

Abstract
Interactions between BabA and Lewis b (Leb) related antigens are the best characterized adhesin-receptor interactions in Helicobacter pylori (H pylori). Several mechanisms for the regulation of BabA expression are predicted, including at both transcriptional and translational levels. The formation of chimeric proteins (babA/B or babB/A chimeras) seems to play an especially important role in translational regulation. Chimeric BabB/A protein had the potential to bind Leb; however, protein production was subject to phase variation through slipped strand mispairing. The babA gene was cloned initially from strain CCUG17875, which contains a silent babA1 gene and an expressed babA2 gene. The sequence of these two genes differs only by the presence of a 10 bp deletion in the signal peptide sequence of babA1 that eliminates its translational initiation codon. However, the babA1 type deletion was found only in strain CCUG17875. A few studies evaluated BabA status by immunoblot and confirmed that BabA-positive status in Western strains was closely associated with severe clinical outcomes. BabA-positive status also was associated with the presence of other virulence factors (e.g. cagA-positive status and vacA s1 genotype). A small class of strains produced low levels of the BabA protein and lacked Leb binding activity. These were more likely to be associated with increased mucosal inflammation and severe clinical outcomes than BabA-positive strains that exhibited Leb binding activity. The underlying mechanism is unclear, and further studies will be necessary to investigate how the complex BabA-receptor network is functionally coordinated during the interaction of H pylori with the gastric mucosa.
Keywords: Helicobacter pylori, BabA, Pathogenesis, Lewis antigens
INTRODUCTION
The adherence of Helicobacter pylori (H pylori) to the gastric mucosa is widely assumed to play an important role in the initial colonization and long-term persistence in the human gastric mucosa. Analysis of the three completed H pylori genomes (strains 26695, J99, and HPAG1) has confirmed the presence of five major outer membrane protein (OMPs) families, which comprise approximately 4% of the H pylori genome. Among the families, members of the large Hop (Helicobacter outer membrane protein) family were the first characterized OMPs in H pylori. Several OMPs in the Hop family have been reported to act as adhesion molecules including the blood group antigen binding adhesin (BabA), sialic acid binding adhesin (SabA), adherence-associated lipoprotein (AlpA and AlpB), outer membrane inflammatory protein (OipA), and HopZ. Lewis b antigen (Leb) and related fucosylated ABO blood group antigens are recognized by BabA[1], whereas sialyl-Lewis x and sialyl-Lewis a antigens (sLex and sLea) are recognized by SabA[2]. The corresponding receptors for AlpAB, OipA, and HopZ remain unknown. To date, BabA-Leb is the best-characterized adhesin-receptor interaction in H pylori. In this review, I summarize recent data giving new insight into BabA and its role in pathogenesis.
IDENTIFICATION OF BABA
It is well known that Leb is the dominant antigen in the gastric mucosa of secretor-positive individuals[3], and the negative secretor status is associated with a Lewis a (Lea)-dominant phenotype in the gastric mucosa (Figure 1). In 1993, two studies showed that H pylori can bind to fucosylated glycoconjugates containing Leb structures on the surface of gastric epithelial cells within human biopsy specimens[4,5]. Studies using transgenic mice expressing the human Leb epitope in gastric epithelial cells indicated that Leb functions as a receptor for an H pylori-specific adhesin and mediates its attachment to the gastric pit and surface mucous cells[6]. Further studies using the same transgenic mice showed that H pylori was adherent to the surface of gastric epithelial cells, resulting in severe chronic gastric inflammation and atrophy; whereas H pylori was localized in the mucous layer in non-transgenic control mice[7].
Figure 1.
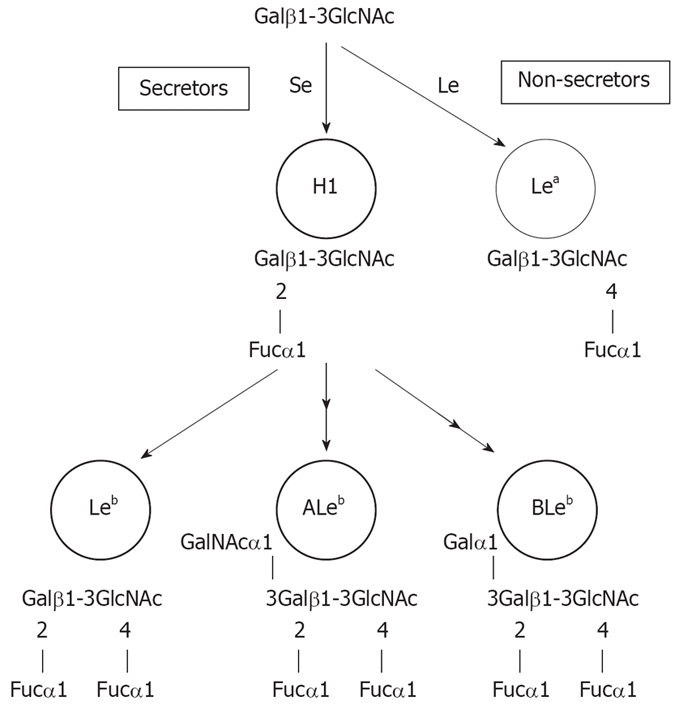
Biosynthetic pathways of Lewis antigens starting from type 1 lacto series core chains. Starting from type 1 core chains, an α1, 2-fucosyltransferase (Se) transfers fucose (Fuc) to the terminal galactose (Gal), resulting in the H-1 antigen (H1). H-1 antigen is a target for GalNAc- or Gal-transferases (in blood group A or B individuals) or remains unmodified (in blood group O individuals). These intermediates the are modified for the fucosylation step by an α1,3/4-fucosyltransferase (Le), resulting in the difucosylated histo-blood group antigens ALeb, ALeb and Leb. Non-secretors are unable to produce an active Se product, and are only targets for the Le gene product Lea.
