

Abstract
Inflammatory bowel disease (IBD), which includes Crohn’s disease (CD) and ulcerative colitis (UC), represents a group of chronic disorders characterized by inflammation of the gastrointestinal tract, typically with a relapsing and remitting clinical course. Mucosal macrophages play an important role in the mucosal immune system, and an increase in the number of newly recruited monocytes and activated macrophages has been noted in the inflamed gut of patients with IBD. Activated macrophages are thought to be major contributors to the production of inflammatory cytokines in the gut, and imbalance of cytokines is contributing to the pathogenesis of IBD. The intestinal inflammation in IBD is controlled by a complex interplay of innate and adaptive immune mechanisms. Cytokines play a key role in IBD that determine T cell differentiation of Th1, Th2, T regulatory and newly described Th17 cells. Cytokines levels in time and space orchestrate the development, recurrence and exacerbation of the inflammatory process in IBD. Therefore, several cytokine therapies have been developed and tested for the treatment of IBD patients.
Keywords: Cytokines, Inflammatory bowel disease, Crohn’s disease, Ulcerative colitis, Inflammation
INTRODUCTION
Inflammatory bowel disease (IBD) comprises two forms, Ulcerative Colitis (UC) and Crohn’s disease (CD). Currently, the pathogenesis of UC and CD is not completely understood, although the chronic relapsing inflammation is thought to be result from a dysregulated, aberrant immune response to intestinal flora in a context of genetic predisposition. In IBD, this loss of immune tolerance toward the enteric flora it is mediated by different molecules.
Cytokines are key signals in the intestinal immune system, and are known to participate in the disruption of the so-called normal state of controlled inflammation (physiological inflammation of the gut)[1]. Cytokines are small peptide proteins produced mainly by immune cells that facilitate communication between cells, stimulate the proliferation of antigen specific effector cells, and mediate the local and systemic inflammation in an autocrine, paracrine, and endocrine pathways[2]. In IBD, the innate immune response plays a critical role. Activated dendritic cells (DC) and macrophages secrete several cytokines that actively regulate the inflammatory response in UC and CD. Once secreted by these antigen presenting cells (APC), these cytokines trigger and differentiate many T cells activating the adaptive immune response. IBD has also a T cell dysregulation where clearance of overreactive and autoreactive cells is disturbed, in addition to an imbalance of Treg/Th1, Th2 and newly described Th17 cells populations in the activated state. The lack of appropriate regulation from T cells, or an over-production of effector T cells, participates in the development and exacerbation of IBD[3].
Altogether, APCs, Th1, Th2, T regulatory cells and most recently characterized Th17 and their cytokine products play a complex role in IBD[4]. These cellular interactions are modulated by both traditionally studied cytokines (such as TNF-α, INF-γ, IL-1, IL-6, IL-4, IL-5, IL10, TGF-β) and others recently characterized (like IL-13, IL-12, IL-18, IL-23), considered to be either pro or anti-inflammatory, as shown in Table 1[5]. Although many common responses in IBD are mediated by cytokines, such as the regulation of the production of inflammatory mediators, reactive oxygen metabolites, nitric oxide, leukotriens, platelet-activating factor, and prostaglandins, activation of the nuclear factor κB (NF-κB) and inhibition of apoptosis, how cytokines determine the nature of the immune response in IBD may be quite different among IBD forms[6]. CD is associated with a Th1 T cell mediated response, characterized by enhanced production of IFN-γ and TNF-α. IL-12 and IL-23 govern the Th1 differentiation which in combination with IL-15, IL-18 and IL-21 will induce the stabilization of polarized Th1. On the other hand, in UC, the local immune response is less polarized, but it is characterized by CD1 reactive natural killer T cell production of IL-13 and Th2 cytokine production, as shown in Figure 1[7].
Table 1.
Role of cytokines and cell lines involved in their production in patients with IBD
| Cytokine | UC | CD | Cells involved in the production |
| TNF-α | Up-regulated | Up-regulated | Macrophages |
| TL1α | Unknown | Up-regulated | Th1 |
| IL-1β | IL-1ra/IL-1 ratio | IL-1ra/IL-1 ratio | Macrophages |
| IL-6 | Up-regulated | Up-regulated | Macrophages, DC, Th17 and others |
| IL-18 | Not | Yes, not in all patients | Macrophages |
| TGF-β | Not clear, maybe defective signalling | Not clear, maybe defective signalling | Th0, Th3, Treg |
| IL-10 | Not clear | Yes, up-regulated | Tr1 and Breg |
| IL-4 | Not clear | Not clear | Th2, NK |
| IL-12 | Up-regulated | Up-regulated | Macrophages, DC |
| IL-23 | Yes | Yes | Macrophages, DC |
| IL-27 | Not clear | Up-regulated | APCs |
| IL-17 | Up-regulated | Up-regulated | Th17 |
| IL-13 | Up-regulated | Not | Th1, NK |
| IL-5 | Up-regulated | Not | Th2, NK |
Figure 1.
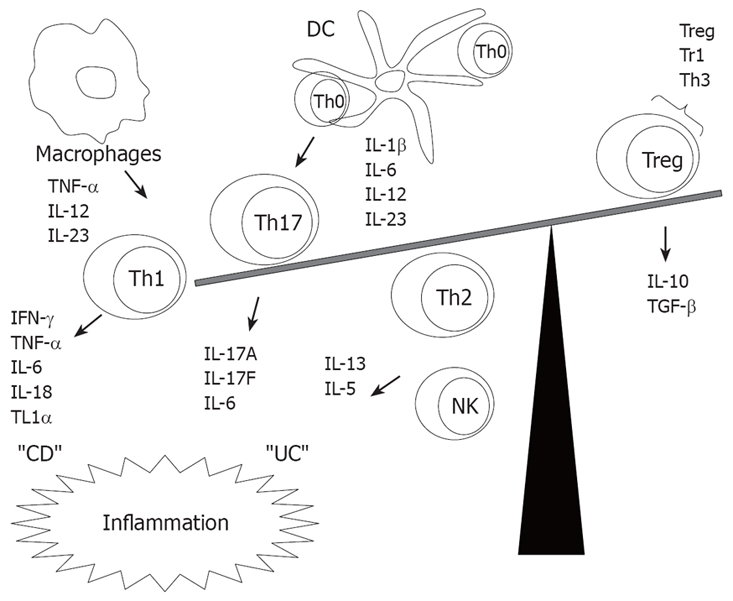
Cytokines imbalance between effector and T regulatory cells in IBD.
