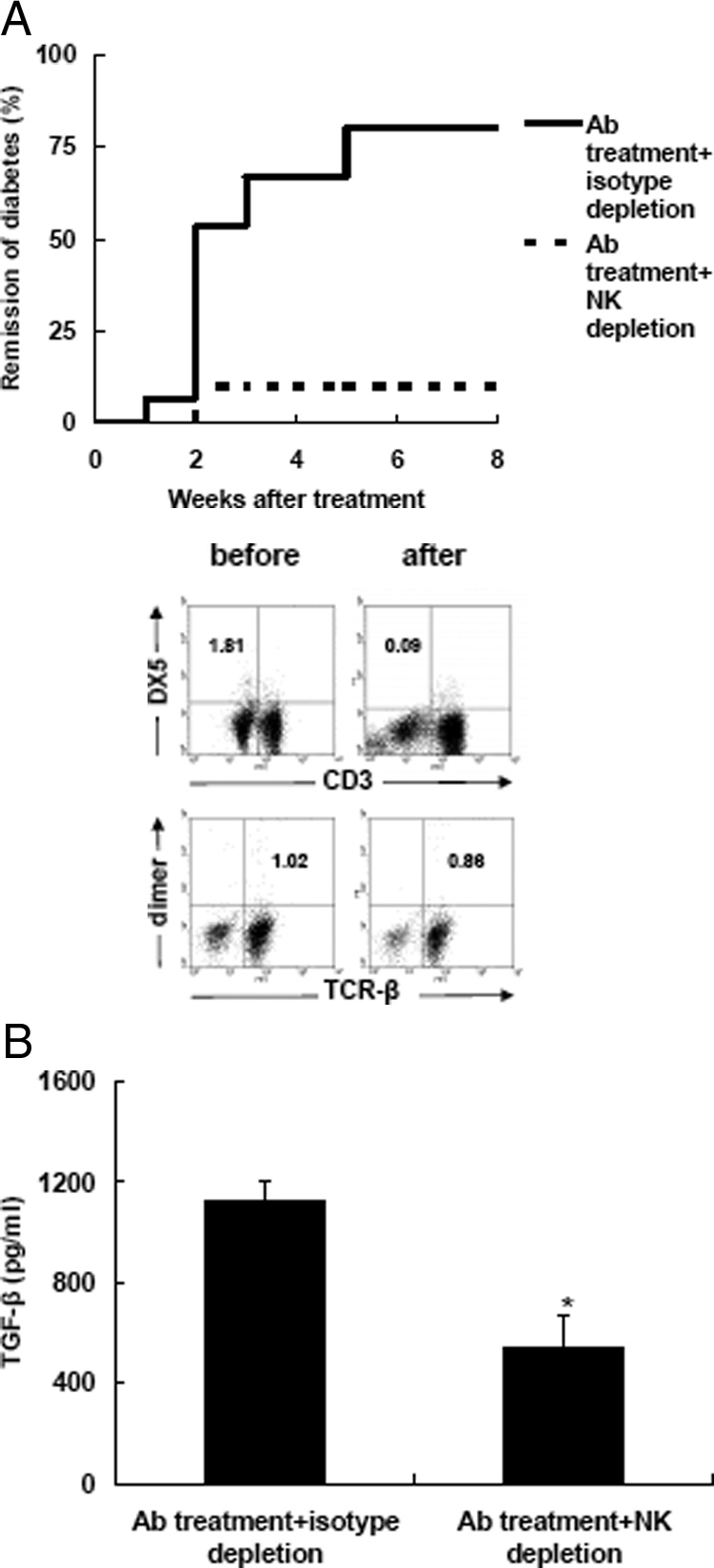
The elimination of AsGM1-positive cells abrogates anti-CD3 F(ab′)2 effects and reduces TGF-β production. A: The effect of in vivo depletion of AsGM1-positive cells on the efficacy of CD3 antibody treatment. Anti-CD3 F(ab′)2-treated remitting NOD mice were treated with anti-AsGM1 antibody or isotype antibodies. The percentages of NK and NKT cells in PLN before and after AsGM1 depletion are shown. Each experimental group consisted of 10 to 15 mice. B: Splenocytes were isolated from mice 4 to 5 weeks after anti-AsGM1 or isotype antibody depletion following anti-CD3 F(ab′)2 treatment, respectively, and stimulated with insulin (40 μg/ml) for 48 hours. The supernatants were collected for detection of TGF-β production. Values are shown as mean ± SD Each group consisted of 4 to 6 mice. *P < 0.05.
