

Abstract
l-colitose and d-perosamine are unusual sugars found in the O-antigens of some Gram-negative bacteria such as Escherichia coli, Vibrio cholerae, and Salmonella enterica, among others. The biosynthetic pathways for these two sugars begin with the formation of GDP-mannose from d-mannose-1-phosphate and GTP followed by the subsequent dehydration and oxidation of GDP-mannose to yield GDP-4-keto-6-deoxymannose. Following the production of GDP-4-keto-6-deoxymannose, the two pathways diverge. In the case of GDP-perosamine biosynthesis, the next step involves an amination reaction at the C-4′ position of the sugar, whereas in GDP-colitose production, the 3′-hydroxyl group is removed. The enzymes catalyzing these reactions are GDP-perosamine synthase and GDP-4-keto-6-deoxymannose-3-dehydratase (ColD), respectively. Both of these enzymes are pyridoxal-5′-phosphate (PLP)-dependent and their three-dimensional structures place them into the well-characterized aspartate aminotransferase superfamily. A comparison of the active site architecture of ColD from Escherichia coli (Strain 5a, type O55:H7) to that of GDP-perosamine synthase from Caulobacter crescentus CB15, suggested that only two mutations would be required to convert ColD into an aminotransferase. Here we present a combined structural and functional analysis of the ColD S187N/H188K mutant protein that, indeed, has been converted from a dehydratase into an aminotransferase.
Most Gram-negative bacteria contain, in addition to the rigid peptidoglycan cell wall, an outer layer referred to as the lipopolysaccharide. It is a complex entity composed of lipid A, a core polysaccharide, and an O-antigen that extends away from the bacterial cell wall. The O-antigens differ among bacteria with respect to sugar content and linkages, are highly immunogenic, and serve as important virulence factors (1). O-antigen differences have been implicated in the degree of pathogenicity, for example as in the Escherichia coli strains O1, O7, and O18 (2, 3). Indeed, serotyping schemes for many Gram-negative bacteria are often based on the variability of the O-antigen. Besides galactose, glucose, rhamnose, and mannose, the O-antigens sometimes contain unusual dideoxy sugars such as l-colitose, d-perosamine, and N-acetyl-d-perosamine. These sugars have been identified in the O-antigens of E. coli (4, 5), Vibrio cholerae (6, 7), and Salmonella enterica (8).
Strikingly, no single organism has been isolated thus far that produces both l-colitose and d-perosamine, but the biosynthetic pathways for these two sugars have much in common. Both compounds are synthesized as GDP-linked sugars from GDP-mannose. As indicated in Scheme 1, both GDP-4-keto-6-deoxymannose-3-dehydratase and GDP-4-keto-6-deoxymannose-4-aminotransferase, hereafter referred to as ColD and GDP-perosamine synthase, respectively, catalyze the third step in either GDP-colitose or GDP-perosamine production. They are pyridoxal-5′-phosphate (PLP)-dependent enzymes that act upon the same substrate, namely GDP-4-keto-6-deoxymannose. ColD is a dehydratase, however, that removes the hexose 3′-hydroxyl group, whereas GDP-perosamine synthase is an aminotransferase that converts the ketone functionality at the hexose C-4′ to an amino group. The enzymes are strikingly similar in structure (9, 10), and thus the features of the active sites that select for one reaction type over another are quite subtle. A ribbon representation of ColD is presented in Figure 1. Both ColD and GDP-perosamine synthase function as dimers, and they belong to the well-characterized aspartate aminotransferase superfamily. In both enzymes, a loop from one subunit folds into the active site of the other subunit to complete the architecture required for cofactor and substrate binding.
Scheme 1.
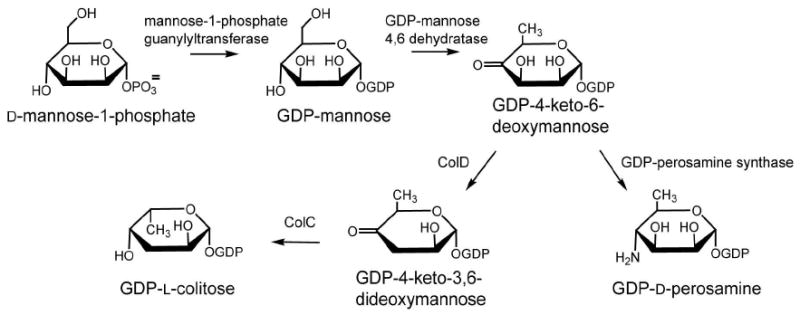
Figure 1.
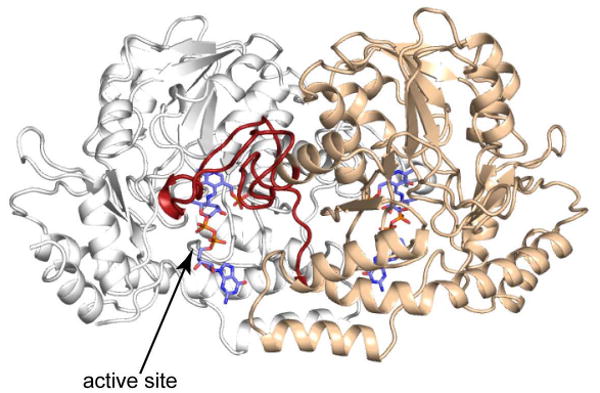
Ribbon representation of ColD. The two subunits of the dimer are color-coded in white and wheat, respectively. The stick models for the sugar analogs trapped as external aldimines indicate the positions of the active sites. A loop from one subunit, indicated in red, folds onto the second subunit of the dimer to form part of the active site.