CLASSICAL PRO-INFLAMMATORY CYTOKINES
Lymphocytes and APCs orchestrate a lot of the inflammation in IBD, mainly the production of TNF-α, a 17-kD pleiotropic cytokine produced by innate immune cells as macrophages, monocytes, and also by differentiated T cells. TNF-α exerts its pro-inflammatory effects through increased production of IL-1β and IL-6, expression of adhesion molecules, proliferation of fibroblasts and procoagulant factors, as well as initiation of cytotoxic, apoptotic, acute-phase responses, and inhibition of apoptosis[8,9]. TNF-α expression in human macrophages was discovered in the colonic tissue and macrophages in both patients with CD and UC[10] and serum levels of TNF-α correlate with clinical and laboratory indices of intestinal disease activity[11]. Clinical studies have reported a dramatic improvement in CD patients treated with anti-TNF-α therapy such as infliximab, adalimumab and certolizumab pegol[12]. Reductions in the number of IFN-γ producing, lamina propria mononuclear cells (LPMC) in colonic biopsies results from anti-TNF-α treated patients[13].
The signalling of TNF-α starts with serum soluble TNF receptorIand II (sTNF-RI, II) levels correlate with disease activity in IBD patients. More specifically, sTNF-RI is up-regulated in the serum of IBD patients compared to healthy controls and could be used as a marker for disease activity[14]. sTNF-RII levels are significantly more elevated in serum from active CD patients as compared to UC and could be used as an additional parameter to discriminate both diseases[12]. Recently TNF receptor type 1-dependent activation of innate responses was shown to reduce intestinal damage-associated mortality[15].
Related to the TNF-α, the TNF-like factor (TL1A) seems to stimulate IFN-γ secretion by binding to the death receptor 3 (DR3). A higher percentage of cells express the TL1A receptor DR3 in mucosal biopsies taken in CD and UC, and increased synthesis of IFN-γ has been observed to correlate with severity of disease in IBD patients[16]. This molecule links TNF related apoptosis in inflammatory intestinal epithelial lesions, tumour-necrosis-factor related apoptosis inducing ligand (TRAIL) messenger RNA and protein were markedly up-regulated in IEC and lamina propria lymphocytes in animal model. Interferon-gamma and TNF-alpha potently induced TRAIL in IEC and TRAIL is highly up-regulated in IEC in inflammatory ileum and colon[9].
In addition to TNF-α, IL-1 seems to be important in the pathogenesis of IBD because of its immunological up-regulatory and pro-inflammatory activities. The IL-1 system consists of IL-1α and IL-1β, both of which are produced by various cell types through the initiation of cyclooxygenase type 2, phospholipase A, and inducible nitric oxide synthase (iNOS)[17]. The IL-1 system can be also highly regulated by IL-1 receptor antagonist (IL-1Ra), as supported by the findings of high plasma and tissue levels of IL-1Ra in patients with IBD, indicating that IL-1Ra may be part of the host mechanism for downregulation of inflammation[18]. The IL-1Ra/IL-1 ratio decreases with increasing IBD activity, while remaining constant in uninvolved CD and inflammatory control specimens: this may contribute to the pathogenesis of chronic gut inflammation[19]. Increased levels of IL-1 in IBD may be result of stimulation of colonic macrophages that can activate interleukin (IL)-1 converting enzyme (ICE) and hence release mature IL-1β into the colonic mucosa[20].
In contrast to other cytokines, IL-6 is a pleiotropic cytokine that exerts its proinflammatory effects largely by means of its soluble IL-6 receptor (sIL-6R). The combination of soluble IL-6 receptor (sIL-6R) and IL-6 stimulates cells that only express gp130 and not IL-6R, a process known as trans-signalling. IL-6 signalling through signal transducer and activator of transcription-3 (STAT3) has been extensively studied[21]. This system plays a central role in several immunologic reactions during the development of IBD, and circulating levels of IL-6 and sIL-6R correlate with many clinical features of CD and UC[22–24]. Blockade of IL-6 trans-signalling causes T-cell apoptosis, indicating that the IL-6-sIL-6R system mediates the resistance of T cells to apoptosis in CD[24]. Yamamoto et al (Kallen KJ[25]) introduced the anti-IL-6 receptor monoclonal antibody to a murine colitis model and found that the treatment with this antibody reduced IFN-γ, TNF-α, and IL-1β mRNA, and suppressed expression of several intracellular adhesion molecules in the colonic vascular endothelium. The anti-IL-6 receptor monoclonal antibody also abrogates murine colitis by effectively blocking the recruitment of leukocytes and increasing T-cell apoptosis[26]. Peripheral immune cells as well as colon epithelial and lamina propria cells with an active form of IL-6/STAT3 system may be responsible of the high correlation with the degree of mucosal inflammation[27]. The signaling of IL-6/STAT3 in the activation of mucosal T cells has been suggested as a major therapeutic target for the future[28]. STAT-3 itself induces the anti-apoptotic factors Bcl-2 and Bcl-xL, thus resulting in T-cell resistance against apoptosis. This circle of T-cell accumulation, mediated by apoptosis resistance, finally leading to chronic inflammation, can be blocked by anti-IL-6 receptor antibodies[29].