In 1998, Ilver et al analyzed the blood group antigen-binding activity by measuring binding of H pylori to 125I-labeled fucosylated blood group antigens[1]. Among 100 H pylori isolates examined, 66% bound the Leb antigen; whereas 95% of the isolates did not bind the related Lea, H-2, Lex, or LeY antigens. The 78 K adhesin recognizing the Leb antigen was detected on the bacterial cell outer membrane and was isolated by a combined ligand identification and purification technique and designated as blood group antigen-binding adhesin (BabA)[1]. Additional analyses revealed two sets of clones that encode two proteins with almost identical NH2-terminal domains and completely identical COOH-terminal domains, but with divergent central domains. The corresponding genes were designated babA and babB; BabA but not BabB had Leb antigen-binding activity. Therefore, the central domain in babA is believed to determine the specificity of receptor binding[1,8–12]; however, the motifs of the babA gene that are involved in binding are still unknown.
FUNCTION OF BABA
BabA originally was defined as an adhesin binding to the Leb antigen. The H-1 antigen is the carbohydrate structure that defines the blood group O phenotype in the ABO blood group system. Leb, which is difucosylated, is formed by the addition of a branched fucose (Fuc) residue to H-1. The antigens that define blood group A and B phenotypes and corresponding antigens in the Lewis blood group system are formed by terminal N-acetylgalactosamine (GalNAc) or galactose (Gal) substitutions of H-1 and Leb [A-1 and A-Lewis b (ALeb), and B-1 and B-Lewis b (BLeb) antigens, respectively; Figure 1].
Recently, Aspholm-Hurtig et al investigated the ability of BabA to bind Leb, ALeb and BLeb[8]. Among 265 Leb-binding H pylori strains from various geographic regions, more than 95% of H pylori strains are “generalists” (able to bind ALeb and BLeb in addition to Leb); whereas a small subset of strains bind exclusively to ALeb, and are called “specialist” strains. The authors proposed that the middle region of BabA was responsible for determining the different binding patterns; however, the specific motifs could not be identified[8]. Interestingly, “specialist” strains originated predominantly from South American individuals (where 60% of strains were classified as “specialist”), who are known to express almost entirely the blood group O phenotype. South American isolates in their study were from Peruvian and Venezuelan Amazon Amerindian populations and also from a Colombian mestizo (mixed Amerindian-European ancestry) population; probably most of these strains came mainly from European strains[13,14]. These data suggest that most specialist babA alleles may have arisen by mutation and/or recombination within the last 500 years. Therefore, the authors propose that such rapid evolution of BabA in response to host mucosal glycosylation patterns would enable the pathogen to adapt to their individual hosts while avoiding host immune responses, and contributes importantly to the extraordinary chronicity of human H pylori infection worldwide.
The mucins secreted by gastric mucous cells form a mucous gel layer covering the gastric mucosa. This gel layer is considered the first line of gastric mucosal defense against luminal noxious agents[15–17], and damage to the mucous gel is thought to precede gastric mucosal injury. The gastric surface mucous cells and gland mucous cells express the secretory mucins MUC5AC and MUC6/MUC5B, respectively[18,19]. The majority of H pylori reside in the gastric mucus overlying the epithelium. It is reported that H pylori could be co-localized with MUC5AC gastric mucin, but not with MUC6-producing cells in the glandular areas, suggesting that adhesion is predominantly toward MUC5AC-specific ligands in gastric mucosa[20]. Subsequently, this binding phenotype could be correlated with the expression of an active BabA protein in H pylori and the presentation of the Leb antigen in the gastric mucin MUC5AC[21,22]. However, since BabA-positive strains also attached to Leb-negative MUC5AC of non-secretors, the involvement of additional epitopes and/or adhesins also must be involved[21]. In addition, binding of H pylori to MUC5B had been described[23] and a recent study confirmed that the binding was predominantly mediated by BabA and to a lesser degree by SabA adhesin[24].
LOCATION OF THE BABA GENE IN THE H PYLORI GENOME
H pylori 26695, J99 and HPAG1 each possess one babA allele (HP1243/JHP0833/HPAG1_0876) and one babB allele (HP0896/JHP1164/HPAG1_0320)[25–27]. Interestingly, the genomic locations of babA and babB genes are completely different among three strains (Figure 2). In strain J99, babA (JHP0833) is downstream of hypD (JHP0835) with a J99-specific gene (JHP0834) intervening, and babB (JHP1164) is downstream of s18 (JHP1165). In strain 26 695, the locations of babA (HP1243) and babB (HP0317) are reversed. The chromosomal locations downstream of hypD and s18 are referred to as locus A and locus B, respectively. In strain 26695, one gene encoding OMPs homologous to babA and babB (HP0317: denoted babC) with unknown function have been identified[27–29]. The location is referred to as locus C; interestingly, in strain HPAG1, the babB gene is located at locus C and babC gene at locus B. The bab genes initially were cloned from the strain CCUG17875, and this strain has two babA genes and one babB gene[1]. Gene inactivation experiments identified that only one gene (denoted babA2) had Leb antigen-binding activity; whereas another gene (babA1) did not; babA1 was located at locus B; however the locus of babA2 was not determined[1].
Figure 2.
