

Abstract
Coenzyme Q10 (CoQ10) is essential for electron transport in the mitochondrial respiratory chain and antioxidant defense. Last year, we reported the first mutations in CoQ10 biosynthetic genes, COQ2, which encodes 4-parahydroxybenzoate: polyprenyl transferase; and PDSS2, which encodes subunit 2 of decaprenyl diphosphate synthase. However, the pathogenic mechanisms of primary CoQ10 deficiency have not been well characterized. In this study, we investigated the consequence of severe CoQ10 deficiency on bioenergetics, oxidative stress, and antioxidant defenses in cultured skin fibroblasts harboring COQ2 and PDSS2 mutations. Defects in the first two committed steps of the CoQ10 biosynthetic pathway produce different biochemical alterations. PDSS2 mutant fibroblasts have 12% CoQ10 relative to control cells and markedly reduced ATP synthesis, but do not show increased reactive oxygen species (ROS) production, signs of oxidative stress, or increased antioxidant defense markers. In contrast, COQ2 mutant fibroblasts have 30% CoQ10 with partial defect in ATP synthesis, as well as significantly increased ROS production and oxidation of lipids and proteins. On the basis of a small number of cell lines, our results suggest that primary CoQ10 deficiencies cause variable defects of ATP synthesis and oxidative stress, which may explain the different clinical features and may lead to more rational therapeutic strategies.—Quinzii, C. M., López, L. C., Von-Moltke, J., Naini, A., Krishna, S., Schuelke, M., Salviati, L., Navas, P., DiMauro, S., Hirano, M. Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency.
Keywords: mitochondria, reactive oxygen species, COQ2, PDSS2
COENZYME Q10 (CoQ10) IS THE predominant human form of endogenous ubiquinone and is synthesized in the mitochondrial inner membrane. CoQ10 is composed of a benzoquinone and a decaprenyl side chain. Whereas the quinone ring is derived from tyrosine or phenylalanine, the isoprenoid side chain is produced by addition of isopentenyl diphosphate molecules, derived from the mevalonate pathway, to farnesyl diphosphate or geranylgeranyl diphosphate in multiple steps catalyzed by decaprenyl diphosphate synthase (Fig. 1). Decaprenyl diphosphate and para-hydroxybenzoate are condensed in a reaction catalyzed by PHB-polyprenyl transferase or COQ2, and the benzoate ring is then modified by at least six enzymes, which catalyze methylation, decarboxylation, and hydroxylation reactions to synthesize CoQ10 (Fig. 1).
Figure 1.

CoQ10 biosynthesis pathway. CoQ10 is composed of a benzoquinone and a decaprenyl side chain. Patient 1 has compound heterozygous mutations in subunit 2 of polyprenyl diphosphate synthase (PDSS2); patients 2 and 3 have a homozygous mutation in COQ2.
In addition to its central role in the mitochondrial respiratory chain as the carrier of electrons from complexes I and II to complex III, CoQ10 participates in other cellular functions (1). In the reduced form (ubiquinol), CoQ10 is one of the most potent lipophilic antioxidants in all cell membranes (2). CoQ10 is also required for pyrimidine nucleoside biosynthesis and may modulate apoptosis and the mitochondrial uncoupling protein (1).