ColD and GDP-perosamine synthase function via ping-pong reaction mechanisms in which glutamate is first utilized in the conversion of the PLP cofactor to its amino derivative, pyridoxamine-5′-phosphate (PMP), releasing α-ketoglutarate. In the second part of the reaction, PMP acts as a nucleophile, reacting with the ketone functionality of GDP-4-keto-6-deoxymannose and forming a ketimine intermediate (Scheme 2). The ketimine is then deprotonated at the cofactor C-4′ position, yielding a resonance-stabilized carbanion. From this point on, however, the mechanisms of these two enzymes diverge. For GDP-perosamine synthesis, the carbanionic intermediate is protonated at the hexose C-4′ position, producing an external aldimine. In contrast, for the dehydratase reaction catalyzed by ColD, the carbanionic intermediate is protonated at the hexose 3′-hydroxyl group, which subsequently departs as water. The dehydration yields a Δ3,4-aminomannoseen intermediate, which is also an external aldimine.
Scheme 2.
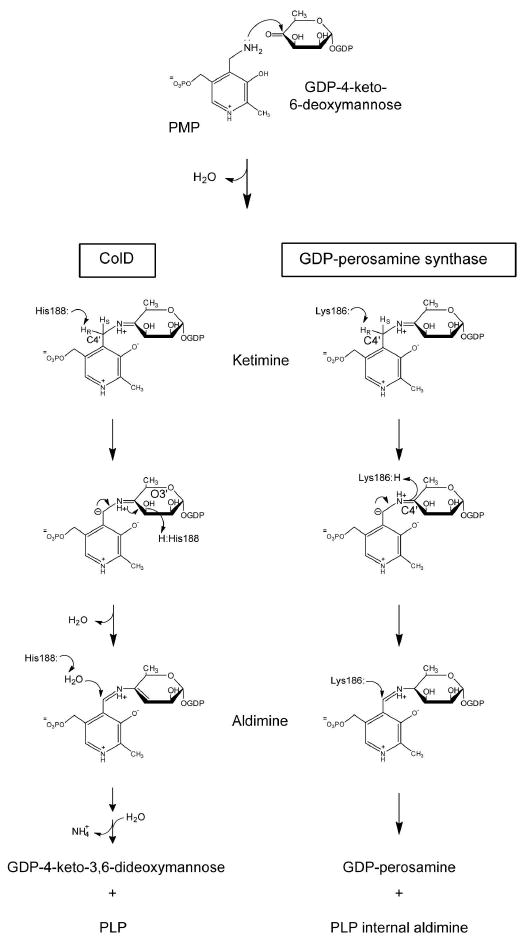
Another difference between the catalytic mechanisms of these two enzymes is the means by which the sugar substrates are cleaved from the cofactor. GDP-perosamine is released via a transimination reaction in which the active site lysine residue displaces the external aldimine, thereby forming the internal aldimine. Contrastingly, ColD activates a water molecule, which conducts a nucleophilic attack at the cofactor C-4′ (Scheme 2). The sugar is then released from the cofactor as an aminomannoseen and the PLP cofactor is regenerated. The aminomannoseen is subsequently protonated and hydrolyzed to yield the final product, with the nitrogen being released as ammonia (11).
ColD is an unusual PLP-dependent enzyme in that it does not utilize an active site lysine residue to covalently attach the PLP cofactor, and thus it cannot form an internal aldimine (11). Instead, ColD has a histidine residue (His 188), which from our previous studies has been shown to be the active site general acid/base responsible for proton transfers throughout the course of the reaction (9, 12, 13). On the basis of these initial studies, we hypothesized that the replacement of an active site lysine with a histidine in ColD was the sole reason for its functioning as a dehydratase rather than an aminotransferase. We reasoned that the difference in side chain length and geometry between histidine and lysine might cause selective protonation of one group of the PLP-sugar intermediate over another group, thereby resulting in one reaction type over the other. Initial site-directed mutagenesis experiments were undertaken in which the active site histidine of ColD was mutated to a lysine (12) and conversely, the active site lysine of GDP-perosamine synthase was changed to a histidine (14). These mutant enzymes were incapable of performing any detectable catalytic reaction on the sugar substrates, clearly demonstrating that the reaction specificity involved more than the simple replacement of the active site general acid/base.
What then are the factors that determine whether these enzymes function as dehydratases or aminotransferases? A superposition of the structures of ColD (from E. coli O55) (13) and GDP-perosamine synthase (from Caulobacter crescentus) (14), both solved in the presence of their sugar substrates, gives some clues (Figure 2). Overall, the active sites are structurally well conserved. However, several important variations are present. Among these is the replacement of Asn 185 in GDP-perosamine synthase with Ser 187 in ColD. This replacement is particularly interesting because Asn 185 in GDP-perosamine synthase is involved in a hydrogen bonding interaction with the hexose ring oxygen of the substrate. In ColD, the serine hydroxyl in this position is too far away to form such an interaction.
Figure 2.