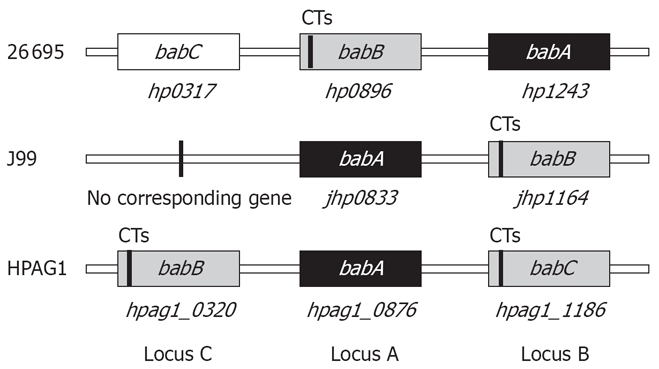
Genomic location of the babA, babB, and babC genes in strains J99, 26695, and HPAG1. CT: CT dinucleotide repeats.
The location of babA and babB in various clinical isolates of H pylori recently has been reported[29,30]. Hennig et al[30] analyzed a panel of 35 H pylori isolates and found that 24 (69%) contained babA sequences. In contrast, the babB gene was identified in 34 strains (97%). The babA gene was located at locus A for 19 strains (54%), at locus B for four strains (11%), and at locus C for three strains (9%). Four strains contain two copies of the babA gene, and the babA sequences found at two loci were identical in three strains and almost identical in one strain (i.e. three substitutions near the 5’ ends of the genes in one strain), indicating that the multiple copies of babA presumably resulted from gene conversion (intragenomic nonreciprocal recombination) events. Importantly, two strains possessed the babA gene; however, the locus could not be identified, suggesting that there are additional unidentified chromosomal loci for babA, although babA may be found in one of three chromosomal loci in most cases.
Colbeck et al[29] analyzed a panel of 44 H pylori strains and found that 32 (73%) contained babA sequences. In contrast, the babB gene was identified in 41 (95%) isolates. The babA gene was located at locus A for 25 strains (57%) and at locus B for 18 strains (41%); locus C was not evaluated. Interestingly, although chromosomal DNA from low-passage-number, single-colony isolates was used, there was a mixed genotype in 30% (13/44) of the isolates, where the population of cells contained both babA and babB at the same locus. As a result, 11 strains were found to contain two copies of the babA gene including eight with mixed babA and babB at locus B.
Overall, from two detailed studies I conclude that the babA gene prefers to be located at locus A, some strains do not possess the babA gene, some strains possess multiple copies of the babA gene, and most strains possess the babB gene. The presence of babB might confer a stronger selective advantage than the presence of babA.
REGULATION OF BABA
Chimera formation between babA and babB
Several different mechanisms for regulation of BabA expression are predicted, including at the transcriptional and translational levels. As for translational regulation, the formation of chimeric proteins seems to play an important role. Chimera formation between babA and babB initially was reported by Pride and Blaser[11] who found that in two of 42 (5%) clinical isolates studied, the 5′ regions of babB were replaced with the first 56 bp of babA (babA/B chimera). In addition, these authors showed that gene conversions frequently (10-3) occur in H pylori, and the events are recA-dependent and DNase-resistant, indicating that they likely result from intragenomic recombination. babA/B chimeras also have been reported experimentally during H pylori infection in Rhesus monkeys[10].
In addition to babA/B chimeras, babB/A chimeras have been observed[9]. A babA2 mutant from strain CCUG17875, defective in Leb-binding, regained its activity by homologous recombination of a silent babA1 gene into the babB locus, resulting in a chimeric babB/A gene. A silent wild-type babA1 gene still was present. The frequency of the babA mutant with Leb-binding was approximately 10-5. Detailed analyses of the chimeric babB/babA gene showed that the first 47 bp were babB-specific, the following 66 bp were shared between both babA and babB, and the remaining sequence was babA-specific. The second crossover event likely occurred within a region where the sequences of the babA1 and the babB locus were identical. The chimeric BabB/A protein has the potential to bind Leb; however, protein production was subject to phase variation through slipped-strand mispairing based on the number of Cysteine-Threonine (CT) dinucleotide repeats in the 5’ region of the babB gene (switch “on” = functional and switch “off” = non-functional).
Initially, only five genes encoding the OMPs in H pylori (oipA, sabA, sabB, babB and hopZ) were reported to undergo phase variations in the 5’ region such that not all strains produce functional proteins[25,27]. However, recent studies confirmed that phase variation is a method of regulating BabA production in some strains[10,29,30]. CT repeats were observed in 13 of 43 (30%) strains[29] and 4 of 22 (18%) strains[30]. Importantly, detailed analyses of the babA gene with CT repeats showed that the signal peptides are closely related to signal peptides of paralogous BabB proteins, whereas sequences further downstream were typical BabA sequences[30]. Taken together, these data suggest that the babA gene with the CT repeat might be the result of the translocation of babA into babB thereby generating a chimeric babB/babA gene. Interestingly, the babC gene in strain HPAG1 possessed CT repeats in the 5’ coding region, whereas the babC gene in strain 26695 did not. These data suggest that babB/C chimera also might have occurred in some strains.
As described above, Colbeck et al found that there were cases with mixed babA and babB genes, especially at locus B[29]. The frequency of babA translocated at the babB locus was between 10-3 and 10-4, which is in agreement with the frequency in strain CCUG17875[9]. Detailed investigation of 10 strains showed that the recombination event was identified from approximately 50 to 200 bp downstream of the ATG in five strains (all recombination occurred at locus B) and upstream of the ATG in the other five strains[29]. In the former case, the resulting gene forms the babB/A chimera, whereas complete recombination occurred in the latter case.
Overall, frequent translocation between babA and babB genes appears to be the main mechanism of regulating BabA expression. Therefore, H pylori uses both antigenic variation and phase variation to regulate babA expression.