Deficiency of CoQ10 has been identified in clinically heterogeneous autosomal recessive diseases, which have been delineated into four major pheno-types: 1) encephalomyopathy characterized by the triad of recurrent myoglobinuria, brain involvement, and ragged-red fibers (3-7); 2) severe infantile multisystemic diseases (8-10); 3) cerebellar ataxia (11-14); and 4) isolated myopathy (15, 16). Within the past two years, the identification of mutations in CoQ10 biosynthetic genes, COQ2, PDSS1, and PDSS2, in patients with infantile-onset diseases has proven the existence of primary CoQ10 deficiencies (17-19), whereas some patients with cerebellar ataxia or isolated myopathies have secondary CoQ10 deficiencies due to mutations in APTX and ETFDH, genes not directly related to ubiquinone synthesis (15, 20, 21). Despite these advances, primary and secondary CoQ10 deficiencies have been defined biochemically and genetically in fewer than 20 patients (22), and their pathogenic mechanisms remain unclear. In skeletal muscle of patients, CoQ10 deficiency has been associated with variable defects of the mitochondrial respiratory chain, increased apoptosis, and up-regulation of antioxidant defenses (5-7, 10, 15, 17). By contrast, studies of cultured fibroblasts from two siblings with infantile-onset CoQ10 deficiency of known genetic etiology showed mild respiratory chain defects, but no evidence of increased superoxide anions, lipid peroxidation, or apoptosis-mediated cell death (23). Moreover, Lopez-Martin and colleagues showed that COQ2 mutant cells require uri-dine to maintain growth and proposed that deficiency of CoQ10 caused a defect of pyrimidines biosynthesis because of the dependence of dihydroorotate dehydrogenase on ubiquinol (24).
Thus, lack of CoQ10 may cause human diseases by one or multiple processes, including reduced respiratory chain activity; enhanced reactive oxygen species (ROS) production, increased ROS susceptibility, or both; or impairment of de novo pyrimidines synthesis.
To further assess these putative pathogenic mechanisms, we studied the oxidative stress and bioenergetics in cultured fibroblasts from 3 patients with CoQ10 deficiency.
MATERIALS AND METHODS
Cell culture
Skin fibroblasts from 3 patients, patient 1 (P1) with PDSS1 mutations (Q322X and S382L) (17), patients 2 and 3 (P2 and P3) with a homozygous COQ2 mutation (Y297C) (10, 19), and 5 controls, were grown in glucose-rich Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1 ml fungizone, and 5 ml penicillin-streptomycin and in RPMI 1640 glucose-free medium supplemented with 10% fetal bovine serum, 25 mM HEPES, 1.5 mM Glutamax, 25 mM galactose, 1 ml fungizone, and 5 ml penicillin-streptomycin. To enhance potential alterations in respiratory chain-dependent ATP synthesis and adenine nucleotide levels due to CoQ10 deficiency, additional cells were grown in RPMI 1640 glucose-free medium supplemented with 10% dialyzed fetal bovine serum (glucose concentration <0.5 μg/ml in medium), 25 mM HEPES, 1.5 mM Glutamax, 25 mM galactose, 1 ml fungizone, and 5 ml penicillin-streptomycin. All cell culture reagents were obtained from Invitrogen (Carlsbad, CA, USA). All patient cell lines at passages 7-10 were grown in triplicate in glucose-rich and galactose media. Because of the poor growth of P3 cells, some experiments were performed only in P1 and P2 fibroblasts.
Cell growth rates
Growth rates were determined on cells from P1, P2, and controls plated in duplicate in 6-well plates, trypsinized, and counted manually after 48, 72, and 96 h of incubation.