In addition, as described above, we also detected cell-bound TGF-β on the surface of both AsGM1-positive cell-depleted and non-depleted splenocytes. In agreement with the above data, we did not find TGF-β expression on the NK cell surface or the alteration of cell-bound TGF-β expression in splenocytes before and after anti-AsGM1 depletion (data not shown). In summary, these data support an immunoregulatory function of NK cells in anti-CD3 F(ab′)2-mediated diabetes reversion and tolerance restoration through producing soluble TGF-β.
NK Cells Induce Effector T Cell Hyporesponse to Islet Autoantigens in a TGF-β-Dependent Manner
For antibody therapy, it is logical to contemplate that the resolution of the inflammatory infiltration is caused by modulation of β-cell autoantigen-reactive diabetogenic T cells. Thus, a plausible hypothesis postulates that the regulatory NK cells induced by antibody treatment maybe inhibit the proliferation of diabetogenic T cells and block diabetes progression. To test this premise, the splenic cells from diabetic mice were pooled and the proliferative response of these diabetogenic cells to antigen stimulation co-cultured with NK cells was assessed. Two critical autoantigens to islet β cells, glutamic acid decarboxylase, and insulin,25,26 as well as control antigens OVA were used. Splenocytes from diabetic mice actively cloned in response to glutamic acid decarboxylase or insulin incubation, showing their effector function to destroy β cells. When added recovered mice-derived NK cells to the culture, this proliferative responses were suppressed strongly, whereas addition of NK cells from control mice did not show such effects (Figure 5). Interestingly, treated NK cells just slightly inhibited responses of effector cells to OVA stimulation, indicating the inhibitory effects of NK cells are β cell antigen-specific, which is consistent with previous observations that anti-CD3 administration blocked β cell-reactive T cell activity specifically instead of rendering general immunosuppression.
Figure 5.

NK cells from anti-CD3 F(ab′)2-treated remitting mice modulate the islet Ag-specific T cell responses of spleen cells. Spleen cells from acutely diabetic NOD mice were co-cultured with the indicated autoantigens or OVA in the presence of purified NK cells from anti-CD3 F(ab′)2-treated or control mice for 72 hours. In some settings, neutralizing anti-TGF-β Abs were added to the culture. As positive controls, OVA-specific splenic T cells were stimulated by anti-CD3 (0.5 μg/ml). During the last 16 hours, the cells were pulsed with 0.5μCi of [3H] thymidine. Proliferation was determined by [3H] thymidine uptake. Data are expressed as stimulation index ± SD of the mean from six mice with a background of 1157 to 2124 counter per minute and tested in triplicate. *P < 0.05; **P < 0.01.
To determine the inhibitory function of NK cells to diabetic cell response is TGF-β-dependent, neutralizing antibodies to TGF-β were added in the co-culture. We found that, blockade of TGF-β bioactivity dramatically abolished NK cell suppressive effects on effector response of diabetogenic cells pulsed by autoantigens (Figure 5). Whereas, addition of isotype Abs had no such effect (data not shown). This indicates that TGF-β is an important mediator in NK cell-mediated suppression of effector cell function.