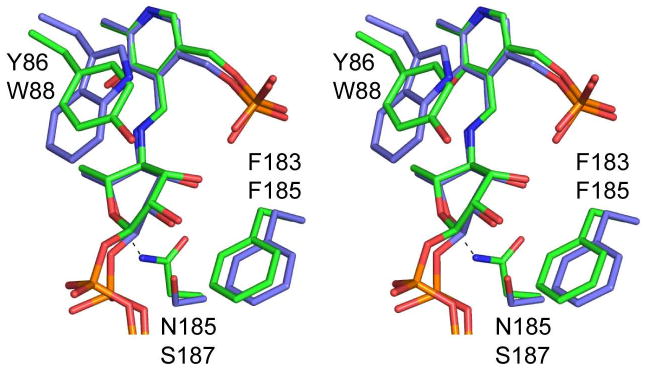
A close-up stereo view of the GDP-perosamine synthase and ColD active sites in the external aldimine forms with their respective sugar products. GDP-perosamine and ColD are highlighted in green and slate filled bonds, respectively. The top and bottom labels correspond to those residues found in GDP-perosamine and ColD. The dashed line indicates the hydrogen bond between Asn 185 in GDP-perosamine synthase and the ring oxygen of the sugar.
Here we describe a combined functional and structural investigation of a double site-directed mutant protein of ColD whereby His 188 was changed to a lysine residue and Ser 187 was changed to an asparagine. Unlike the simple replacement of His 188 for a lysine, we demonstrate that by changing both residues we were able to confer sugar aminotransferase activity onto the ColD scaffold. Details concerning the structure of the double mutant protein and its kinetic parameters are presented.
Materials and Methods
Site-Directed Mutagenesis, Protein Expression, and Purification of ColD
The S187N mutation was introduced using methods identical or similar to those described within the QuikChange site-directed mutagenesis kit (Stratagene). The ColD gene used for this study had already been mutagenized to H188K and inserted into the pET28t plasmid (12, 14). The S187N/H188K mutant protein was expressed and purified in a manner identical to that previously described for the wild-type enzyme (9). The enzyme was dialyzed against 25 mM Tris-HCl (pH 8.0) and 100 mM NaCl. The protein concentration was determined by measuring the absorbance at 280 nm, with an extinction coefficient of 0.96 (mg/mL)-1 cm-1 as calculated with the Expasy Proteomics Server (http://www.expasy.ch/).
Functional Assays
Typical reaction mixtures contained 50 mM HEPES (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 2 mM GDP-mannose, 10 mM glutamate, 50 μM NADP, 50 μM PLP, 5 μM GDP-mannose-4, 6-dehydratase, and 3 μM ColD. The reactions were incubated for 2-4 hours at 25-37°C and were subsequently filtered through an Amicon membrane to remove the enzymes. The reaction mixture was then purified with an ÄKTA Purifier HPLC system (G.E. Healthcare) equipped with a 1 mL Resource Q anion exchange column. The samples were loaded onto the column in water and eluted with a linear gradient to 50% 1M NH4HCO3 (pH 8.5). Sample peaks with retention times similar to those of authentic samples of GDP-perosamine or the ColD product, GDP-4-keto-3,6-dideoxymannose, were collected and analyzed by ESI mass spectrometry.
Steady State Kinetic Experiments
Steady state kinetic parameters for the ColD S187N/H188K mutant enzyme were determined via a discontinuous HPLC-based assay as previously described (14). The concentration of glutamate ranged from 0.05 mM to 5.0 mM and the concentration of GDP-4-keto-6-deoxymannose ranged from 0.01 mM to 0.10 mM. Substrate inhibition was observed with GDP-4-keto-6-deoxymannose concentrations over 0.15 mM, so those concentrations were not surveyed. The data were fitted to the following equation: v0 = (VmaxAB)/(KaB + KbA + AB).
Crystallization of the ColD S187N/H188K Enzyme
Conditions for the crystallization of the ColD S187N/H188K mutant enzyme were surveyed by the hanging drop method of vapor diffusion utilizing a screen developed in the laboratory that tests for various pH values, precipitants, and additives. Crystals suitable for an x-ray diffraction analysis were grown via batch methods by mixing 20 μL of a protein solution with 20 μL of the precipitant solution. The concentration of the protein solution was typically 15 mg mL-1, and it contained 2 mM PLP and 2 mM α-ketoglutarate. Precipitant solutions consisted of 20-25% poly(ethylene glycol) 3400, 100 mM MES (pH 6.0), and 200 mM MgCl2. Crystals grew to maximum dimensions of 0.8 × 0.8 × 0.8 mm. These crystals belonged to the P1 space group with unit cell dimensions of a = 69.7 Å, b = 114.7 Å, c = 114.6 Å, α = 80.0°, β = 76.2° and γ = 76.3°, and they contained four dimers in the asymmetric unit. Each subunit contains 388 amino acid residues.
Structural Analysis of the ColD S187N/H188K Mutant Enzyme
A single crystal of the double mutant protein was taken directly from the mother liquor and mounted in a quartz capillary tube. X-ray data were collected at room temperature using a Bruker AXS Platinum 135 CCD detector controlled with the Proteum software suite (Bruker AXS, Inc.). The x-ray source was CuKα radiation from a Rigaku RU200 x-ray generator operated at 50 kV and 90 mA. The data were processed with SAINT (Bruker AXS, Inc.) and internally scaled with SADABS (Bruker AXS, Inc). Relevant x-ray data collection statistics are presented in Table 1.