Mitochondrial bioenergetics
For the ATP synthesis assay, cells were grown in 15-cm-diameter plates until confluent, then collected in PBS using a scraper. After centrifugation at 5000 rpm for 3 min, the cells were suspended in 350 μl of respiration buffer (150 mM KCl, 25 mM Tris-HCl, 2 mM EDTA, 0.1% BSA, 10 mM potassium phosphate, 0.1 mM MgCl2, pH 7.4), permeabilized with digitonin (50-75 μg/mg prot) for 1 min with gentle agitation and washed with 1 ml of respiration buffer (25). After centrifugation at 800 g for 5 min, pellets were suspended in 350 μl of respiration buffer and divided into two tubes with 160 μl each. Oligomycin (2 μg/ml) was added to one tube and respiration buffer to the other. Pyruvate (1 mM) and malate (1 mM) were added to both tubes, and ATP synthesis was induced by adding ADP (0.1 M) (25). After 1 min, the reaction was stopped with 0.5 M PCA, vortexed for 30 s, and centrifuged at 13,000 rpm for 10 min at 4°C. The pellets were stored at -80°C for protein determination. Adenine nucleotides were measured in the resultant supernatants into an Alliance HPLC (Waters Corporation, Milford, MA, USA) with an Alltima C18NUC reverse-phase column (Alltech Associates, Deerfield, IL, USA) (26). After stabilizing the column with the mobile phase, we injected samples (50 μl) into the HPLC system. The mobile phase consisted of 0.2 M ammonium phosphate buffer, pH 3.5 (phase A), and 30% methanol in 0.2 M ammonium phosphate buffer, pH 3.5 (phase B). The following time schedule for the binary gradient was used: 35 min, 100% A (18 min at 0.5 ml/min and then 1 ml/min), 5 min 0% to 100% B and then 100% B for 15 min (1 ml/min), 5 min 0% to 100% A (1 ml/min), and then 15 min with 100% A (10 min at 1 ml/min and then 0.5 ml/min) (26). Standard curves for AMP, ADP, and ATP were constructed with 15 μM, 30 μM, and 60 μM of each nucleotide. Absorbances of the samples were measured with an UV detector at 260 nm wavelength, and the concentration of each nucleotide in the samples was calculated based on the peak area. Adenine nucleotide levels were expressed in nanomoles per milligram protein. ATP synthesis was expressed in nanomoles per minute per milligram protein. Adenine nucleotides concentrations were measured as described (17, 25, 26) with slight modifications. Briefly, the cells were grown in 10-cm-diameter plates until confluent and then collected in ice-cold PBS using a scraper. After centrifugation at 5,000 rpm for 3 min at 4°C, pellets were suspended in 200 μl of ice-cold 0.5 M PCA, vortexed for 30 s, and centrifuged at 13,000 rpm for 10 min at 4°C. The pellets were stored at -80°C for protein measurement and supernatants were used for adenine nucleotide determination.
Cell membrane potential measurement
To estimate cell membrane potential, P1, P2, and control fibroblasts were exposed to MitoTracker Red CMXRos and tetramethylrhodamine, ethyl ester, perchlorate (TMRE) (Molecular Probes, Invitrogen Corp., Carlsbad, CA, USA). Approximately 1 × 106 cells were trypsinized, incubated with 50 nM MitoTracker for 30 min at 37°C, washed twice with PBS, and resuspended in 500 μl of PBS. The same number of cells were trypsinized, incubated with 50 nM TMRE for 20 min at 37°C, washed twice with PBS, and resuspended in 500 μl of PBS. Cytofluorometric analysis was performed using a FACSCalibur cell analyzer equipped with a 488-nm At-kt laser, and fluorescence was measured using the FL2 channel. Data were acquired using Cell Pro Quest and analyzed using Flowjo software (Becton Dickinson, Franklin Lakes, NJ, USA).
Phosphofructokinase activity and extracellular lactate measurement
To measure activity of phosphofructokinase (PFK), cell extracts (0.3- 0.8 mg protein) were added to a reaction mix containing 40 mM Tris-HCl (pH 8.0), 6 mM MgCl2, 2 mM ATP, 200 μM NADH, 0.001- 0.01 U aldolase, 0.375-10 U glycerophosphate dehydrogenase, 3.75-100 U of triosephosphate isomerase, and 3 mM fructose-6-phosphate. Oxidation of NADH was assessed by absorbance at 340 nm for 3 min at 30°C. The results were expressed in nanomoles of oxidized NADH/min/mg prot.