In addition, as described above, to clarify whether NK or DX5+ DCs were regulators to suppress Ag-specific proliferative response of lymphocytes, we further examined the suppressive capacity of NK cells and DX5+ DCs in our system. We found that DX5+ DCs did not inhibit the colonization of diabetic splenocytes in response to autoantigen stimulation and blockade of TGF-β activity did not affect this process. In contrast, CD3−CD11c−DX5+ cells (NK cells) showed suppressive function in TGF-β-dependent manner (see supplemental Figure S3B at http://ajp.amjpathol.org). As controls, control mice-derived NK or DX5+ DCs did not show regulatory activity (data not shown). Taken together, these data indicated that NK cells rather than DX5+ DCs were key mediators in suppressing lymphocyte proliferation in autoantigen-specific manners.
NK Cells Prevent the Transfer of Diabetes into NOD.scid Mice
Subsequently, we examined in vivo protective function of NK cells using the co-transfer model. Diabetic cells (1 × 107) were transfused into young NOD.scid mice with NK cells from treated or control mice. The results showed that as few as 2.5 × 105 NK cells from treated remitting mice were sufficient to inhibit the onset of transferred diabetes, whereas the similar or even higher number (5 × 105) of control mice-derived NK cells failed to prevent diabetes development. Interestingly, blockade of TGF-β bioactivity remarkably abrogated the protective function of NK cells from remitting mice (Figure 6). Consistent with in vitro data as described above, TGF-β neutralization did not affect the effect of control NK cells (data not shown). These data clearly highlight a TGF-β-dependent protective role of NK cells after CD3-specific antibody treatment.
Figure 6.
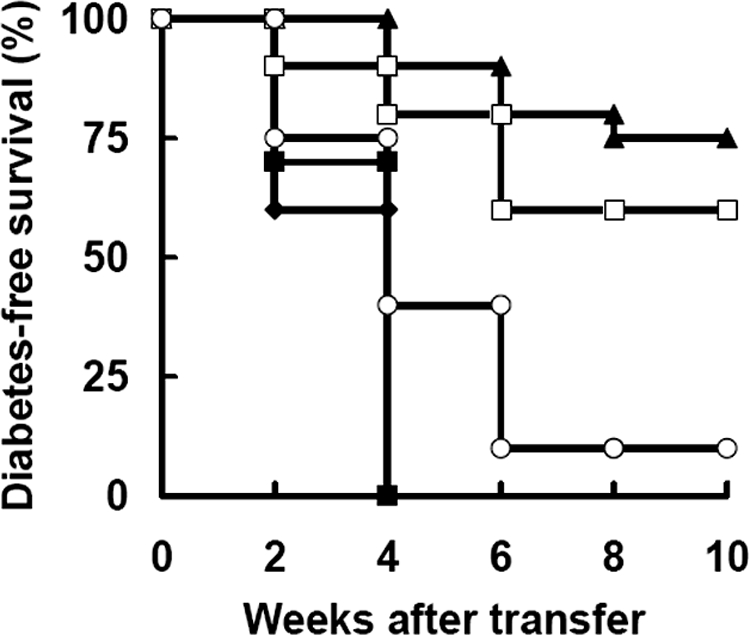
Expanded NK cells suppress the transfer of diabetes. NOD.scid females were injected with diabetic splenocytes (1 × 107) alone (closed diamond, n = 10) or mixed with isolated NK cells from untreated (5 × 105, closed square, n = 10), anti-CD3 F(ab′)2-treated mice (5 × 105, closed triangle, n = 10; 2.5 × 105, open square, n = 10). In some experiments, NOD.scid mice receiving the cocktail of diabetic spleen cells and treated mice-derived NK cells (2.5 × 105) were infused with anti-TGF-β Abs simultaneously (open circle, n = 10). The development of diabetes in the recipients was monitored weekly. Data were pooled from two separate experiments.
Further, we evaluated the protective effects of two subsets residing in CD3−DX5+ population (NK and DX5+ DCs) in this cotransfer model. CD3−CD11c−DX5+ population (NK cells) actively prevented diabetes transfer in a TGF-β-dependent manner. In contrast, DX5+ DCs did not block the onset of transferred diabetes (see supplemental Figure S3C at http://ajp.amjpathol.org). These data clearly suggested that NK cells, but not DX5+ DCs, modulated overt autoimmune response to homeostasis in a TGF-β-dependent fashion.