Table 1. X-ray Data Collection and Least-Squares Refinement Statistics.
| Space Group | P1 |
|---|---|
| Unit Cell Dimensions | a = 69.7 Å, b = 114.7 Å, c = 114.6 Å, α = 80.0°, β = 76.2°, γ = 76.3°. |
| Resolution Limits (Å) | 50.0 − 2.2 (2.3 − 2.2) b |
| Number of Independent Reflections | 152774 (15786) |
| Completeness (%) | 90.8 (75.2) |
| Redundancy | 2.96 (1.33) |
| Avg I/Avg σ(I) | 7.8 (2.0) |
| Rsym (%)a | 11.1 (30.5) |
| cR-factor (overall) %/no. of reflections | 18.0/152769 |
| R-factor (working) %/no. of reflections | 17.7/137469 |
| R-factor (free) %/no. of reflections | 27.2/15300 |
| No. Protein Atomsd | 24782 |
| No. Heteroatomse | 423 |
| Average B values | |
| Protein Atoms (Å2) | 43.3 |
| Ligands (Å2) | 55.5 |
| Solvent (Å2) | 34.4 |
| Weighted root-mean-square deviations from ideality | |
| Bond Lengths (Å) | 0.012 |
| Bond Angles (deg) | 1.8 |
| Trigonal Planes (Å) | 0.009 |
| General Planes (Å) | 0.016 |
| Torsional Angles (deg)f | 20.0 |
| Ramachandran Statistics Core, Allowed, Generously Allowed, Disallowed (%) | 84.8, 13.8, 0.6, 0.8 |
Rsym = (• | I - Ī | / • I) × 100.
Statistics for the highest resolution bin from 2.3 - 2.2Å.
R-factor = (• | FO - FC | / • | FO |) × 100 where FO is the observed structure-factor amplitude and Fc is the calculated structure-factor amplitude.
The average B-factor for the protein atoms includes the PLP cofactors.
Heteroatoms include 6 α-ketoglutarate molecules and 363 water molecules.
The torsional angles were not restrained during the refinement.
The structure was solved by molecular replacement using the program Phaser (15, 16), with the ColD H188K monomer serving as a search model (PDB accession number 2R0T) (12). Cofactors, substrates, solvents, and residues at the sites of the mutations were omitted from the search model. Eight monomers, corresponding to four dimers, were placed in the asymmetric unit, yielding an initial R-factor of 29%. Manual model adjustments and solvent placements were performed with Coot (17), and least-squares refinement of the model was carried out with the software package TNT (18). The final model had an R-factor of 18.0% for all measured x-ray data to 2.2 Å. Relevant refinement and Ramachandran statistics are presented in Table 1. In the original wild-type ColD structure, Met 193, Val 334, and Asn 354 adopted φ,ψ angles that were outside of the allowed regions of the Ramachandran plot, but the electron densities for these residues were unambiguous (9). The dihedral angles for these three residues were located near the “nucleophile elbow” region of the Ramachandran plot with φ = ∼60° and ψ = ∼-100°. Likewise, the double mutant protein also adopts these φ,ψ angles, which results in 0.8% of the residues lying within the theoretically disallowed region of the Ramachandran plot.
Results and Discussion
The activity of the ColD S187N/H188K mutant enzyme was assessed via an HPLC-based assay in which reaction mixtures were separated and components with retention times similar to authentic samples of GDP-4-keto-3,6-dideoxymannose or GDP-perosamine were analyzed by ESI mass spectrometry. The presence of GDP-perosamine (587 amu) or GDP-4-keto-3,6-dideoxymannose (570 amu) in a given sample was used as confirmation of GDP-perosamine synthase or ColD activity, respectively.
As noted in the introduction, our previous studies showed that the ColD H188K single mutant enzyme lacked any detectable catalytic activity with its sugar substrate. Our attention then turned to the residue immediately N-terminal to His 188. In ColD, this residue is Ser 187, whereas in GDP-perosamine synthase, it is Asn 185 (Figure 2). Ser 187 in ColD does not interact directly with the sugar substrate, whereas Asn 185 in GDP-perosamine synthase participates in a hydrogen bond with the ring oxygen of the hexose moiety. We reasoned that this hydrogen bond plays a critical role in positioning the hexose for aminotransferase activity. As such, we constructed a single site-directed mutant of ColD, which replaced Ser 187 for an asparagine, but still retained His 188. Presented in Figure 3 is the HPLC trace from the reaction containing the ColD S187N single mutant enzyme. Peak 1 corresponds to NADPH that is left over from the synthesis of GDP-4-keto-6-deoxymannose. The ESI mass spectrum of Peak 2 corresponds to the wild-type ColD product, GDP-4-keto-3,6-dideoxymannose (570 amu). Peak 3 contains GDP (442 amu), a breakdown product of the sugar substrates. From these data it can be concluded that the ColD S187N single mutant enzyme retains wild-type ColD activity (i.e. it is a sugar dehydratase).
Figure 3.