IL-18
IL-18 is produced by intestinal epithelial cells and was originally identified as an IFN-γ inducing factor, shares similarities with the IL-1 family in terms of its structure, processing, receptor, signal transduction pathway, and pro-inflammatory properties[30]. Recent studies have shown that the balance between this pleiotropic pro-inflammatory cytokine and its natural inhibitor, IL-18-binding protein (IL-18BP), may contribute to the pathogenesis of IBD[31]. A local increase of IL-18 expression has been demonstrated in chronic lesions of CD compared with uninvolved areas or normal controls[32], and an increase in IL-18 was also shown to be accompanied by marked increases in IL-18 receptor-positive immune cells as well as intense transcription of IL-18 induced by cytokines, such as IFN-γ, IL-1β, and TNF-α[33]. In a recent study has been reported that IL-18 up-regulation may be found only in a minority of patients with CD[34]. Furthermore, in the presence of IL-18, T cells from the inflamed CD tissue have been shown to produce less IL-10 than control tissue[35]. Although recombinant IL-18 alone induces significant proliferative responses in freshly isolated mucosal lymphocytes from CD patients[36,37], a synergy between IL-12 and IL-18 in activated macrophages may be a regulatory mechanism driving lamina propria lymphocytes toward a Th1 response in IBD[38]. It has been reported that cytokine IL-12 may act in synergy with IL-18 to promote the induction of IFN-γ, leading to severe gut inflammation in mice[39]. The development of Th1 CD4+ T cells in the intestinal mucosa is driven by IL-12, produced from activated macrophages, and IL-18, produced from activated macrophages and colonic epithelial cells. The synergistic effect is mainly caused by mechanisms involving the up-regulation of the IL-18 receptor by IL-12[40].
ANTI-INFLAMMATORY AND IMMUNOM ODULATORY CYTOKINES
IL-10
IL-10 is an anti-inflammatory cytokine that inhibits both antigen presentation and subsequent release of pro-inflammatory cytokines, thereby attenuating mucosal inflammation. The pivotal role played by IL-10 within the mucosal immune system has been extensively studied in the chronic ileo-colitis that develops in gene-targeted IL-10 knockout mice and by its therapeutic efficacy in several animal models of colitis[41]. An inactivation of IL-10 in mice results in an increased production of IL-12 and IFN-γ[42,43]. Inflamed tissues and granulomas of CD show low IL-10[44]. Melgar et al[45] reported a highly significant increase in IL-10 mRNA levels in T lymphocytes and in IL-10-positive cells in the colons of UC patients. Recently production of IL-10 by regulatory T cells has been implicated as important issue in IBD[46]. Other regulatory cells that may participate in UC through the production of IL-10 are a regulatory B cells subtype called Bregs[47]. The importance of IL-10 production by B cells has been evidenced in IBD models and in humans[48,49], Mizoguchi et al showed that Bregs can be responsible for the suppression and/or recovery form acquired immune mediated inflammations by mechanisms that include IL-10 and TGF-β1 in IBD[47].
IL-4 and TGF-β
Overall, anti-inflammatory cytokines whose roles are less well characterized in IBD include IL-4 and TGF-β. IL-4 is a stimulatory molecule for B and T cells, and has known immunosuppressive effects in the intestine[50]. T-cell receptor alpha chain-deficient mice (TCR -/-) treated with anti-IL-4 monoclonal antibody showed a decrease in Th2-type mRNA cytokine production and an increase in expression of IFN-γ, suggesting that IL-4 plays a major role in inducing Th2-type CD4+ cells in the gut to shift towards a Th1 response[51]. Also another study showed that development of colitis in the TCR -/- mice depends on IL-4 rather than IFN-γ[52]. A study reported that the administration of IL-4 led to a significant reduction of the vascular endothelial growth factor (VEGF) production by peripheral blood mononuclear cells in active CD and UC patients[53].
Similarly, TGF-β is an inhibitory cytokine recognized as a key regulator of immunological homeostasis and inflammatory responses. Reduced TGF-β activity is considered to be responsible for the development of autoimmune disorders in several pathologic conditions including IBD[54]. Defective transforming growth factor TGF-β1 signaling due to high levels of Smad7 is a feature of IBD[55]. UC patients have exhibited increased production of TGF-β1 by LPMC as compared with both CD patients and controls, highlighting that although TGF-β acts on the systemic immune system to promote a potent immunosuppressive effect, locally TGF-β may demonstrate pro-inflammatory properties[56]. Evidence suggests that TGF-β can act in concert with epidermal, insulin-like, fibroblast growth factors, as well as VEGF to protect host tissue from luminal challenges and facilitate repair of mucosal injury in IBD[57,58]. As future therapy, the inhibition of Smad7 may reestablish TGF-β1 function and the suppression of colitis as proven in experimental models of colitis[59].
IL-12 and related cytokines
IL-12 and IL-23 belong to the IL-12 family of pro-inflammatory heterodimeric cytokines and comprises IL-12p40/IL-12p35 and IL-12p40/IL-23p19 subunits[60]. They are mainly produced by activated APCs and accessory cells such as DC and phagocytes[61]. The receptors for these cytokines are also heterodimeric IL-12 binds an IL-12Rβ-IL-12Rβ2 heterodimer, whereas IL-23 binds an IL-12Rβ1-IL-23R heterodimer[60]. The receptors for both IL-12 and IL-23 are mainly expressed on T cells, NK cells, and NKT cells. However, low levels of the receptor for IL-23 are also expressed on monocytes, macrophages, and DCs[61]. Both cytokines activate TYK2 and JAK2 as well as STAT1, STAT3, STAT4, and STAT5[60]. Although IL-12 activates STAT4 most efficiently, IL-23 preferentially activates STAT3[60]. Despite the similarities in receptor subunit and signaling, recent studies have shown that IL-12 and IL-23 drive divergent immunological pathways.
The expression of IL-12 is up-regulated in both active UC and CD biopsies and it correlates with activity index score[62]. Levels of IL12p40 and IL12Rβ2 are higher in early rather than in late CD suggesting that IL12-mediated modulation is strongly dependent on the stage of disease[63]. In particular, to drive adaptive immune responses, DCs (that sense the nature of the micro-organisms in the intestine) are key producers of IL-12 in IBD[64].
In animal models, IL-23 showed to be essential for manifestation of chronic intestinal inflammation, whereas IL-12 is not. A critical target of IL-23 is a unique subset of tissue-homing memory T cells, which are specifically activated by IL-23 to produce the pro-inflammatory mediators IL-17 and IL-6[65].