Genomic mutations in the coding region of the babA gene
The babA gene initially was cloned from strain CCUG17875, which contains a silent babA1 gene and an expressed babA2 gene[1]. The sequence of these two genes differed only by the presence of a 10 bp deletion in the signal peptide sequence of babA1 that eliminates its translational initiation codon. However, my group recently found that all 80 strains from a panel of Western and East Asian isolates contained an intact ATG start codon in the babA gene[31], and another group also reported the absence of the babA1 type deletion[11,12,29,30,32]. Overall, the absence of a translation initiation codon, as described for babA1 from CCUG17875, should be rare. Point mutations leading to stop codon, deletion and insertion in other parts of the babA gene also are not common; Hennig et al found one of 24 babA-positive strains (4%) contained a frameshift mutation that prevented expression of a full-length BabA protein (amino acid position at 55)[30].
Transcriptional regulation of BabA
Transcriptional regulation of BabA also has been reported. Backstrom et al found that only babA2, but not babA1 was transcribed in strain CCUG17875[9]. Their analyses showed that babA transcription seemed to be regulated by the number of adenine [poly(A)] nucleotides within the -10 to -35 region of the babA promoter. The -10 and -35 region of the babA2 sequences are highly homologous to the consensus for E.coli σ70 promoter sequences. This region was stable when the number of adenines was 10 (babA2) but would become non-functional when the number was 14 (babA1). The authors hypothesized that the poly(A) sequences between the -10 and the -35 sites could be prone to slippage mutations that allow changes in the level of transcription of downstream genes. However, other studies could not confirm that the -10 to -35 spacing played an important role in regulating babA expression[30,31]. Further studies will be necessary to fully interrogate the roles of transcriptional regulation of BabA.
Overall, there are several predicted mechanisms that may control BabA expression in some strains; however, there are many cases that remain unexplained. H pylori strains that do not produce BabA can be divided into five types, as shown in Table 1.
Table 1.
Five major types of H pylori strains that do not produce BabA
| babA gene | Status |
| Negative | Include babA/B chimeras |
| Present | Regulated by slipped strand repairing and the status is “off” (probably equal to babB/A chimeras) |
| Present | Lack a translation initiation codon (single case of babA1 in strain CCUG17875) |
| Present | Have a frameshift mutation(s) causing non-productive translation |
| Present | Without apparent mutations and without a hypothesis for the lack of expression |
RELATIONSHIP BETWEEN BABA AND LEB BINDING ACTIVITY
My group recently examined BabA protein and Leb binding activity for 80 strains (40 from Japan and 40 from Colombia)[31]. BabA protein was measured by immunoblot analyses using anti-BabA antiserum (AK277), and Leb binding activity was measured by binding of H pylori to 125I-labeled fucosylated blood group antigens. H pylori strains were divided into two major groups: BabA-positive (76 strains) or BabA-negative (four strains). Semi-quantitative analyses of the BabA-positive strains allowed the BabA-positive strains to be classified into two distinct groups: those with high levels of BabA expression (68 strains) or those with low levels of BabA expression (eight strains). All of the 68 strains that exhibited Leb binding activity produced high levels of BabA. The low and non-producer strains did not exhibit Leb binding activity. Based on this finding, my group classified the strains into three distinct groups based on their expression levels of BabA: (1) BabA-high producers (BabA-H), which produce BabA protein at high enough levels to mediate Leb binding, (2) BabA-low producers (BabA-L), which produce a small amount of BabA but not enough to mediate Leb binding, and (3) BabA-negative strains, which do not produce any BabA protein.
BABA, LEB BINDING ACTIVITY AND CLINICAL OUTCOMES
There currently are only a few studies that correlate the importance of BabA with clinical outcomes using immunoblot analyses[31,33,34]. My group recently performed large scale studies of 520 geographically diverse patients presenting with different clinical symptoms to evaluate BabA status by immunoblot analysis[31]. A total of 250 isolates from Western countries (150 strains from Colombia, 100 from the U.S.) and 270 isolates from East Asia (150 from Korea and 120 from Japan) were studied. All strains from East Asia expressed BabA protein. Twenty-four (9.8%) of Western strains were BabA-negative and were associated with milder gastric injury and lower H pylori density than BabA-positive status. BabA-negative status was inversely correlated with cagA status or vacA s1 genotype (i.e. only one (4.2%) and none (0%) of these BabA-negative strains were cagA- or vacA s1-positive, respectively). This is in agreement with previous studies that the cagA status was related to the presence of Leb binding activity[1] and the presence of the babA gene[30].
Importantly, a small class of strains were BabA-positive but produced low levels of the BabA protein and lacked Leb binding activity (BabA-L)[31]. Although these strains were functionally BabA-negative and were typically CagA-positive, they were more likely to be associated with duodenal ulcer, gastric cancer, and increased mucosal inflammation and atrophy than BabA-positive strains that exhibited in vitro Leb binding activity (BabA-H strains) and BabA-negative strains. This finding suggests that either in vitro Leb binding activity does not accurately reflect the severity of mucosal damage or that the clinical outcome or in vitro binding activity does not accurately reflect in vivo conditions. The underlying reason why strains with BabA-L status were more highly correlated with severe diseases than strains with BabA-H status is unknown, and it remains unclear whether expressing low levels of BabA have a direct role in the pathogenesis of gastroduodenal diseases. It is possible that BabA expression is influenced by the intragastric environment and that the phenotype of the BabA-L strains is an epiphenomenon rather than a cause of disease. It is possible that strong Leb binding activity is associated with an inappropriate immune response resulting in severely inflamed mucosa. If so, the ability to change the BabA status from a high producer to low producer (i.e. Leb binding to Leb non-binding) would be advantageous for the organism, and a low producer might reflect an adaptation of H pylori that enhances survival in inflamed gastric mucosa. It also is possible that BabA expression down-regulates the proinflammatory effects of other putative virulence factors, such as the cag PAI and OipA.