Extracellular lactate levels were measured as described previously with minor modifications. Briefly, after cells were grown in 15-cm-diameter plates to 80% confluence, culture medium was changed, and after 72 h, 200 μl of the medium was removed, treated with 200 μl of 1 M PCA, and centrifuged at 25,000 g for 10 min at 4°C. The supernatant was neutralized by adding 28 μl of 5 M potassium carbonate and centrifuged at 12,000 g for 10 min at 4°C. The supernatant was used to measure lactate spectrophotometrically using Lactate Assay Kit (BioVision, Inc., Mountain View, CA, USA). The lactate levels in unused medium was subtracted from the measured values. The results were expressed in nanomoles per milligram protein.
Oxidative stress analyses
To estimate production of ROS, CoQ10-deficient and control fibroblasts were exposed to MitoSOX Red, a fluorochrome specific to anion superoxide produced in the inner mitochondrial compartment (Molecular Probes, Invitrogen Corp., Carlsbad, CA, USA) (27, 28). Approximately 1 × 106 cells were trypsinized, incubated with MitoSox for 30 min at 37°C, washed twice with PBS and resuspended in 500 μl of PBS. Cytofluorometric analysis was performed as described above. MitoSOX intensity was also monitored by fluorescence microscopy (IX70 inverted system microscope; Olympus, Tokyo, Japan). Cells grown on microscope slides in 6-well plates for 24 h were incubated with MitoSox for 30 min at 37°C, washed twice in PBS, fixed with 4% paraformaldehyde in PBS for 0.5-1 h at room temperature, and washed twice with PBS, incubated for 10 min at 37°C with MitoTracker Green (Molecular Probes, Invitrogen Corp.) to label mitochondria, and washed again before mounting. A control cell line treated with 0.5 μM antimycin A for 15 h before the MitoSOX staining was used as a positive control for increased ROS production (29).
To determine the level of oxidative damage of proteins, we performed Western blot analysis of carbonyls group content in protein using the OxyBlot kit (Chemicon, Millipore Corp, Billerica, MA, USA). In brief, 20 μg of proteins was incubated with 2,4-dinitrophenylhydrazine to form 2,4-dinitrophenyl hydrazone derivates. 2,4-Dinitrophenyl-derivatized proteins were separated on a 12% polyacrylamide gel and transferred to polyvinyl prolidone membranes for 30 min at 50 V in Mini Trans-Blot cell apparatus (Bio-Rad, Hercules, CA, USA). Blots were hybridized with rabbit anti-DNP antibody and goat anti-rabbit horseradish peroxide conjugated secondary antibody (Chemicon, Millipore Corp.) and visualized by autoradiography using Supersignal West Dura Extended Duration Substrate (Pierce, Rockford, IL, USA). The blots were subsequently immunostained with an anti-acid maltase antibody for normalization of total proteins levels. Quantitative analysis of the blots was carried out using Image J software (http:rsb.info.nih.gov/ij/) and the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA).
For lipid peroxidation (LPO) measurements, confluent cells were collected in PBS from 15-cm-diameter plates using scrapers. After centrifugation at 5000 rpm for 5 min, cells were suspended in 20 mM Tris-HCl buffer, pH 7.4, containing 5 mM butylated hydroxytoluene, and sonicated to lyse the cell. To remove large particles, the samples were centrifuged at 3000 g for 10 min at 4°C. Aliquots of the supernatants were either stored at -80°C for total protein determination or used for LPO. Bioxytech LPO-568 assay kit was used to determine both malondialdehyde (MDA) and 4-hydroxyalkenals (4HDA) (Oxis International, Foster City, CA, USA) (30). Concentrations of LPO were normalized per milligram protein.