Recently, another IL-12-related cytokine, IL-27, was described. IL-27 consists of EBI3, an IL-12p40-related protein, and p28, a newly discovered IL-12p35-related polypeptide. Mucosal expression of IL-23p19 and IL-27p28 transcripts correlate with the inflammatory activity in IBD both CD and UC. Particularly, IL-27p28 transcripts and EBI3 transcripts were significantly elevated only in active CD[66].
IL-17 and Th17 cells
Recently, a new T cell subset named “Th17”, characterized by the production of IL-17, was identified as an important player in inflammatory responses[67]. Sequencing the human genome resulted in the discovery of an additional five members of the IL-17 family that were consecutively named IL-17B to IL-17F. IL-17A is exclusively produced by Th17 cells[68]. The production of IL-17 relies on STAT3 activation triggered by IL-23[69]. IL-17 in general induces the recruitment of immune cells to peripheral tissues, a response that requires NF-κB activation after IL-17 receptor engagement[70,71]. IL-17 also leads to the induction of many pro-inflammatory factors, including TNF-α, IL-6, and IL-1β, suggesting an important role for IL-17 in localizing and amplifying inflammation[72–74]. Furthermore, TNF-α and IL-6, which are both produced by Th17 cells, not only support Th17 cell development but also synergize with IL-17 to enhance the production of pro-inflammatory mediators[74]. Regulatory T cells CD4+CD25-Foxp3- could be a source of Th17 cells[75]. In human cells, IL-1, IL-6, and IL-23 promote human CD4+ to Th17 differentiation, but TGF-β1 is not needed like in mouse[76]. IL-17 as well as Th17 cells have both been found to be elevated in serum and intestinal tissue of IBD patients. IL-17 was not detected in inactive patients tissue as well as other colitis[77].
IL-13 and T cell response in UC
UC is characterized by a Th2 immune response in which IL-13, which is produced by specialized cells such as NK T-cells, was identified as an important effector cytokine[78]. In UC, IL-13 may impair epithelial barrier function by affecting epithelial apoptosis, tight junctions, and restitution velocity[79]. Both discoveries were made by determining the cytokine profile of LPMC isolated from tissue recovered from colonic resection from UC and CD patients. It was found that LPMC from UC patients secreted high amounts of Th2 cytokines IL-13 and IL-5[78,79]. This research group found that the IL-13 and IL-5 LPMC cells bear NK specific markers CD161 and recognize CD1d, indicating that they are NK T-cells[78]. These NK T-cells are considered “non-classical”. The NK T-cells isolated from UC patients exhibited cytotoxicity towards an epithelial cell line (HT-29)[78]. This cell population possibly could be the cells causing epithelial cell cytotoxicity in UC described in the 1980s[80]. IL-13 signalling through the IL-13α2 receptor (IL-13Rα) in general is involved in induction of TGF-β1 production and fibrosis[81]. The signalling through IL-13Rα was important in the fibrosis caused by TGF-β1 in an animal model[82]. However, the extent to which this leads to the ultimate cascade of inflammation in UC remains to be determined.
NOVEL CYTOKINES INVOLVED IN IBD
Other cytokines like IL-21 and IL-22, which have been implicated in the pathophysiology of inflammatory and autoimmune diseases such as asthma, arthritis and lupus, play also an important role in IBD. IL-21 is a T cell derived cytokine member of the common gamma-chain-dependent cytokine family, which in general acts on intestinal epithelium helping to maintain the ongoing Th1 inflammation by inducing the production of IFN-γ[83,84]. IL-21 also has been shown to enhance the expansion of NK cells[85]. IL-21 is expressed by immune T and B cells and non-immune cells like fibroblasts, where it activates the metalloproteinase 1 production, and signalling through its receptor IL-21R it activates STAT-3 in T cells[86]. IL-21, like IL-6 and IL-23 is also involved in Th17 cell differentiation[87] and it is over-expressed in both CD and UC, with higher levels being found in CD[88].
IL-22 was originally described as an IL-9-induced gene and was named as IL-10-related T cell-derived inducible factor (IL-TIF)[89]. This cytokine shows 22% amino acid identity with IL-10 and belongs to a family of cytokines with limited homology to IL-10. IL-22 binds at the cell surface to a receptor complex composed of two chains belonging to the class II cytokine receptor family (CRF2): IL-22R1 and IL-10R2[90,91]. In the intestinal cells, particularly innate immune cells, the binding of IL-22 to its respective R1 chain induces a conformational change that enables IL-10R2 to interact with the newly formed ligand-receptor complexes. This in turn, activates a signal transduction cascade that results in rapid activation of several transcription factors, including STAT1/3 proteins[91]. The principal sources of IL-22 are natural killer and activated T and B cells. Th17 has proven a very important role in this matter[92]. IL-22 has proinflammatory functions in IEC and is upregulated in CD both in tissue and in serum[93,94]. Surprisingly, in a murine model of UC, Sugimoto et al demonstrated a novel protective role for IL-22, in which IL-22 attenuates in the intestine inflammation by inducing mucin membrane bound production by goblet cells[93,94]. Another recent paper showed that IL23R genotypes affect IL-22 serum concentrations, linking for the first time genetic CD susceptibility to Th17 cell function[94].
POTENTIAL BIOLOGICAL THERAPIES DIRECTED TO CYTOKINES
Controlling the expression, production and activity of IL-23 as well as IL-17 is an approach that would allow the development of a novel treatment strategy with more anti-inflammatory efficacy and potentially with less suppressive effects on host defenses[95]. There are different biologic therapies directed to several cytokines tested in patients with IBD: fontolizumab (anti-interferon γ) is a humanized monoclonal antibody to interferon gamma. A small phase 2 study of fontolizumab at subcutaneous doses of 10 mg/kg in patients with moderate to severe CD demonstrated efficacy and safety[96]. A randomized clinical trial of 79 patients with CD receiving 1 mg or 3 mg of anti-IL-12 monoclonal antibody versus placebo demonstrated a response in 75% of CD patients compared with 25% in the placebo group[97]. Other antibodies have been generated against to T-cell subsets blockade including CD3+ cells (visilizumab) and CD25+ cells (daclizumab and basiliximab) for UC. Pilot studies have shown promising results in steroid-resistant UC patients[98]. IL-6 participates in a variety of critical functions, including T cell growth and differentiation, as well as B-cell proliferation. In a pilot study, where patients with active CD were treated with an antibody directed against the IL-6 receptor (Atlizumab), 80% responded at the full dose compared with 31% in the placebo group[99]. IL-11 is produced by cells of mesenchymal origin. A placebo controlled trial of subcutaneous IL-11 in patients with active CD did not demonstrate clear efficacy[100].