DETECTION OF FUNCTIONAL BABA GENE
Most previous studies evaluating BabA (babA) status have used PCR techniques based on detection of the 10 bp deletion to distinguish between the babA2 and babA1 genes (Table 2)[35–53]. However, as described above, strains carrying the prototypical silent babA1 gene are very rare, and in addition, the BabA protein levels often do not match the presence of the babA (babA2) gene[31]. Current terminology for babA1 and babA2 in the literature is confusing, and many researchers mistakenly understand that H pylori strains that do not produce BabA are either babA gene-negative or babA1-positive (= babA gene lacking a translation initiation codon). However, only one case with babA1 has been reported, and BabA non-producing strains also usually possess non-functional silent babA gene sequences (i.e. 2, 4, and 5 in Table 2). Unfortunately, current PCR methods regard non-functional babA status as babA2-positive. In addition, a recent study confirmed that the PCR method used to detect babA2 with only one primer pair previously designed yielded many false-negative results, probably due to sequence variation among strains[31].
Table 2.
PCR-based genotyping for the babA2 gene in H pylori positive cases n (%)
|
Prevalence of babA2 gene |
||||||||||
| Study | Year | Population | Number studied | Total | Gastritis | PUD | Cancer | MALT | Duodenitis | Related to diseases |
| Western countries | ||||||||||
| Gerhard et al | 1999 | Germany | 114 | 82 (72) | 18 (51) | 23 (100) | 21 (78) | 20 (69) | Yes | |
| Prinz et al1, 2 | 2001 | Germany | 145 | 57 (39) | 57 (39) | - | ||||
| Rad et al1 | 2002 | Germany | 141 | 54 (38) | 54 (38) | - | ||||
| Zambon et al | 2003 | Italy | 167 | 60 (36) | 26 (28) | 20 (49) | 14 (42) | Yes | ||
| Oleastro et al | 2003 | Portugal | 140 | 45 (32) | 24 (23) | 21 (58) | Yes | |||
| Podzorski et al | 2003 | USA | 61 | 33 (36) | 22 (36) | - | ||||
| Oliveira et al | 2003 | Brazil | 208 | 96 (46) | 24 (32) | 43 (54) | 29 (56) | Yes | ||
| Rad et al3 | 2004 | Germany | 207 | 73 (35) | 73 (35) | - | ||||
| Lehours et al | 2004 | France | 82 | 40 (49) | 21 (54) | 19 (44) | No | |||
| Gatti et al | 2005 | Brazil | 89 | 42 (47) | 37 (53) | 3 (20) | 1 (100) | 1 (33) | No | |
| Olfat et al | 2005 | Germany | 92 | 41 (45) | 19 (28) | 22 (88) | Yes | |||
| Sweden | 74 | 33 (45) | 21 (48) | 12 (40) | No | |||||
| Portugal | 91 | 31 (34) | 12 (20) | 19 (63) | Yes | |||||
| Finland | 57 | 34 (60) | 12 (46) | 22 (71) | Borderline (P = 0.06) | |||||
| Gatti et al | 2006 | Brazil | 94 | 38 (40) | 18 (41) | 20 (40) | No | |||
| Asian countries | ||||||||||
| Mizushima et al | 2001 | Japan | 179 | 152 (85) | 34 (81) | 73 (85) | 36 (90) | 9 (82) | No | |
| Yu et al | 2002 | China | 104 | 83 (80) | 83 (80) | - | ||||
| Lai et al | 2002 | Taiwan | 101 | 101 (100) | 41 (100) | 46 (100) | 14 (100) | No | ||
| Han et al | 2004 | China | 141 | 90 (64) | 28 (65) | 50 (65) | 12 (57) | Yes (DU vs GU) | ||
| Zheng et al | 2006 | China | 72 | 28 (39) | 11 (39) | 17 (40) | No | |||
| Lee et al | 2006 | Korea | 135 | 83 (61) | 64 (57) | 19 (86) | Yes | |||
| Erzin et al | 2006 | Turkey | 91 | 49 (54) | 7 (23) | 12 (43) | 24 (73) | Yes | ||
PUD: Peptic ulcer disease; MALT: Mucosal-associated lymphoid tissue; DU: Duodenal ulcer; GU: Gastric ulcer.
88% had German nationality and 12% were from other European countries;
Samples were examined from the antrum and the corpus, and the corpus data are presented (in the antrum, 55 were babA2-positive);
89% had German nationality and 11% were from other southern European countries.
Only a few studies have used a forward primer that is within the promoter region of the babA gene, a region that is identical to the sequence of babA2 but different from that of babA1 in strain CCUG17875[32,54,55]; however, recent analyses showed that the primers could also detect babB gene[31]. Overall, the information gained from currently used PCR-based methods must be interpreted with caution. In addition, I propose that researchers should not use current PCR-based methods in future studies.
Nonetheless, approximately half of the studies have suggested a correlation between babA2-positive H pylori in Western countries and increased risk of developing significant clinical outcomes[38,44–46,52] and are in agreement with protein data as described above[31,33,34]. The prevalence of clinical isolates with a non-functional babA2 gene without production of BabA protein may be low and negligible in some studies.