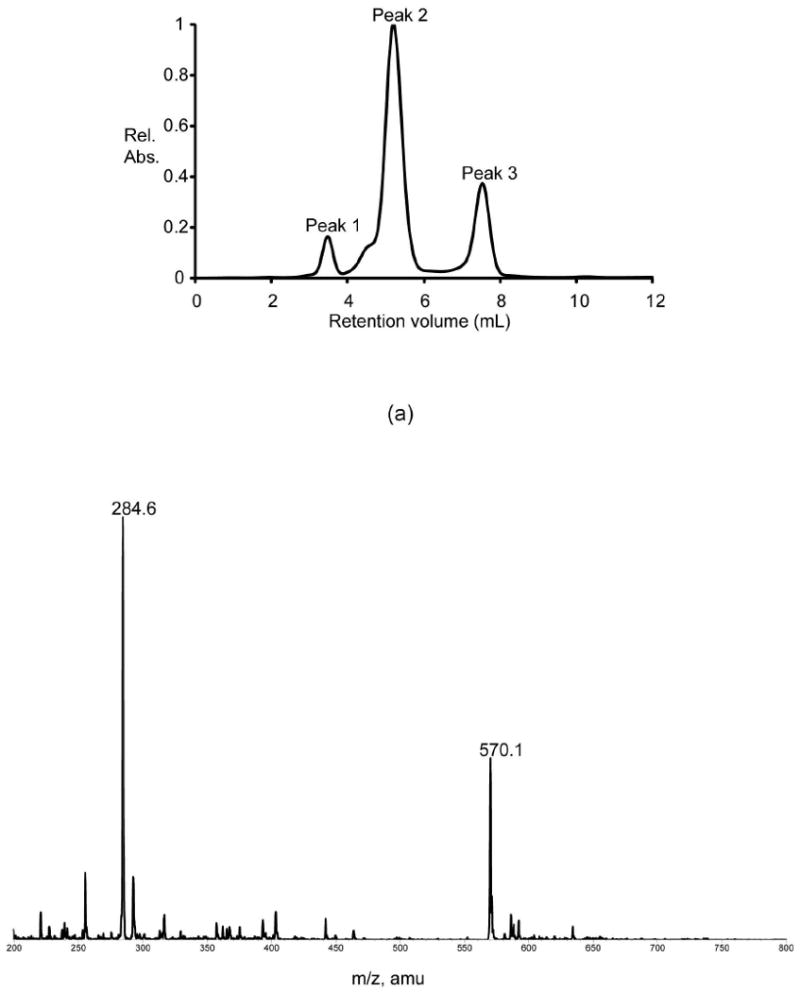
Activity assay for the S187N mutant protein. (a) The HPLC elution profile (monitored at 253 nm) is shown when the S187N mutant protein was assayed. Shown in (b) is the ESI mass spectrum corresponding to Peak 2 in the elution profile given in (a). The peaks at 570.1 and 284.6 correspond to the singly and doubly charged ions of GDP-4-keto-3,6-dideoxymannose.
We next suspected that perhaps a combination of both the active site general acid/base and the residue immediately adjacent to it act in concert to select a particular reaction type. With this in mind, the ColD S187N/H188K mutant enzyme was constructed, purified, and assayed. The HPLC trace of the reaction with this double mutant protein is presented in Figure 4. Again, Peak 1 is NADPH and the ESI mass spectrum of Peak 2 shows unreacted GDP-4-keto-6-deoxymannose (586 amu) and a small amount of GDP-4-keto-3,6-dideoxymannose (570 amu). Importantly, Peak 3 contains GDP-perosamine (587 amu) as well as GDP (442 amu). These data indicate that the ColD S187N/H188K mutant enzyme functions as a sugar-4-aminotransferase. It should be noted that the presence of the ColD wild-type product, GDP-4-keto-3,6-deoxymannose, indicates that the double mutant enzyme retains some wild-type ColD activity. However, given that the peak corresponding to the wild-type ColD product (570 amu) is barely above experimental background, it is clear that sugar-4-aminostransferase activity is the dominant reaction type for ColD S187N/H188K mutant enzyme.
Figure 4.
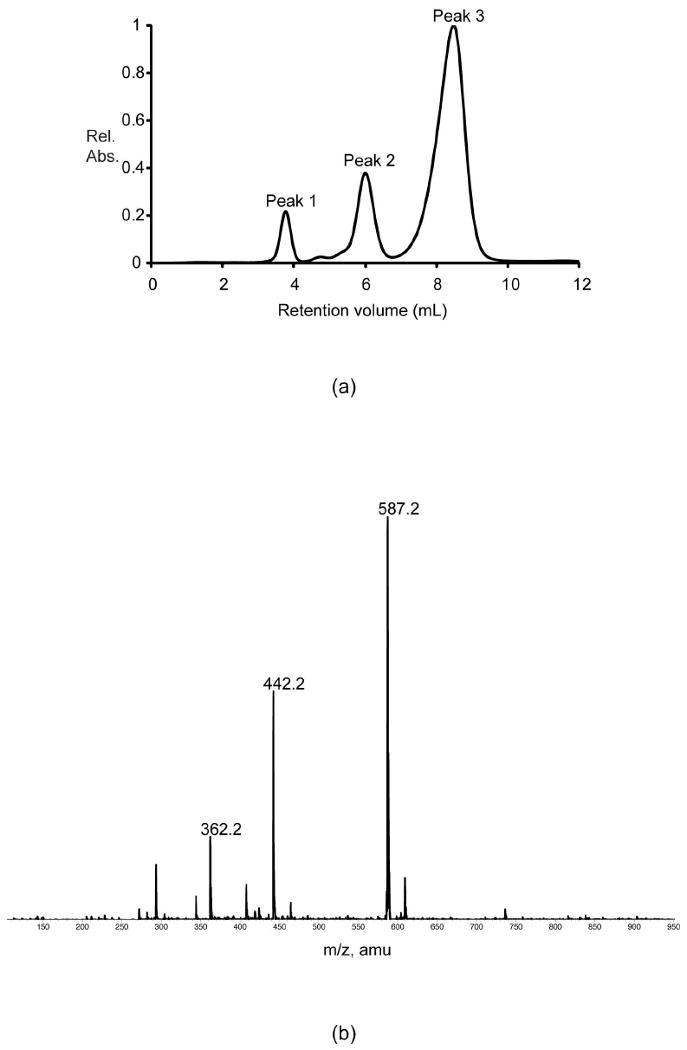
Activity assay for the S187N/H188K mutant protein. (a) The HPLC elution profile (monitored at 253 nm) is shown when the double mutant protein was assayed. Shown in (b) is the ESI mass spectrum corresponding to Peak 3 in the elution profile presented in (a). The peak at 587.2 corresponds to GDP-perosamine whereas the remaining the peaks at 442.2 and 362.2 correspond to GDP and GMP, respectively due to parent ion fragmentation.