CONCLUSION
Cytokines are important in the pathogenesis of IBD and their manipulation has successfully reduced disease severity and maintained remission. Following the discovery of novel cytokines and the role they may play in gut mucosal immunity, as well as the emergence of new concepts and changing paradigms in IBD pathogenesis, the roles of several cytokines have been elucidated and tested in both preclinical animal models and clinical trials of patients with IBD. Complementary to this, proof of concept for new cytokine targets is rapidly developing, with the possibility of future cytokine-based therapies that may offer greater specificity and decreased toxicity for the treatment of IBD. In addition, further applications of cytokine-based therapies in human clinical trials and preclinical animal studies are ongoing.
Peer reviewers: Atsushi Mizoguchi, Assistant Professor, Department of Experimental Pathology, Massachusetts General Hospital, Simches 8234, 185 Cambridge Street, Boston MA 02114, United States; Emiko Mizoguchi, Professor, Department of Medicine, Gastrointestinal Unit, Massachusetts General Hospital, GRJ 702, 55 Fruit Street, Boston MA 02114, United States; Bret Lashner, PhD, Cleveland Clinic, 9500 Euclid Ave, Cleveland OH 44195, United States
S- Editor Li DL L- Editor Negro F E- Editor Ma WH
References
- 1.Jump RL, Levine AD. Mechanisms of natural tolerance in the intestine: implications for inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:462–478. doi: 10.1097/00054725-200407000-00023. [DOI] [PubMed] [Google Scholar]
- 2.Neuman MG. Immune dysfunction in inflammatory bowel disease. Transl Res. 2007;149:173–186. doi: 10.1016/j.trsl.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Leon F, Smythies LE, Smith PD, Kelsall BL. Involvement of dendritic cells in the pathogenesis of inflammatory bowel disease. Adv Exp Med Biol. 2006;579:117–132. doi: 10.1007/0-387-33778-4_8. [DOI] [PubMed] [Google Scholar]
- 4.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 5.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2000;51:289–298. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 6.Ince MN, Elliott DE. Immunologic and molecular mechanisms in inflammatory bowel disease. Surg Clin North Am. 2007;87:681–696. doi: 10.1016/j.suc.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Monteleone G, Fina D, Caruso R, Pallone F. New mediators of immunity and inflammation in inflammatory bowel disease. Curr Opin Gastroenterol. 2006;22:361–364. doi: 10.1097/01.mog.0000231808.10773.8e. [DOI] [PubMed] [Google Scholar]
- 8.Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 9.Begue B, Wajant H, Bambou JC, Dubuquoy L, Siegmund D, Beaulieu JF, Canioni D, Berrebi D, Brousse N, Desreumaux P, et al. Implication of TNF-related apoptosis-inducing ligand in inflammatory intestinal epithelial lesions. Gastroenterology. 2006;130:1962–1974. doi: 10.1053/j.gastro.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 10.Stucchi A, Reed K, O'Brien M, Cerda S, Andrews C, Gower A, Bushell K, Amar S, Leeman S, Becker J. A new transcription factor that regulates TNF-alpha gene expression, LITAF, is increased in intestinal tissues from patients with CD and UC. Inflamm Bowel Dis. 2006;12:581–587. doi: 10.1097/01.MIB.0000225338.14356.d5. [DOI] [PubMed] [Google Scholar]
- 11.Reimund JM, Wittersheim C, Dumont S, Muller CD, Baumann R, Poindron P, Duclos B. Mucosal inflammatory cytokine production by intestinal biopsies in patients with ulcerative colitis and Crohn’s disease. J Clin Immunol. 1996;16:144–150. doi: 10.1007/BF01540912. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto-Furusho JK. Innovative therapeutics for inflammatory bowel disease. World J Gastroenterol. 2007;13:1893–1896. doi: 10.3748/wjg.v13.i13.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baert FJ, Rutgeerts PR. Anti-TNF strategies in Crohn’s disease: mechanisms, clinical effects, indications. Int J Colorectal Dis. 1999;14:47–51. doi: 10.1007/s003840050182. [DOI] [PubMed] [Google Scholar]
- 14.Spoettl T, Hausmann M, Klebl F, Dirmeier A, Klump B, Hoffmann J, Herfarth H, Timmer A, Rogler G. Serum soluble TNF receptor I and II levels correlate with disease activity in IBD patients. Inflamm Bowel Dis. 2007;13:727–732. doi: 10.1002/ibd.20107. [DOI] [PubMed] [Google Scholar]
- 15.Mizoguchi E, Hachiya Y, Kawada M, Nagatani K, Ogawa A, Sugimoto K, Mizoguchi A, Podolsky DK. TNF receptor type I-dependent activation of innate responses to reduce intestinal damage-associated mortality. Gastroenterology. 2008;134:470–480. doi: 10.1053/j.gastro.2007.11.055. [DOI] [PubMed] [Google Scholar]
- 16.Bamias G, Martin C 3rd, Marini M, Hoang S, Mishina M, Ross WG, Sachedina MA, Friel CM, Mize J, Bickston SJ, et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J Immunol. 2003;171:4868–4874. doi: 10.4049/jimmunol.171.9.4868. [DOI] [PubMed] [Google Scholar]
- 17.Dinarello CA. The IL-1 family and inflammatory diseases. Clin Exp Rheumatol. 2002;20:S1–S13. [PubMed] [Google Scholar]
- 18.Ashwood P, Harvey R, Verjee T, Wolstencroft R, Thompson RP, Powell JJ. Functional interactions between mucosal IL-1, IL-ra and TGF-beta 1 in ulcerative colitis. Inflamm Res. 2004;53:53–59. doi: 10.1007/s00011-003-1219-z. [DOI] [PubMed] [Google Scholar]