Analyses of antioxidants defense
Glutathione reductase (GRd) and glutathione peroxidase (GPx) activities were assayed by following the oxidation of NADPH at 340 nm (31). Cells from one confluent 10-cm plate (~2-3×106) were collected using trypsin and stored at -80°C. On the day of the assay, the frozen pellet was resuspended in 350 μl of lysis solution (0.25 M sucrose; 10 mM Tris/HCl, pH 7.5; 1 mM EDTA-K2; 0.5 mM PMSF; 0.5 mM dTT; 0.1% Nonidet), sonicated briefly, and centrifuged at 15,000 g for 25 min at 4°C. Supernatants were used for GPx and GRd assays, and protein determination. To assay GPx activity, 850 μl of working solution (50 mM KH2PO4; 3 mM EDTA-K2, pH 7.0; 2 mM KCN; 2 mM reduced glutathione (GSH); 0.2 mM NADPH; and 2 U/ml GRd) and 125 μl of sample supernatant were combined in a 1-ml quartz cuvette. After a 3-min equilibration period at 30°C, the reaction was started by the addition of 25 μl of 160 μM cumene hydrogen peroxide, and the change in absorbance was recorded for 3 min. For the GRd assay, 840 μl of phosphate-EDTA buffer (100 mM KH2PO4, 0.5 mM EDTA-K2, pH 7.6) was combined with 10 μl of 10 mM NADPH and 125 μl of sample supernatant. After a 3-min equilibration period at 30°C, 25 μl of 40 mM oxidized glutathione (GSSG) was added, and the change in absorbance over time was recorded for an additional 3 min. Specific activities of GPx and GRd were expressed in nanomole-oxidized NADPH per minute per protein.
GSH and GSSG were measured indirectly by following the formation of 2-nitro-5-thiobenzoic acid (TNB) at 412 nm (31). Cells from one confluent 10-cm plate (~2-3×106 fibroblasts) were collected using trypsin, and stored at -80°C. On the day of the assay, the frozen pellet was resuspended in 750 μl of 6% metaphosphoric acid, vortexed, incubated at room temperature for 10 min, and centrifuged at 13,000 rpm for 10 min at 4°C. Pellets were stored at -80°C for protein determination, and the super-natants were used to measure GSH. For GSSG determination, 150 μl of supernatant was mixed with 3 μl of 2-vinylpyridine and 9 μl of triethanolamine and incubated at room temperature for 60 min. For total glutathione (T-GSH) concentration, the acidic sample supernatant was first diluted 1:6 in phosphate-EDTA buffer (100 mM K2HPO4, 5 mM EDTA-K2, pH 7.4). For both T-GSH and GSSG determination, 100 μl of sample, 750 μl of phosphate buffer, 50 μl of 10 mM DTNB, and 80 μl of 5 mM NADPH were added to a 1-ml cuvette. After a 3-min equilibration period at 30°C, 20 μl (1.35 U) of GRd was added, and the change in absorption over time was measured for an additional 3 min. Absolute concentrations were determined using a standard curve of GSH (0.5 μg/ml, 1 μg/ml, 2 μg/ml, and 4 μg/ml prepared in 6% metaphosphoric acid and diluted in phosphate buffer). GSH concentration was calculated as T-GSH-GSSG; concentrations of T-GSH, GSH, and GSSG were expressed in nanomoles per milligram protein.
Screening for CoQ4
To identify CoQ4 in the mutant skin fibroblasts, we followed two strategies. First, when 15-cm diameter culture plates were confluent, cells were collected, and all forms of CoQ were extracted with hexane and identified by EQ-HPLC using a reverse-phase column and an isocratic mobile phase consisting of 87% methanol, 1.5% 2-propanol, 1.5% acetic acid, and 50 mM sodium acetate. To identify peaks, a standard sample with 25 ng/ml of each CoQ4, CoQ9, and CoQ10 was formulated. Second, we measured the incorporation of 14C-PHB (450 Ci/mol) into all forms of ubiquinone in cultured fibroblasts. Briefly, 0.02 μCi/ml of 14C-PHB was added to cultured cells and after 48 h, fibroblasts were collected and ubiquinones were extracted with hexane. Radiolabeled ubiquinones were isolated by HPLC with a C18 reverse-phase column and were collected and measured in a scintillation counter.
Statistical analysis
Control data are expressed as the mean ± sd of 5 different samples in triplicate experiments. Patient data are expressed as the mean ± sd of triplicate experiments. Two-tailed Student's t test was used to compare the mean between groups.