CONCLUSION
Several different mechanisms for regulation of BabA expression are predicted, including at both the transcriptional and translational levels. The formation of chimeric proteins seems to play an especially important role in translational regulation. The chimeric BabB/A protein has the potential to bind Leb; however, the production was subject to phase variation through slipped-strand mispairing. Currently used PCR-based methods to evaluate BabA status do not take this mechanism of regulation into account, and information gained from currently used PCR-based methods must be interpreted with caution. I strongly recommend that researchers should not use PCR-based methods in their future studies. Recent studies evaluating BabA status by immunoblot confirmed that BabA-positive status in Western strains was closely associated with severe gastric injury, high H pylori density, and severe clinical outcomes. A small class of strains produced low levels of the BabA protein and lacked Leb binding activity. Surprisingly, they were more likely to be associated with increased mucosal inflammation, atrophy, and severe clinical outcomes than BabA-positive strains that exhibit Leb binding activity. The underlying reason is unclear, and further studies will be necessary to investigate how the complex BabA-receptor network is functionally coordinated during the interaction of H pylori with the gastric mucosa.
Supported by (in part) National Institutes of Health Grants, R01 DK62813
Peer reviewer: Özlem Yilmaz, PhD, Associate Professor of Microbiology, Dokuz Eylul University, School of Medicine, Department of Microbiology and Clinical Microbiology, Inciralti 35340, Izmir, Turkey
S- Editor Zhong XY L- Editor Li M E- Editor Ma WH
References
- 1.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 2.Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakamoto S, Watanabe T, Tokumaru T, Takagi H, Nakazato H, Lloyd KO. Expression of Lewisa, Lewisb, Lewisx, Lewisy, siayl-Lewisa, and sialyl-Lewisx blood group antigens in human gastric carcinoma and in normal gastric tissue. Cancer Res. 1989;49:745–752. [PubMed] [Google Scholar]
- 4.Boren T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 5.Falk P, Roth KA, Boren T, Westblom TU, Gordon JI, Normark S. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. Proc Natl Acad Sci USA. 1993;90:2035–2039. doi: 10.1073/pnas.90.5.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falk PG, Bry L, Holgersson J, Gordon JI. Expression of a human alpha-1,3/4-fucosyltransferase in the pit cell lineage of FVB/N mouse stomach results in production of Leb-containing glycoconjugates: a potential transgenic mouse model for studying Helicobacter pylori infection. Proc Natl Acad Sci USA. 1995;92:1515–1519. doi: 10.1073/pnas.92.5.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guruge JL, Falk PG, Lorenz RG, Dans M, Wirth HP, Blaser MJ, Berg DE, Gordon JI. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc Natl Acad Sci USA. 1998;95:3925–3930. doi: 10.1073/pnas.95.7.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, Vikstrom S, Sjostrom R, Linden S, Backstrom A, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305:519–522. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- 9.Backstrom A, Lundberg C, Kersulyte D, Berg DE, Boren T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc Natl Acad Sci USA. 2004;101:16923–16928. doi: 10.1073/pnas.0404817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solnick JV, Hansen LM, Salama NR, Boonjakuakul JK, Syvanen M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc Natl Acad Sci USA. 2004;101:2106–2111. doi: 10.1073/pnas.0308573100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pride DT, Blaser MJ. Concerted evolution between duplicated genetic elements in Helicobacter pylori. J Mol Biol. 2002;316:629–642. doi: 10.1006/jmbi.2001.5311. [DOI] [PubMed] [Google Scholar]
- 12.Pride DT, Meinersmann RJ, Blaser MJ. Allelic Variation within Helicobacter pylori babA and babB. Infect Immun. 2001;69:1160–1171. doi: 10.1128/IAI.69.2.1160-1171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kersulyte D, Mukhopadhyay AK, Velapatino B, Su W, Pan Z, Garcia C, Hernandez V, Valdez Y, Mistry RS, Gilman RH, et al. Differences in genotypes of Helicobacter pylori from different human populations. J Bacteriol. 2000;182:3210–3218. doi: 10.1128/jb.182.11.3210-3218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaoka Y, Orito E, Mizokami M, Gutierrez O, Saitou N, Kodama T, Osato MS, Kim JG, Ramirez FC, Mahachai V, et al. Helicobacter pylori in North and South America before Columbus. FEBS Lett. 2002;517:180–184. doi: 10.1016/s0014-5793(02)02617-0. [DOI] [PubMed] [Google Scholar]
- 15.Ota H, Katsuyama T. Alternating laminated array of two types of mucin in the human gastric surface mucous layer. Histochem J. 1992;24:86–92. doi: 10.1007/BF01082444. [DOI] [PubMed] [Google Scholar]
- 16.Allen A, Flemstrom G, Garner A, Kivilaakso E. Gastroduodenal mucosal protection. Physiol Rev. 1993;73:823–857. doi: 10.1152/physrev.1993.73.4.823. [DOI] [PubMed] [Google Scholar]