In order to assess how well the ColD S187N/H188K mutant enzyme binds its substrate and catalyzes its reaction, the steady-state kinetic parameters were determined and are presented in Table 2. Also presented in Table 2 are the steady-state kinetic parameters for the C. crescentus wild-type GDP-perosamine synthase reaction (14). The Km for the ColD double mutant protein is somewhat perturbed relative to wild-type GDP-perosamine synthase (0.050 mM versus 0.013 mM, respectively). The kcat for the overall reaction, however, is perturbed to a greater extent, 0.028 s-1 versus 2.7 s-1 for wild-type GDP-perosamine synthase. The overall net effect is that the catalytic efficiency of the ColD double mutant protein is reduced by a factor of ∼400 relative to that of the wild-type GDP-perosamine synthase. Note that the Km for glutamate with the ColD double mutant enzyme is strikingly low compared to wild-type GDP-perosamine synthase. This occurs because aminotransfer to the sugar has become the rate-limiting step, and therefore much less glutamate is required for the reaction to proceed at the reduced rate. This phenomenon is common among ping-pong mechanisms in which active site mutations have been made or alternative substrates are used (14, 19).
Table 2. Steady-State Kinetic Parameters for Wild-Type GDP-Perosamine Synthase and the ColD Double Mutant Protein.
| Enzyme |
Km (GDP-4-keto-6-deoxymannose) (mM) |
Km (glutamate) (mM) |
kcat (s-1) |
kcat/Km (sugar) (M-1 s-1) |
|---|---|---|---|---|
| GDP-perosamine synthase (C. crescentus) | 0.013 ± 0.006 | 4.6 ± 1.4 | 2.7 ± 0.6 | (2.1 ± 1.1) × 105 |
| ColD S187N/H188K mutant protein (E. coli O55) | 0.05 ± 0.01 | 0.17 ± 0.03 | 0.028 ± 0.003 | (5 ± 2) × 102 |
The three-dimensional structure of the ColD S187N/H188K mutant enzyme was subsequently solved to assess its active site geometry. Crystals used for this analysis contained eight subunits in the asymmetric unit and diffracted to 2.2-Å resolution. Overall the electron density for the active site was best defined in subunit F in the x-ray coordinate file, and thus the following discussion refers only to it. Note, however, that the α-carbons for the other subunits superimpose onto subunit F with root-mean-square deviations of between 0.30 and 0.36 Å. Electron density corresponding to the sites of the mutations is presented in Figure 5a. The electron density is not as well ordered as observed, for example, in the wild-type structures for ColD or GDP-perosamine synthase. Indeed, the introduction of these mutations leads to conformational flexibility within the active site region, which may in part account for the lower catalytic efficiency of the ColD double mutant protein versus wild-type GDP-perosamine synthase.
Figure 5.
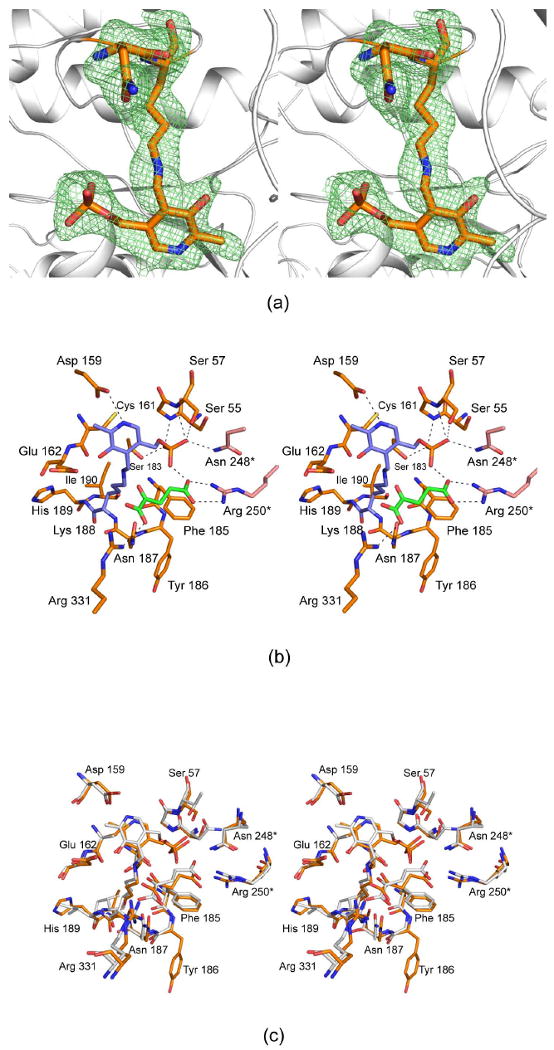
The structure of the S187N/H188K mutant protein. (a) Electron density corresponding to the sites of the mutation is shown. The map was contoured at ∼2σ and calculated with coefficients of the form (Fo - Fc), where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude. (b) A close-up view of the active site is displayed. Those residues located within ∼3.4 Å of Asn 187 and the internal aldimine (at Lys 188) are shown. The dashed lines indicate possible hydrogen bonding interactions. The internal aldimine is depicted in filled slate bonds whereas the α-ketoglutarate is highlighted in the green filled bonds. Two residues, Asn 248* and Arg 250*, are contributed by the second subunit of the dimer, and hence they are displayed in salmon. (c) A superposition of the active sites for the ColD double mutant protein and wild-type GDP-perosamine synthase is shown. ColD and GDP-perosamine synthase are colored in gold and white, respectively. The residues labeled correspond to ColD.