RESULTS
Patients and fibroblasts
Patient 1 (P1) had Leigh syndrome and nephropathy due to compound heterozygous mutations in the PDSS2 gene, which encodes subunit 2 of decaprenyl diphosphate synthase, the first committed enzyme of the CoQ10 biosynthetic pathway (17). P1 fibroblasts had 12% CoQ10 and 28% residual CII+III activity. Patients 2 and 3 (P2 and P3) were siblings with nephropathy and encephalopathy carrying a homozygous mutation in the COQ2 gene, which encodes 4-parahydroxybenzoate: polyprenyl transferase (19). P2 and P3 cells with 30% CoQ10 had 48% residual CII+III activity (24).
Cell growth rate
In galactose media, growth rate was reduced in P2 fibroblasts, but comparable in P1 and control cells (Fig. 2).
Figure 2.
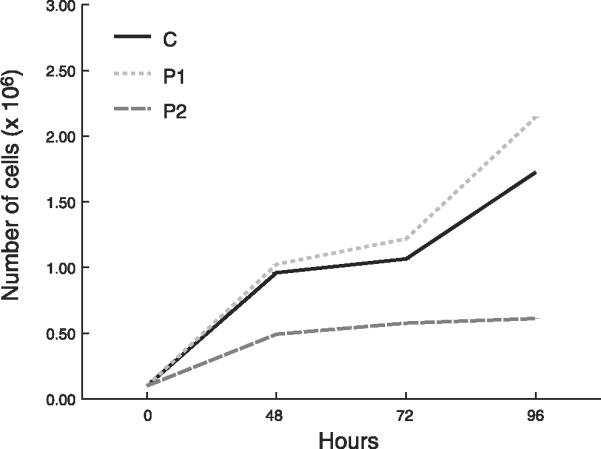
Cell growth rates in galactose medium. Control (C) data are expressed as the mean ± SD of 5 different samples in triplicate experiments. Patient data are expressed as the mean ± sd of triplicate experiments. *P < 0.05 vs. control.
Bioenergetics
In galactose media, mitochondrial ATP synthesis was significantly reduced in the cells from all three patients (P1-P3); P1 fibroblasts showed greater reductions (51% decrease) than P2 (32% decrease) and P3 cells (26% decrease) (Fig. 3). By contrast, in galactose medium, steady-state levels of total adenine nucleotides (AN) and ATP were not significantly decreased in P1 but were reduced in P2 and P3 (Fig. 4A, B). ATP/ADP ratio, a value that reflects the state of mitochondrial ATP production, was significantly decreased in the cells from all three patients (P<0.05), but the decreases were more dramatic in P2 and P3 than P1 fibroblasts (Fig. 4C).
Figure 3.
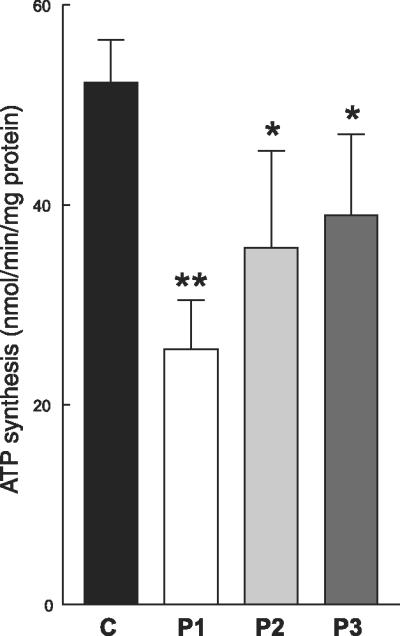
ATP synthesis in fibroblasts cultured in galactose medium. In respiration-dependent (galactose) media, mitochondrial ATP synthesis was significantly reduced in the cells from all three patients (P1-3); P1 fibroblasts showed greater reductions (51% decrease) than P2 (32% decrease) and P3 cells (26% decrease), reflecting the respiratory chain defect due to CoQ10 deficiency. Control data are expressed as the mean ± SD of 5 different samples in triplicates experiments. Patient data are expressed as the mean ± SD of triplicate experiments.*P < 0.05 vs. control; **P < 0.01 vs. control.