NKT Cells from Remitting Mice Modify NK Cells to Produce TGF-β
Our previous study has proved NKT cells as key regulators for the restoration of immune homeostasis after anti-CD3 F(ab′)2 treatment.5 Hepatic NKT cells secrete high levels of regulatory T helper (Th)2 cytokines (ie, IL-4 and IL-10) and drive peripheral pathogenic state toward a homeostatic phenotype. Strikingly, NKT cells themselves, whether from CD3 antibody-treated or control NOD mice, did not produce TGF-β. It is reported that NKT cells are able to induce a population of Gr-1+ myeloid cells with characteristics of producing TGF-β in a tumor recurrence model.27 Thus we suspect that CD3 antibody treatment expands a subset of regulatory NKT cells, thereby these NKT cells may modulate NK cells to obtain a protective phenotype and restrain the destructive function of autoreactive T cells. To test this, the NKT cells were isolated from the liver of antibody-treated remitting NOD mice and co-cultured with NK (CD3−CD11c−DX5+) cells from acutely diabetic NOD mice for 3 days. In addition, to clarify the mechanisms by which NKT cells regulate NK cells, neutralizing antibodies to IL-4/IL-10 were added into the culture in some setting. The results indicated that, in contrast to those from diabetic NOD mice, remitting mice-derived regulatory NKT cells had ability to induce NK cells to produce TGF-β (Figure 7). Interestingly, it was repeatedly verified that these modified NK cells did not express membrane-bound TGF-β (data not shown). Moreover, this effect of NKT cells on NK cells depended on IL-4 and/or IL-10 secreted by NKT cells as blockade of IL-4 and/or IL-10 bioactivity abolished TGF-β production by NK cells (Figure 7). These data suggest that the protective NK cell population in recovered mice may be regulated by NKT cells in a Th2 cytokine-dependent manner, which is consistent with the finding that protection against diabetes by CD3 antibody treatment was mediated by IL-4/IL-10.
Figure 7.

Regulatory NKT cells modulate NK cells to produce TGF-β in a Th2 cytokine-dependent fashion. NK (CD3−CD11c−DX5+) cells from acutely diabetic NOD mice were cultured alone or with autologous NKT cells (dNKT cells) or NTK cells from recovered mice (rNKT cells) at the ratio of 2:1 for 3 days. In some experiments, neutralizing Abs to IL-4/IL-10 (5 μg/ml) or isotypes were added in the culture. TGF-β production in the supernatants was determined by enzyme-linked immunosorbent assay. Values are shown as mean ± SD of five mice. *P < 0.05; **P < 0.01.
Discussion
In treating overt autoimmune disorders, anti-CD3 monoclonal antibody is a promising candidate to re-establish immune homeostasis.2,28,29,30 Our previous work has demonstrated that a short-time administration of anti-CD3 antibodies resulted in permanent restoration of normoglycemia and immune balance. In surveying the underlying mechanism, we found that NKT cells and TGF-β were critical components in regulating overresponsive autoimmunity. However, the cellular source of TGF-β and how NKT cells exert influence on TGF-β production is still elusive. In this study, we provide evidence that NK cells play a regulatory role in immunomodulatory function of CD3-specific antibody on overt diabetes in NOD mice. Anti-CD3 treatment primes NKT cell response to produce Th2 cytokines IL-4/IL-10, which drive NK cells to produce TGF-β thereby inhibit peripheral pathogenic cells as well as restore immune homeostasis.