- 17.Ho SB, Takamura K, Anway R, Shekels LL, Toribara NW, Ota H. The adherent gastric mucous layer is composed of alternating layers of MUC5AC and MUC6 mucin proteins. Dig Dis Sci. 2004;49:1598–1606. doi: 10.1023/b:ddas.0000043371.12671.98. [DOI] [PubMed] [Google Scholar]
- 18.Ho SB, Shekels LL, Toribara NW, Kim YS, Lyftogt C, Cherwitz DL, Niehans GA. Mucin gene expression in normal, preneoplastic, and neoplastic human gastric epithelium. Cancer Res. 1995;55:2681–2690. [PubMed] [Google Scholar]
- 19.Van De Bovenkamp JH, Korteland-Van Male AM, Buller HA, Einerhand AW, Dekker J. Infection with Helicobacter pylori affects all major secretory cell populations in the human antrum. Dig Dis Sci. 2005;50:1078–1086. doi: 10.1007/s10620-005-2708-4. [DOI] [PubMed] [Google Scholar]
- 20.Van den Brink GR, Tytgat KM, Van der Hulst RW, Van der Loos CM, Einerhand AW, Buller HA, Dekker J. H pylori colocalises with MUC5AC in the human stomach. Gut. 2000;46:601–607. doi: 10.1136/gut.46.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van de Bovenkamp JH, Mahdavi J, Korteland-Van Male AM, Buller HA, Einerhand AW, Boren T, Dekker J. The MUC5AC glycoprotein is the primary receptor for Helicobacter pylori in the human stomach. Helicobacter. 2003;8:521–532. doi: 10.1046/j.1523-5378.2003.00173.x. [DOI] [PubMed] [Google Scholar]
- 22.Linden S, Nordman H, Hedenbro J, Hurtig M, Boren T, Carlstedt I. Strain- and blood group-dependent binding of Helicobacter pylori to human gastric MUC5AC glycoforms. Gastroenterology. 2002;123:1923–1930. doi: 10.1053/gast.2002.37076. [DOI] [PubMed] [Google Scholar]
- 23.Namavar F, Sparrius M, Veerman EC, Appelmelk BJ, Vandenbroucke-Grauls CM. Neutrophil-activating protein mediates adhesion of Helicobacter pylori to sulfated carbohydrates on high-molecular-weight salivary mucin. Infect Immun. 1998;66:444–447. doi: 10.1128/iai.66.2.444-447.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walz A, Odenbreit S, Mahdavi J, Boren T, Ruhl S. Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology. 2005;15:700–708. doi: 10.1093/glycob/cwi049. [DOI] [PubMed] [Google Scholar]
- 25.Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 26.Oh JD, Kling-Backhed H, Giannakis M, Xu J, Fulton RS, Fulton LA, Cordum HS, Wang C, Elliott G, Edwards J, et al. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: evolution during disease progression. Proc Natl Acad Sci USA. 2006;103:9999–10004. doi: 10.1073/pnas.0603784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 28.Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun. 2000;68:4155–4168. doi: 10.1128/iai.68.7.4155-4168.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colbeck JC, Hansen LM, Fong JM, Solnick JV. Genotypic profile of the outer membrane proteins BabA and BabB in clinical isolates of Helicobacter pylori. Infect Immun. 2006;74:4375–4378. doi: 10.1128/IAI.00485-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hennig EE, Allen JM, Cover TL. Multiple chromosomal loci for the babA gene in Helicobacter pylori. Infect Immun. 2006;74:3046–3051. doi: 10.1128/IAI.74.5.3046-3051.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujimoto S, Olaniyi Ojo O, Arnqvist A, Wu JY, Odenbreit S, Haas R, Graham DY, Yamaoka Y. Helicobacter pylori BabA expression, gastric mucosal injury, and clinical outcome. Clin Gastroenterol Hepatol. 2007;5:49–58. doi: 10.1016/j.cgh.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheu BS, Sheu SM, Yang HB, Huang AH, Wu JJ. Host gastric Lewis expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut. 2003;52:927–932. doi: 10.1136/gut.52.7.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamaoka Y, Souchek J, Odenbreit S, Haas R, Arnqvist A, Boren T, Kodama T, Osato MS, Gutierrez O, Kim JG, et al. Discrimination between cases of duodenal ulcer and gastritis on the basis of putative virulence factors of Helicobacter pylori. J Clin Microbiol. 2002;40:2244–2246. doi: 10.1128/JCM.40.6.2244-2246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, El-Zimaity HM, Reddy R, Arnqvist A, Graham DY. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut. 2006;55:775–781. doi: 10.1136/gut.2005.083014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erzin Y, Koksal V, Altun S, Dobrucali A, Aslan M, Erdamar S, Dirican A, Kocazeybek B. Prevalence of Helicobacter pylori vacA, cagA, cagE, iceA, babA2 genotypes and correlation with clinical outcome in Turkish patients with dyspepsia. Helicobacter. 2006;11:574–580. doi: 10.1111/j.1523-5378.2006.00461.x. [DOI] [PubMed] [Google Scholar]
- 36.Gatti LL, Fagundes e Souza EK, Leite KR, Bastos EL, Vicentini LR, Silva LC, Smith Mde A, Payao SL. cagA vacA alelles and babA2 genotypes of Helicobacter pylori associated with gastric disease in Brazilian adult patients. Diagn Microbiol Infect Dis. 2005;51:231–235. doi: 10.1016/j.diagmicrobio.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 37.Gatti LL, Modena JL, Payao SL, Smith Mde A, Fukuhara Y, Modena JL, de Oliveira RB, Brocchi M. Prevalence of Helicobacter pylori cagA, iceA and babA2 alleles in Brazilian patients with upper gastrointestinal diseases. Acta Trop. 2006;100:232–240. doi: 10.1016/j.actatropica.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 38.Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci USA. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han YH, Liu WZ, Zhu HY, Xiao SD. Clinical relevance of iceA and babA2 genotypes of Helicobacter pylori in a Shanghai population. Chin J Dig Dis. 2004;5:181–185. doi: 10.1111/j.1443-9573.2004.00175.x. [DOI] [PubMed] [Google Scholar]