- 19.Dionne S, D’Agata ID, Hiscott J, Vanounou T, Seidman EG. Colonic explant production of IL-1and its receptor antagonist is imbalanced in inflammatory bowel disease (IBD) Clin Exp Immunol. 1998;112:435–442. doi: 10.1046/j.1365-2249.1998.00595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAlindon ME, Hawkey CJ, Mahida YR. Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut. 1998;42:214–219. doi: 10.1136/gut.42.2.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K, et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med. 2001;193:471–481. doi: 10.1084/jem.193.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitsuyama K, Toyonaga A, Sasaki E, Ishida O, Ikeda H, Tsuruta O, Harada K, Tateishi H, Nishiyama T, Tanikawa K. Soluble interleukin-6 receptors in inflammatory bowel disease: relation to circulating interleukin-6. Gut. 1995;36:45–49. doi: 10.1136/gut.36.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinisch W, Gasche C, Tillinger W, Wyatt J, Lichtenberger C, Willheim M, Dejaco C, Waldhor T, Bakos S, Vogelsang H, et al. Clinical relevance of serum interleukin-6 in Crohn's disease: single point measurements, therapy monitoring, and prediction of clinical relapse. Am J Gastroenterol. 1999;94:2156–2164. doi: 10.1111/j.1572-0241.1999.01288.x. [DOI] [PubMed] [Google Scholar]
- 24.Van Kemseke C, Belaiche J, Louis E. Frequently relapsing Crohn’s disease is characterized by persistent elevation in interleukin-6 and soluble interleukin-2 receptor serum levels during remission. Int J Colorectal Dis. 2000;15:206–210. doi: 10.1007/s003840000226. [DOI] [PubMed] [Google Scholar]
- 25.Kallen KJ. The role of transsignalling via the agonistic soluble IL-6 receptor in human diseases. Biochim Biophys Acta. 2002;1592:323–343. doi: 10.1016/s0167-4889(02)00325-7. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto M, Yoshizaki K, Kishimoto T, Ito H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol. 2000;164:4878–4882. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- 27.Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 28.Carey R, Jurickova I, Ballard E, Bonkowski E, Han X, Xu H, Denson LA. Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:446–457. doi: 10.1002/ibd.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mudter J, Neurath MF. Apoptosis of T cells and the control of inflammatory bowel disease: therapeutic implications. Gut. 2007;56:293–303. doi: 10.1136/gut.2005.090464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lebel-Binay S, Berger A, Zinzindohoue F, Cugnenc P, Thiounn N, Fridman WH, Pages F. Interleukin-18: biological properties and clinical implications. Eur Cytokine Netw. 2000;11:15–26. [PubMed] [Google Scholar]
- 31.Leach ST, Messina I, Lemberg DA, Novick D, Rubenstein M, Day AS. Local and systemic interleukin-18 and interleukin-18-binding protein in children with inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:68–74. doi: 10.1002/ibd.20272. [DOI] [PubMed] [Google Scholar]
- 32.Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF Jr, Foley E, Moskaluk CA, Bickston SJ, Cominelli F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol. 1999;162:6829–6835. [PubMed] [Google Scholar]
- 33.Pages F, Lazar V, Berger A, Danel C, Lebel-Binay S, Zinzindohoue F, Desreumaux P, Cellier C, Thiounn N, Bellet D, et al. Analysis of interleukin-18, interleukin-1 converting enzyme (ICE) and interleukin-18-related cytokines in Crohn's disease lesions. Eur Cytokine Netw. 2001;12:97–104. [PubMed] [Google Scholar]
- 34.Schmidt C, Giese T, Goebel R, Schilling M, Marth T, Ruether A, Schreiber S, Zeuzem S, Meuer SC, Stallmach A. Interleukin-18 is increased only in a minority of patients with active Crohn’s disease. Int J Colorectal Dis. 2007;22:1013–1020. doi: 10.1007/s00384-007-0282-2. [DOI] [PubMed] [Google Scholar]
- 35.Maerten P, Shen C, Colpaert S, Liu Z, Bullens DA, van Assche G, Penninckx F, Geboes K, Vanham G, Rutgeerts P, et al. Involvement of interleukin 18 in Crohn‘s disease: evidence from in vitro analysis of human gut inflammatory cells and from experimental colitis models. Clin Exp Immunol. 2004;135:310–317. doi: 10.1111/j.1365-2249.2004.02362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corbaz A, ten Hove T, Herren S, Graber P, Schwartsburd B, Belzer I, Harrison J, Plitz T, Kosco-Vilbois MH, Kim SH, et al. IL-18-binding protein expression by endothelial cells and macrophages is up-regulated during active Crohn’s disease. J Immunol. 2002;168:3608–3616. doi: 10.4049/jimmunol.168.7.3608. [DOI] [PubMed] [Google Scholar]
- 37.Kanai T, Watanabe M, Okazawa A, Sato T, Yamazaki M, Okamoto S, Ishii H, Totsuka T, Iiyama R, Okamoto R, et al. Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn’s disease. Gastroenterology. 2001;121:875–888. doi: 10.1053/gast.2001.28021. [DOI] [PubMed] [Google Scholar]
- 38.Okazawa A, Kanai T, Nakamaru K, Sato T, Inoue N, Ogata H, Iwao Y, Ikeda M, Kawamura T, Makita S, et al. Human intestinal epithelial cell-derived interleukin (IL)-18, along with IL-2, IL-7 and IL-15, is a potent synergistic factor for the proliferation of intraepithelial lymphocytes. Clin Exp Immunol. 2004;136:269–276. doi: 10.1111/j.1365-2249.2004.02431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K. Regulation of interferon-gamma production by IL-12 and IL-18. Curr Opin Immunol. 1998;10:259–264. doi: 10.1016/s0952-7915(98)80163-5. [DOI] [PubMed] [Google Scholar]
- 40.Micallef MJ, Tanimoto T, Kohno K, Ikegami H, Kurimoto M. Interleukin 18 induces a synergistic enhancement of interferon gamma production in mixed murine spleen cell-tumor cell cultures: role of endogenous interleukin 12. Cancer Detect Prev. 2000;24:234–243. [PubMed] [Google Scholar]
- 41.Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev. 2007;59:1073–1083. doi: 10.1016/j.addr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 43.Rennick DM, Fort MM. Lessons from genetically engineered animal models. XII. IL-10-deficient (IL-10(-/-) mice and intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2000;278:G829–G833. doi: 10.1152/ajpgi.2000.278.6.G829. [DOI] [PubMed] [Google Scholar]