Indeed, the role of NK cells in autoimmune diseases remains controversial.31 On one hand, NK cells exhibit a pathogenic function in diabetes model induced by anti-CTLA-4 Ab.32 In contrast, NK cells can participate in the control of autoimmune diabetes by complete freund’s.33 Similar to diabetes, these controversies are also present in experimental autoimmune encephalomyelitis. Zhang et al34 found that NK cells were protective in mice model. However, Shi et al35 found that NK cells contributed to the disease promotion in the same model. This dogma might be attributed to different roles of NK cells in different stages of disease progression and different settings. In our system, we found the development of a protective subset of NK cells in the modified microenvironment by anti-CD3 F(ab′)2 treatment. These cells displayed a NK3-like phenotype, equivalent to the Th3 profile.36
Intriguingly, a previous study demonstrated that, in the RIP-LCMV mouse model, prevention from type 1 diabetes by blockade of CD40-CD40L interaction was mediated by a highly potent, bitypic cell population sharing phenotypic and functional properties of both NK and DCs, namely DX5+ DCs.24 One considers that NK cell-mediated protection in our model may be contaminated by DX5+ DCs. Therefore, CD3−DX5+ populations were divided into CD3−CD11c−DX5+ (NK) and CD3−CD11c+DX5+ (DX5+ DCs) subsets and their inhibitory effects in vitro and in vivo were tested, respectively. The results showed that key players in modifying autoimmunity to immune balance were NK cells rather than DX5+ DCs (see supplemental Figure S3 at http://ajp.amjpathol.org). This discrepancy can be resolved by that DX5+ DCs might play a role under certain circumstances, especially in virus-induced autoimmune diseases.
Subsequently, the studies on the involving mechanisms revealed that TGF-β-dependent pathway was a major regimen for NK cells to regulate harmful response. NK cells actively inhibited effector T cell proliferation in vitro and their attack to islets. Once TGF-β was neutralized, strikingly, this effect disappeared. This is consistent with strong immunoregulatory roles of TGF-β in controlling lymphocyte proliferation, differentiation, and survival.37 Noticeably, these modified NK cells did not express membrane-bound TGF-β, indicating that soluble TGF-β is the dominant type in mediating NK function. Apart from inhibitory function on T cell expansion by secreting TGF-β, NK cells are known to capable of killing target cells by cytotoxic activity. In experimental autoimmune encephalomyelitis, one mechanism of NK cells to modulate autoimmunity is to directly destroy autoreactive T cells in a perforin or Fas/FasL-dependent manner.38 Thus, one considers whether it is also available in anti-CD3 F(ab′)2-mediated diabetes reversion. However, our and other researchers’ studies have observed that pathogenic cells against islets are still present in anti-CD3 F(ab′)2-treated remitting mice (39, our unpublished data). Moreover, soluble TGF-β reportedly is able to reduce NK cell cytotoxicity and perforin gene transcription.40 Thus, cytotoxicity of NK cells seems to be dispensable for suppressing autoimmune T cell proliferation in our system.
It is apparent that the anti-CD3 F(ab′)2 antibody has no direct effect on NK cells which lack corresponding antigens. What is responsible for bridging anti-CD3 F(ab′)2 effects and NK cell modulation? Here we focus on NKT cells which were shown to be polarized to Th2 phenotype in cytokine pattern after CD3 antibody treatment in our previous observation.5 These Th2-like NKT subsets can influence T cell differentiation, driving effector cells toward a non-pathogenic Th2 profile. It is believed that NKT cells can also affect other cell types by producing cytokines and/or direct cell contact, including NK cells.41 Indeed, the data presented in this report support this premise. These modified NKT cells could prompt NK populations to display a regulatory phenotype (Figure 7). Similar to these data, a previous study proposed that activated NKT subsets had potentials to induce the development of TGF-β-producing Gr-1+ myeloid cells in a tumor recurrence model.27 Consequently, the plausible scenario underlying anti-CD3 F(ab′)2 therapeutic effects is that, anti-CD3 F(ab′)2 antibody treatment drives NKT cells mainly residing in the liver to show a Th2 profile, thereafter these regulatory NKT lymphocytes modify NK subsets to a TGF-β-secreting phenotype, as well as TGF-β is sufficient to inhibit the effector response of diabetogenic cells and restore immune homeostasis,42 although the details in vivo need further elucidation.