A close-up view of the ColD double mutant protein active site is presented in Figure 5b. The phosphoryl moiety of the PLP is anchored to the protein via hydrogen bonds from the backbone amide groups of Gly 56 and Ser 57 and the side chains of Ser 57, Ser 183, and Asn 248. In addition, the pyridoxal ring sits within hydrogen bonding distance to the side chain of Asp 159. For this structural analysis, the mutant enzyme was crystallized in the presence of α-ketoglutarate. The side chain carboxylate of α-ketoglutarate forms a salt bridge with the guanidinium group of Arg 250 whereas its α-carboxylate interacts with the guanidinium group of Arg 331. Note that one of the carboxylate oxygens of the α-ketoglutarate is located within ∼2.3 Å of a PLP phosphoryl oxygen thus suggesting that a proton is shared between these two atoms. A superposition of the active sites for the ColD double mutant protein and wild-type GDP-perosamine synthase is presented in Figure 5c, and as can be seen, they are remarkably similar.
A question that might be asked is why the ColD double mutant enzyme possesses GDP-perosamine synthase activity whereas the single mutant protein, H188K, does not. A major difference between the ColD and GDP-perosamine synthase catalytic mechanisms is the placement of a proton on the appropriate atom (Scheme 2). It is likely that the hydrogen bond that forms between the hexose ring oxygen and the asparagine residue forces the sugar into an orientation that favors protonation by the lysine Nζ at the hexose C-4′. The fact that the ColD S187N single mutant enzyme retains wild-type ColD activity suggests that the histidine side chain is too short or possesses improper geometry to place the proton on the sugar C-4′ position, despite the hydrogen bonding interaction between the sugar and the asparagine. As a consequence, the proton is placed on the hexose-3-hydroxyl group thereby leading to dehydratase activity.
Given that we were able to confer aminotransferase activity upon ColD, we were curious as to whether we could convert GDP-perosamine synthase into a dehydratase through site-directed mutagenesis. The GDP-perosamine synthase K186H single mutant enzyme had no detectable catalytic activity with its sugar substrate. Interestingly, none of the other GDP-perosamine synthase mutants (N185S, K186H/N185S, K186H/N185S/Y221F, and K186H/N185S/Y221F/M215W) tested thus far possess sugar-3-dehydratase activity (data not shown). The ability to perform a dehydration reaction most likely requires changes in residues not immediately surrounding the sugar substrate. Indeed, dehydratase activity requires the activation of a water molecule to attack the aminomannoseen-PLP intermediate, which is not required for aminotransferase activity.
In 2003, Yamada et al., put forth a hypothesis to explain why serine dehydratase removes the hydroxyl group of serine, whereas similar enzymes function as aminotransferases (20). Their explanation was based upon differences in active site residues, including the identity of the side chain that interacts with the nitrogen of the pyridoxal ring (cysteine in serine dehydratase and aspartate in aminotransferases) and the polarities of the environments in which the PLP phosphates and substrate carboxylate groups are situated. This hypothesis, however, is not applicable to the reaction specificities of ColD and GDP-perosamine synthase, since both enzymes position an aspartate residue within hydrogen bonding distance to the pyridoxal ring nitrogen (Figure 5). Additionally, in both enzymes, the PLP cofactors and the substrates reside in environments of similar polarity.
Very recently, Smith et al., accomplished a remarkable functional change with the E1 dehydratase, which is involved in the 3-deoxygenation of a CDP-glucose derivative during the production of CDP-ascarylose in Yersinia pseudotuberculosis (21). E1, like ColD, utilizes PLP and contains an active site histidine (His 220) instead of a lysine, but unlike ColD, E1 contains a [2Fe-2S] cluster and requires the NADH-dependent accessory protein E3 to complete its dehydration reaction. Furthermore, E1 does not require glutamate to convert the PLP cofactor into PMP, but instead directly releases the sugar product as a ketone, leaving the cofactor in the PMP form, ready for the next catalytic cycle. Four site-directed mutations (D194H/Y217H/H220K/F345H) were performed on E1, converting this enzyme from a PMP-dependent dehydratase to a PLP-dependent aminotransferase. Furthermore, since there is no overall reduction of the substrate, the accessory protein E3 is not required for the aminotransferase activity.
In summary, the results described here demonstrate that in the case of ColD, as opposed to that observed for E1 of the CDP-ascarylose biosynthetic pathway, only two site-directed mutations are required to convert it into an aminotransferase. Interestingly, however, it has not yet been possible to convert GDP-perosamine synthase into a dehydratase. Most likely this is because sugar dehydration is a much more difficult process, and quite subtle factors may be involved. The work presented here raises an intriguing question regarding the evolution of PLP-dependent enzymes. Even though wild-type ColD acts as a dehydratase on its sugar substrate, it also performs an aminotransferase reaction during the generation of PMP by converting l-glutamate to α-ketoglutarate. Thus, ColD contains the active site machinery to perform aminotransferase reactions and the double mutant enzyme presented here simply allows this reaction type to be performed upon the sugar substrate as well. In contrast, GDP-perosamine synthase is strictly an aminotransferase, and thus it has not evolved the machinery to perform a dehydration reaction, which may explain why it has proven more challenging to produce a mutant GDP-perosamine synthase with dehydratase activity. Additional site-directed mutagenesis experiments with GDP-perosamine synthase are presently underway.
Acknowledgments
We thank Dr. W. W. Cleland for helpful discussions.