- 44.Schreiber S, Heinig T, Thiele HG, Raedler A. Immu-noregulatory role of interleukin 10 in patients with inflammatory bowel disease. Gastroenterology. 1995;108:1434–1444. doi: 10.1016/0016-5085(95)90692-4. [DOI] [PubMed] [Google Scholar]
- 45.Melgar S, Yeung MM, Bas A, Forsberg G, Suhr O, Oberg A, Hammarstrom S, Danielsson A, Hammarstrom ML. Over-expression of interleukin 10 in mucosal T cells of patients with active ulcerative colitis. Clin Exp Immunol. 2003;134:127–137. doi: 10.1046/j.1365-2249.2003.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Latinne D, Fiasse R. New insights into the cellular immunology of the intestine in relation to the pathophy-siology of inflammatory bowel diseases. Acta Gastroenterol Belg. 2006;69:393–405. [PubMed] [Google Scholar]
- 47.Mizoguchi A, Bhan AK. A case for regulatory B cells. J Immunol. 2006;176:705–710. doi: 10.4049/jimmunol.176.2.705. [DOI] [PubMed] [Google Scholar]
- 48.Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16:219–230. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- 49.Goetz M, Atreya R, Ghalibafian M, Galle PR, Neurath MF. Exacerbation of ulcerative colitis after rituximab salvage therapy. Inflamm Bowel Dis. 2007;13:1365–1368. doi: 10.1002/ibd.20215. [DOI] [PubMed] [Google Scholar]
- 50.Rogler G, Andus T. Cytokines in inflammatory bowel disease. World J Surg. 1998;22:382–389. doi: 10.1007/s002689900401. [DOI] [PubMed] [Google Scholar]
- 51.Iijima H, Takahashi I, Kishi D, Kim JK, Kawano S, Hori M, Kiyono H. Alteration of interleukin 4 production results in the inhibition of T helper type 2 cell-dominated inflammatory bowel disease in T cell receptor alpha chain-deficient mice. J Exp Med. 1999;190:607–615. doi: 10.1084/jem.190.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mizoguchi A, Mizoguchi E, Bhan AK. The critical role of interleukin 4 but not interferon gamma in the pathogenesis of colitis in T-cell receptor alpha mutant mice. Gastroenterology. 1999;116:320–326. doi: 10.1016/s0016-5085(99)70128-9. [DOI] [PubMed] [Google Scholar]
- 53.Griga T, Hebler U, Voigt E, Tromm A, May B. Interleukin-4 inhibits the increased production of vascular endothelial growth factor by peripheral blood mononuclear cells in patients with inflammatory bowel disease. Hepatogastroenterology. 2000;47:1604–1607. [PubMed] [Google Scholar]
- 54.Marek A, Brodzicki J, Liberek A, Korzon M. TGF-beta (transforming growth factor-beta) in chronic inflammatory conditions - a new diagnostic and prognostic marker? Med Sci Monit. 2002;8:RA145–RA151. [PubMed] [Google Scholar]
- 55.Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest. 2001;108:601–609. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Del Zotto B, Mumolo G, Pronio AM, Montesani C, Tersigni R, Boirivant M. TGF-beta1 production in inflammatory bowel disease: differing production patterns in Crohn’s disease and ulcerative colitis. Clin Exp Immunol. 2003;134:120–126. doi: 10.1046/j.1365-2249.2003.02250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanazawa S, Tsunoda T, Onuma E, Majima T, Kagiyama M, Kikuchi K. VEGF, basic-FGF, and TGF-beta in Crohn’s disease and ulcerative colitis: a novel mechanism of chronic intestinal inflammation. Am J Gastroenterol. 2001;96:822–828. doi: 10.1111/j.1572-0241.2001.03527.x. [DOI] [PubMed] [Google Scholar]
- 58.Lawrance IC, Maxwell L, Doe W. Inflammation location, but not type, determines the increase in TGF-beta1 and IGF-1 expression and collagen deposition in IBD intestine. Inflamm Bowel Dis. 2001;7:16–26. doi: 10.1097/00054725-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 59.Boirivant M, Pallone F, Di Giacinto C, Fina D, Monteleone I, Marinaro M, Caruso R, Colantoni A, Palmieri G, Sanchez M, et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology. 2006;131:1786–1798. doi: 10.1053/j.gastro.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 60.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O’Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202:139–156. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- 61.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 62.Nielsen OH, Kirman I, Rudiger N, Hendel J, Vainer B. Upregulation of interleukin-12 and -17 in active inflammatory bowel disease. Scand J Gastroenterol. 2003;38:180–185. doi: 10.1080/00365520310000672. [DOI] [PubMed] [Google Scholar]
- 63.Kugathasan S, Saubermann LJ, Smith L, Kou D, Itoh J, Binion DG, Levine AD, Blumberg RS, Fiocchi C. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut. 2007;56:1696–1705. doi: 10.1136/gut.2006.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Latinne D, Fiasse R. New insights into the cellular immunology of the intestine in relation to the pathophy-siology of inflammatory bowel diseases. Acta Gastroenterol Belg. 2006;69:393–405. [PubMed] [Google Scholar]
- 65.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmidt C, Giese T, Ludwig B, Mueller-Molaian I, Marth T, Zeuzem S, Meuer SC, Stallmach A. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn’s disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 68.Paradowska A, Masliniski W, Grzybowska-Kowalczyk A, Lacki J. The function of interleukin 17 in the pathogenesis of rheumatoid arthritis. Arch Immunol Ther Exp (Warsz) 2007;55:329–334. doi: 10.1007/s00005-007-0032-8. [DOI] [PubMed] [Google Scholar]
- 69.Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, Jin HT, Min SY, Ju JH, Park KS, et al. STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J Immunol. 2006;176:5652–5661. doi: 10.4049/jimmunol.176.9.5652. [DOI] [PubMed] [Google Scholar]