It is worthy to note that previous reports demonstrated increased proportion of CD4+CD25+ regulatory T cells in remitting mice.13 Whereas, we did not find this change by detecting the expression of Foxp3 protein in CD4 T cells. These observations are in agreement with the data presented in this study, given that in some setting NK cells could suppress the induction/expansion of CD4+CD25+ regulatory T cells.43
In summary, this study underlines the importance of TGF-β-secreting NK cells in regulating autoimmunity in anti-CD3 F(ab′)2-mediated diabetes reversion and tolerance restoration. These regulatory NK cells efficiently blocked self-attack activity of pathogenic cells and the development of overt diabetes in a TGF-β-dependent manner. These findings may have important implications for the understandings of involving mechanisms in immunomodulation of anti-CD3 F(ab′)2 antibody on autoimmune diseases.
Supplementary Material
Acknowledgments
We are thankful to Ming Yu for excellent technical assistance.
Footnotes
Address reprint requests to Prof. Yan Li, Department of Molecular Immunology, Institute of Basic Medical Sciences, Taiping Road, No. 27, Beijing 100850, People’s Republic of China. E-mail: liyan62033@yahoo.com.cn.
Supported by grants from the National Key Basic Research Program of China (2007CB512406) and the National Natural Science Foundation of China (30801029).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Cameron MJ, Meagher C, Delovitch TL. Failure in immune regulation begets IDDM in NOD mice. Diabetes Metab Rev. 1998;14:177–185. doi: 10.1002/(sici)1099-0895(199806)14:2<177::aid-dmr209>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA. 1994;91:123–127. doi: 10.1073/pnas.91.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, De Block C, Seigneurin JM, De Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, Frewin M, Waldmann H, Bach JF, Pipeleers D, Chatenoud L. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A single course of anti-CD3 monoclonal antibody hOKT3γ1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54:1763–1769. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Han G, Wang J, Wang R, Xu R, Shen B, Qian J, Li Y. Active tolerance induction and involvement of CD1d-restricted NKT cells in anti-CD3 F(ab′)2 treatment reversed new onset diabetes in NOD mice. Am J Pathol. 2008;172:972–979. doi: 10.2353/ajpath.2008.070159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Han G, Wang J, Wang R, Xu R, Shen B, Qian J, Li Y. Essential roles of TGF-β in anti-CD3 antibody therapy: reversal of diabetes in nonobese diabetic mice independent of Foxp3+CD4+ regulatory T cells. J Leukoc Biol. 2008;83:280–287. doi: 10.1189/jlb.0707498. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Biology of natural killer cells. Adv Immuol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos FJ. Regulation of adaptive immmunity by natural killer cells. Immunol Res. 1998;17:303–312. doi: 10.1007/BF02786453. [DOI] [PubMed] [Google Scholar]
- Kelly JM, Takeda K, Darcy PK, Yagita H, Smith MJ. A role of IFN-γ in primary and secondary immunity generated by NK cell-sensitive tumor-expressing CD80 in vivo. J Immunol. 2002;168:4472–4479. doi: 10.4049/jimmunol.168.9.4472. [DOI] [PubMed] [Google Scholar]
- Poulton LD, Smyth MJ, Hawke CG, Silveira P, Shepherd D, Naidenko OV, Godfrey DI, Baxter AG. Cytometric and functional analyses of NK and NKT cell deficiencies in NOD mice. Int Immunol. 2001;13:887–896. doi: 10.1093/intimm/13.7.887. [DOI] [PubMed] [Google Scholar]
- Johansson SE, Hall H, Björklund J, Höglund P. Broadly impaired NK cell function in nonobese diabetic mice is partially restored by NK cell activation in vivo and by IL-12/IL-18 in vitro. Int Immunol. 2004;16:1–11. doi: 10.1093/intimm/dxh002. [DOI] [PubMed] [Google Scholar]