Abbreviations
- ESI
electrospray ionization
- GDP
guanosine 5′-diphosphate
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- HPLC
high performance liquid chromatography
- MES
2-(N-morpholino)ethanesulfonic acid
- NADP
nicotinamide adenine dinucleotide phosphate
- PLP
pyridoxal-5′-phosphate
- PMP
pyridoxamine 5′-phosphate
- Tris
tris-(hydroxymethyl)aminomethane
Footnotes
This paper is dedicated in memory of Michael A. Toomey and Joseph A. Holden
This research was supported by an NIH grant (DK47814 to H. M. H.)
X-ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, N. J. (accession no. 3GR9).
References
- 1.Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Wang Q, Reeves PR, Wang L. Structure and genetics of Shigella O antigens. FEMS Microbiol Rev. 2008;32:627–653. doi: 10.1111/j.1574-6976.2008.00114.x. [DOI] [PubMed] [Google Scholar]
- 2.Pluschke G, Mayden J, Achtman M, Levine RP. Role of the capsule and the O antigen in resistance of O18:K1 Escherichia coli to complement-mediated killing. Infect Immun. 1983;42:907–913. doi: 10.1128/iai.42.3.907-913.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Achtman M, Pluschke G. Clonal analysis of descent and virulence among selected Escherichia coli. Annu Rev Microbiol. 1986;40:185–210. doi: 10.1146/annurev.mi.40.100186.001153. [DOI] [PubMed] [Google Scholar]
- 4.Edstrom RD, Heath EC. Isolation of colitose-containing oligosaccharides from the cell wall lipopolysaccharide of Escherichia coli. Biochem Biophys Res Commun. 1965;21:638–643. doi: 10.1016/0006-291x(65)90534-6. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu T, Yamasaki S, Tsukamoto T, Takeda Y. Analysis of the genes responsible for the O-antigen synthesis in enterohaemorrhagic Escherichia coli O157. Microb Pathog. 1999;26:235–247. doi: 10.1006/mpat.1998.0253. [DOI] [PubMed] [Google Scholar]
- 6.Albermann C, Piepersberg W. Expression and identification of the RfbE protein from Vibrio cholerae O1 and its use for the enzymatic synthesis of GDP-D-perosamine. Glycobiology. 2001;11:655–661. doi: 10.1093/glycob/11.8.655. [DOI] [PubMed] [Google Scholar]
- 7.Hisatsune K, Kondo S, Isshiki Y, Iguchi T, Kawamata Y, Shimada T. O-antigenic lipopolysaccharide of Vibrio cholerae O139 Bengal, a new epidemic strain for recent cholera in the Indian subcontinent. Biochem Biophys Res Commun. 1993;196:1309–1315. doi: 10.1006/bbrc.1993.2395. [DOI] [PubMed] [Google Scholar]
- 8.Xiang SH, Haase AM, Reeves PR. Variation of the rfb gene clusters in Salmonella enterica. J Bacteriol. 1993;175:4877–4884. doi: 10.1128/jb.175.15.4877-4884.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cook PD, Thoden JB, Holden HM. The structure of GDP-4-keto-6-deoxy-d-mannose-3-dehydratase: a unique coenzyme B6-dependent enzyme. Protein Sci. 2006;15:2093–2106. doi: 10.1110/ps.062328306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook PD, Holden HM. GDP-perosamine synthase: structural analysis and production of a novel trideoxysugar. Biochemistry. 2008;47:2833–2840. doi: 10.1021/bi702430d. [DOI] [PubMed] [Google Scholar]
- 11.Alam J, Beyer N, Liu HW. Biosynthesis of colitose: expression, purification, and mechanistic characterization of GDP-4-keto-6-deoxy-d-mannose-3-dehydrase (ColD) and GDP-l-colitose synthase (ColC) Biochemistry. 2004;43:16450–16460. doi: 10.1021/bi0483763. [DOI] [PubMed] [Google Scholar]
- 12.Cook PD, Holden HM. A Structural Study of GDP-4-Keto-6-Deoxy-d-Mannose-3-Dehydratase: Caught in the Act of Geminal Diamine Formation. Biochemistry. 2007;46:14215–14224. doi: 10.1021/bi701686s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook PD, Holden HM. GDP-4-keto-6-deoxy-d-mannose 3-dehydratase, accommodating a sugar substrate in the active site. J Biol Chem. 2008;283:4295–4303. doi: 10.1074/jbc.M708893200. [DOI] [PubMed] [Google Scholar]
- 14.Cook PD, Carney AE, Holden HM. Accommodation of GDP-linked sugars in the active site of GDP-perosamine synthase. Biochemistry. 2008;47:10685–10693. doi: 10.1021/bi801309q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;D57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 16.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D. 2004;D60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 17.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 18.Tronrud DE, Ten Eyck LF, Matthews BW. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr Sect A. 1987;43:489–501. [Google Scholar]
- 19.Matthews RG. Flavins and Flavoproteins. Walter de Gruyter & Co.; New York: 1990. [Google Scholar]
- 20.Yamada T, Komoto J, Takata Y, Ogawa H, Pitot HC, Takusagawa F. Crystal structure of serine dehydratase from rat liver. Biochemistry. 2003;42:12854–12865. doi: 10.1021/bi035324p. [DOI] [PubMed] [Google Scholar]
- 21.Smith P, Szu PH, Bui C, Liu HW, Tsai SC. Structure and mutagenic conversion of E1 dehydrase: at the crossroads of dehydration, amino transfer, and epimerization. Biochemistry. 2008;47:6329–6341. doi: 10.1021/bi702449p. [DOI] [PMC free article] [PubMed] [Google Scholar]