- 40.Lai CH, Kuo CH, Chen YC, Chao FY, Poon SK, Chang CS, Wang WC. High prevalence of cagA- and babA2-positive Helicobacter pylori clinical isolates in Taiwan. J Clin Microbiol. 2002;40:3860–3862. doi: 10.1128/JCM.40.10.3860-3862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee HS, Choe G, Kim WH, Kim HH, Song J, Park KU. Expression of Lewis antigens and their precursors in gastric mucosa: relationship with Helicobacter pylori infection and gastric carcinogenesis. J Pathol. 2006;209:88–94. doi: 10.1002/path.1949. [DOI] [PubMed] [Google Scholar]
- 42.Lehours P, Menard A, Dupouy S, Bergey B, Richy F, Zerbib F, Ruskone-Fourmestraux A, Delchier JC, Megraud F. Evaluation of the association of nine Helicobacter pylori virulence factors with strains involved in low-grade gastric mucosa-associated lymphoid tissue lymphoma. Infect Immun. 2004;72:880–888. doi: 10.1128/IAI.72.2.880-888.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizushima T, Sugiyama T, Komatsu Y, Ishizuka J, Kato M, Asaka M. Clinical relevance of the babA2 genotype of Helicobacter pylori in Japanese clinical isolates. J Clin Microbiol. 2001;39:2463–2465. doi: 10.1128/JCM.39.7.2463-2465.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oleastro M, Gerhard M, Lopes AI, Ramalho P, Cabral J, Sousa Guerreiro A, Monteiro L. Helicobacter pylori virulence genotypes in Portuguese children and adults with gastroduodenal pathology. Eur J Clin Microbiol Infect Dis. 2003;22:85–91. doi: 10.1007/s10096-002-0865-3. [DOI] [PubMed] [Google Scholar]
- 45.Olfat FO, Zheng Q, Oleastro M, Voland P, Boren T, Karttunen R, Engstrand L, Rad R, Prinz C, Gerhard M. Correlation of the Helicobacter pylori adherence factor BabA with duodenal ulcer disease in four European countries. FEMS Immunol Med Microbiol. 2005;44:151–156. doi: 10.1016/j.femsim.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Oliveira AG, Santos A, Guerra JB, Rocha GA, Rocha AM, Oliveira CA, Cabral MM, Nogueira AM, Queiroz DM. babA2- and cagA-positive Helicobacter pylori strains are associated with duodenal ulcer and gastric carcinoma in Brazil. J Clin Microbiol. 2003;41:3964–3966. doi: 10.1128/JCM.41.8.3964-3966.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Podzorski RP, Podzorski DS, Wuerth A, Tolia V. Analysis of the vacA, cagA, cagE, iceA, and babA2 genes in Helicobacter pylori from sixty-one pediatric patients from the Midwestern United States. Diagn Microbiol Infect Dis. 2003;46:83–88. doi: 10.1016/s0732-8893(03)00034-8. [DOI] [PubMed] [Google Scholar]
- 48.Prinz C, Schoniger M, Rad R, Becker I, Keiditsch E, Wagenpfeil S, Classen M, Rosch T, Schepp W, Gerhard M. Key importance of the Helicobacter pylori adherence factor blood group antigen binding adhesin during chronic gastric inflammation. Cancer Res. 2001;61:1903–1909. [PubMed] [Google Scholar]
- 49.Rad R, Gerhard M, Lang R, Schoniger M, Rosch T, Schepp W, Becker I, Wagner H, Prinz C. The Helicobacter pylori blood group antigen-binding adhesin facilitates bacterial colonization and augments a nonspecific immune response. J Immunol. 2002;168:3033–3041. doi: 10.4049/jimmunol.168.6.3033. [DOI] [PubMed] [Google Scholar]
- 50.Rad R, Dossumbekova A, Neu B, Lang R, Bauer S, Saur D, Gerhard M, Prinz C. Cytokine gene polymorphisms influence mucosal cytokine expression, gastric inflammation, and host specific colonisation during Helicobacter pylori infection. Gut. 2004;53:1082–1089. doi: 10.1136/gut.2003.029736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu J, Leung WK, Go MY, Chan MC, To KF, Ng EK, Chan FK, Ling TK, Chung SC, Sung JJ. Relationship between Helicobacter pylori babA2 status with gastric epithelial cell turnover and premalignant gastric lesions. Gut. 2002;51:480–484. doi: 10.1136/gut.51.4.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zambon CF, Navaglia F, Basso D, Rugge M, Plebani M. Helicobacter pylori babA2, cagA, and s1 vacA genes work synergistically in causing intestinal metaplasia. J Clin Pathol. 2003;56:287–291. doi: 10.1136/jcp.56.4.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng PY, Tang FA, Qi YM, Li J. Association of peptic ulcer with increased expression of Lewis antigens, but not vacuolating cytotoxin activity or babA2 gene status, in Helicobacter pylori strains from China. Chin J Dig Dis. 2006;7:61–65. doi: 10.1111/j.1443-9573.2006.00246.x. [DOI] [PubMed] [Google Scholar]
- 54.Lai CH, Poon SK, Chen YC, Chang CS, Wang WC. Lower prevalence of Helicobacter pylori infection with vacAs1a, cagA-positive, and babA2-positive genotype in erosive reflux esophagitis disease. Helicobacter. 2005;10:577–585. doi: 10.1111/j.1523-5378.2005.00363.x. [DOI] [PubMed] [Google Scholar]
- 55.Mattar R, dos Santos AF, Eisig JN, Rodrigues TN, Silva FM, Lupinacci RM, Iriya K, Carrilho FJ. No correlation of babA2 with vacA and cagA genotypes of Helicobacter pylori and grading of gastritis from peptic ulcer disease patients in Brazil. Helicobacter. 2005;10:601–608. doi: 10.1111/j.1523-5378.2005.00360.x. [DOI] [PubMed] [Google Scholar]