- Zhou R, Wei H, Tian Z. NK3-like NK cells are involved in protective effect of polyinosinic-polycytidylic acid on type 1 diabetes in nonobese diabetic mice. J Immunol. 2007;178:2141–2147. doi: 10.4049/jimmunol.178.4.2141. [DOI] [PubMed] [Google Scholar]
- Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-β-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+CD25+ T regulatory cells control anti-islet CD8+ T cells through TGF-β–TGF-β receptor interactions in type 1 diabetes. Proc Natl Acad Sci USA. 2003;100:10878–10883. doi: 10.1073/pnas.1834400100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, Strober W. TGF-β1 play an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol. 2004;172:834–842. doi: 10.4049/jimmunol.172.2.834. [DOI] [PubMed] [Google Scholar]
- Horwitz DA, Gray JD, Ohtsuka K. Role of NK cells and TGF-β in the regulation of T-cell-dependent antibody production in health and autoimmune disease. Microbes Infect. 1999;1:1305–1311. doi: 10.1016/s1286-4579(99)00253-1. [DOI] [PubMed] [Google Scholar]
- Gray JD, Hirokawa M, Ohtsuka K, Horwitz DA. Generation of an inhibitory circuit involving CD8+ T cells. IL-2, and NK cell-derived TGF-β: contrasting effects of anti-CD2 and anti-CD3. J Immunol. 1998;160:2248–2254. [PubMed] [Google Scholar]
- Brunda MJ. Interleukin-12. J Leukoc Biol. 1994;55:280–288. doi: 10.1002/jlb.55.2.280. [DOI] [PubMed] [Google Scholar]
- Xia Z, Xu L, Zhong W, Wei J, Li N, Shao J, Li Y, Yu S, Zhang Z. Heme oxygenase-1 attenuates ovalbumin-induced airway inflammation by up-regulation of Foxp3 T-regulatory cells, interleukin-10, and membrane-bound transforming growth factor-β1. Am J Pathol. 2007;171:1904–1914. doi: 10.2353/ajpath.2007.070096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroukhova M, Qi Z, Oriss TB, Dixon-McCarthy B, Ray P, Ray A. Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-β. J Clin Invest. 2006;116:996–1004. doi: 10.1172/JCI26490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroukhova M, Segiun-Devaux C, Oriss TB, Dixon-McCarthy B, Yang L, Ameredes BT, Corcoran TE, Ray A. Tolerance induced by inhaled antigen involves CD4+ T cells expressing membrane-bound TGF-β and FOXP3. J Clin Invest. 2004;114:28–38. doi: 10.1172/JCI20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, Pertel T, Carnaud C, Bluestone JA, Lanier LL. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- Homann D, Jahreis A, Wolfe T, Hughes A, Coon B, van Stipdonk MJ, Prilliman KR, Schoenberger SP, von Herrath MG. CD40L blockade prevents autoimmune diabetes by induction of bitype NK/DC regulatory cells. Immunity. 2002;16:403–415. doi: 10.1016/s1074-7613(02)00290-x. [DOI] [PubMed] [Google Scholar]
- Kim J, Richter W, Aanstoot H-J, Shi Y, Fu Q, Rajotte R, Warnock G, Baekkeskov S. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. 1993;42:1799–1808. doi: 10.2337/diab.42.12.1799. [DOI] [PubMed] [Google Scholar]
- Lernmark A, Agardh CD. Immunomodulation with human recombinant autoantigens. Trends Immunol. 2005;26:608–612. doi: 10.1016/j.it.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, Letterio JJ, Paul WE, Berzofsky JA. Transforming growth factor-β production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–1752. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran GT, Carter N, He XY, Spicer TS, Plain KM, Nicolls M, Hall BM, Hodgkinson SJ. Reversal of experimental allergic encephalomyelitis with non-mitogenic, non-depleting anti-CD3 mAb therapy with a preferential effect on T(h)1 cells that is augmented by IL-4. Int Immunol. 2001;13:1109–1120. doi: 10.1093/intimm/13.9.1109. [DOI] [PubMed] [Google Scholar]