- 70.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 71.Witowski J, Ksiazek K, Jorres A. Interleukin-17: a mediator of inflammatory responses. Cell Mol Life Sci. 2004;61:567–579. doi: 10.1007/s00018-003-3228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, Gaffen SL. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- 75.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 76.Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res. 2008;41:87–102. doi: 10.1007/s12026-007-8014-9. [DOI] [PubMed] [Google Scholar]
- 77.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 80.Strober W, James SP. The immunologic basis of inflammatory bowel disease. J Clin Immunol. 1986;6:415–432. doi: 10.1007/BF00915248. [DOI] [PubMed] [Google Scholar]
- 81.Fichtner-Feigl S, Fuss IJ, Young CA, Watanabe T, Geissler EK, Schlitt HJ, Kitani A, Strober W. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol. 2007;178:5859–5870. doi: 10.4049/jimmunol.178.9.5859. [DOI] [PubMed] [Google Scholar]
- 82.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 83.Fantini MC, Monteleone G, Macdonald TT. New players in the cytokine orchestra of inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1419–1423. doi: 10.1002/ibd.20212. [DOI] [PubMed] [Google Scholar]
- 84.Fina D, Caruso R, Pallone F, Monteleone G. Interleukin-21 (IL-21) controls inflammatory pathways in the gut. Endocr Metab Immune Disord Drug Targets. 2007;7:288–291. doi: 10.2174/187153007782794308. [DOI] [PubMed] [Google Scholar]
- 85.de Rham C, Ferrari-Lacraz S, Jendly S, Schneiter G, Dayer JM, Villard J. The proinflammatory cytokines IL-2, IL-15 and IL-21 modulate the repertoire of mature human natural killer cell receptors. Arthritis Res Ther. 2007;9:R125. doi: 10.1186/ar2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Caruso R, Fina D, Peluso I, Stolfi C, Fantini MC, Gioia V, Caprioli F, Del Vecchio Blanco G, Paoluzi OA, Macdonald TT, et al. A functional role for interleukin-21 in promoting the synthesis of the T-cell chemoattractant, MIP-3alpha, by gut epithelial cells. Gastroenterology. 2007;132:166–175. doi: 10.1053/j.gastro.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 87.Chen Z, Laurence A, O’Shea JJ. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin Immunol. 2007;19:400–408. doi: 10.1016/j.smim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Monteleone G, Monteleone I, Fina D, Vavassori P, Del Vecchio Blanco G, Caruso R, Tersigni R, Alessandroni L, Biancone L, Naccari GC, et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterology. 2005;128:687–694. doi: 10.1053/j.gastro.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 89.Dumoutier L, Louahed J, Renauld JC. Cloning and characterization of IL-10-related T cell-derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. J Immunol. 2000;164:1814–1819. doi: 10.4049/jimmunol.164.4.1814. [DOI] [PubMed] [Google Scholar]
- 90.Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, Wood WI, Goddard AD, Gurney AL. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J Biol Chem. 2000;275:31335–31339. doi: 10.1074/jbc.M005304200. [DOI] [PubMed] [Google Scholar]
- 91.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, Tsujikawa T, Kitoh K, Kim-Mitsuyama S, Takayanagi A, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology. 2005;129:969–984. doi: 10.1053/j.gastro.2005.06.071. [DOI] [PubMed] [Google Scholar]
- 93.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schmechel S, Konrad A, Diegelmann J, Glas J, Wetzke M, Paschos E, Lohse P, Goke B, Brand S. Linking genetic susceptibility to Crohn's disease with Th17 cell function: IL-22 serum levels are increased in Crohn's disease and correlate with disease activity and IL23R genotype status. Inflamm Bowel Dis. 2008;14:204–212. doi: 10.1002/ibd.20315. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Z, Hinrichs DJ, Lu H, Chen H, Zhong W, Kolls JK. After interleukin-12p40, are interleukin-23 and interleukin-17 the next therapeutic targets for inflammatory bowel disease? Int Immunopharmacol. 2007;7:409–416. doi: 10.1016/j.intimp.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 96.Hommes DW, Mikhajlova TL, Stoinov S, Stimac D, Vucelic B, Lonovics J, Zakuciova M, D'Haens G, Van Assche G, Ba S, et al. Fontolizumab, a humanised anti-interferon gamma antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn's disease. Gut. 2006;55:1131–1137. doi: 10.1136/gut.2005.079392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, et al. Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 98.Creed TJ, Probert CS, Norman MN, Moorghen M, Shepherd NA, Hearing SD, Dayan CM. Basiliximab for the treatment of steroid-resistant ulcerative colitis: further experience in moderate and severe disease. Aliment Pharmacol Ther. 2006;23:1435–1442. doi: 10.1111/j.1365-2036.2006.02904.x. [DOI] [PubMed] [Google Scholar]
- 99.Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, Matsumoto T, Yamamura T, Azuma J, Nishimoto N, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology. 2004;126:989–996; discussion 947. doi: 10.1053/j.gastro.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 100.Herrlinger KR, Witthoeft T, Raedler A, Bokemeyer B, Krummenerl T, Schulzke JD, Boerner N, Kueppers B, Emmrich J, Mescheder A, et al. Randomized, double blind controlled trial of subcutaneous recombinant human interleukin-11 versus prednisolone in active Crohn’s disease. Am J Gastroenterol. 2006;101:793–797. doi: 10.1111/j.1572-0241.2005.00356.x. [DOI] [PubMed] [Google Scholar]