- Utset TO, Auger JA, Peace D, Zivin RA, Xu D, Jolliffe L, Alegre ML, Bluestone JA, Clark MR. Modified anti-CD3 therapy in psoriatic arthritis: a phase I/II clinical trial. J Rheumatol. 2002;29:1907–1913. [PubMed] [Google Scholar]
- Steffens S, Burger F, Pelli G, Dean Y, Elson G, Kosco-Vilbois M, Chatenoud L, Mach F. Short-term treatment with anti-CD3 antibody reduces the development and progression of atherosclerosis in mice. Circulation. 2006;114:1977–1984. doi: 10.1161/CIRCULATIONAHA.106.627430. [DOI] [PubMed] [Google Scholar]
- Johansson S, Berg L, Hall H, Hoglund P. NK cells: elusive players in autoimmunity. Trends Immunol. 2005;26:613–618. doi: 10.1016/j.it.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Poirot L, Benoist C, Mathis D. Natural killer cells distinguish inncuous and destructive forms of pancreatic islet autoimmunity. Proc Natl Acad Sci USA. 2004;101:8102–8107. doi: 10.1073/pnas.0402065101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IF, Qin H, Trudeau J, Dutz J, Tan R. Regulation of autoimmune diabetes by complete Freund’s adjuvant is mediated by NK cells. J Immunol. 2004;172:937–942. doi: 10.4049/jimmunol.172.2.937. [DOI] [PubMed] [Google Scholar]
- Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitisby natural killer (NK) cells. J Exp Med. 1997;186:1677–1687. doi: 10.1084/jem.186.10.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi FD, Takeda K, Akira S, Sarventick N, Ljunggren HG. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-γ by NK cells. J Immunol. 2000;165:3099–3104. doi: 10.4049/jimmunol.165.6.3099. [DOI] [PubMed] [Google Scholar]
- Fukaura H, Kent SC, Pietrusewicz MJ, Khoury SJ, Weiner HL, Hafler DA. Induction of circulating myelin basic protein and proteolipid protein-specific transforming growth factor-β1-secreting Th3 T cells by oral administration of myelin in multiple sclerosis patients. J Clin Invest. 1996;98:70–77. doi: 10.1172/JCI118779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AL, Flavell RA. Transforming growth factor-β regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Smeltz RB, Wolf NA, Swanborg RH. Inhibition of autoimmune T cell responses in the DA rat by bone marrow-derived NK cells in vitro: implications for autoimmunity. J Immunol. 1999;163:1390–1397. [PubMed] [Google Scholar]
- Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol. 1997;158:2947–2954. [PubMed] [Google Scholar]
- Smyth MJ, Strobl SL, Young HA, Ortaldo JR, Ochoa AC. Regulation of lymphokine-activated killer activity and pore-forming protein gene expression in human peripheral blood CD8+ T lymphocytes. Inhibition by transforming growth factor-β. J Immunol. 1991;146:3289–3297. [PubMed] [Google Scholar]
- Bach JF, Bendelac A, Brenner MB, Cantor H, De Libero G, Kronenberg M, Lanier LL, Raulet DH, Shlomchik MJ, von Herrath MG. The role of innate immunity in autoimmunity. J Exp Med. 2004;200:1527–1531. doi: 10.1084/jem.20042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Yang H, Kim IS, Saint-Hilaire F, Thomas DA, De BP, Ozkaynak E, Muthukumar T, Hancock WW, Crystal RG, Suthanthiran M. Systemic transforming growth factor-β1 gene therapy induces Foxp3+ regulatory T cells, restores self-tolerance, and facilitates regeneration of β cell function in overtly diabetic nonobese diabetic mice. Transplantation. 2005;79:1091–1096. doi: 10.1097/01.tp.0000161223.54452.a2. [DOI] [PubMed] [Google Scholar]
- Brillard E, Pallandre J, Chalmers D, Ryffel B, Radlovic A, Seilles E, Rohrlich PS, Pivot X, Tiberghien P, Saas P, Borg C. Natural killer cells prevent CD28-mediated Foxp3 transcription in CD4+CD25− T lymphocytes. Exp Hematol. 2007;35:416–425. doi: 10.1016/j.exphem.2006.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.