Figure 4.

Adenine nucleotide levels. In galactose medium, steady-state levels of total adenine nucleotides (ANs) and ATP were not significantly decreased in P1 but were reduced in P2 and P3 (A, B). ATP/ADP ratio, a value that reflects the state of mitochondrial ATP production, was significantly decreased in the cells from all three patients (P<0.05), but the decreases were more dramatic in P2 and P3 than P1 fibroblasts (C). In glucose-rich media, P2 but not P1 cells showed decreased ATP/ADP ratio, whereas neither P1 nor P2 fibroblasts showed significant decreases in total ANs or ATP (D,E). ANs were measured in P1 cells grown in media supplemented with 10% dialyzed FBS and demonstrated decreased levels of ATP, ATP/ADP ratio, and AN compared with cells cultured in galactose or glucose-rich media, indicating that glucose in undialyzed FBS provided substrate for anaerobic glycolysis (A-C). Control (C) data are expressed as the mean ± SD of 5 different samples in triplicate experiments. Patient data are expressed as the mean ± SD of triplicate experiments.*P < 0.05 vs. control; **P < 0.01 vs. control.
In glucose-rich media, P2, but not P1 cells showed decreased ATP/ADP ratio, whereas neither P1 nor P2 fibroblasts showed significant decreases in total adenine nucleotides or ATP (Fig. 4D-F).
To assess whether anaerobic glycolysis in FBS-containing medium could account for the normal levels of AN and ATP despite impaired respiratory chain activity (demonstrated by decreased ATP synthesis in galactose medium), adenine nucleotides were measured in P1 cells grown in media supplemented with 10% dialyzed FBS (containing less than 0.5 μg/ml of glucose) and demonstrated decreased levels of ATP, ATP/ADP ratio, and AN compared with cells cultured in galactose or glucose-rich media (Fig. 4A-C)
Cell membrane potential measurement
Flow cytometry analysis of cells stained with either Mitotracker Red CMXRos (Fig. 5) or TMRE (data not shown) revealed a subpopulation of P2 cells with decreased membrane potential in galactose medium, whereas in glucose-rich medium, both P1 and P2 cells showed subpopulations with decreased membrane potentials. In both patient fibroblast lines, the major peak of Mitotracker Red CMXRos (Fig. 5) or TMRE staining overlapped with the control peak.
Figure 5.

Assessment of mitochondrial membrane potential with MitoTracker Red. Flow cytometry of P1 and P2 cells revealed subpopulations with decreased fluorescent intensity compared with the control cells cultured in glucose medium (bottom panels), as indicated by minor peaks to the left of the major peak. P2 cells cultured in galactose also showed a subpopulation with decreased mitochondrial membrane potential.
Phosphofructokinase activity and lactate measurement
PFK activity was 20% higher in P1 cells than in controls (P>0.05), but comparable in P2 and control cells cultured in galactose and glucose-rich media. In both galactose and glucose-rich media, levels of extracellular lactate were significantly higher in P1 cells but not in P2 fibroblasts compared to control cells (Fig. 6).
Figure 6.

Phosphofructokinase (PFK) activity and lactate level in cells. PFK activity and lactate level were increased in P1 cells, indicating up-regulation of the rate-limiting step of glycolysis. Control (C) data are expressed as the mean ± SD of 5 different samples in triplicate experiments. Patient data are expressed as the mean ± SD of triplicate experiments. *P < 0.05 vs. control
ROS production and oxidative stress
MitoSOX Red stain revealed increased superoxide anion in COQ2 mutant fibroblasts but not in PDSS2 mutant cells nor in all five control cell lines cultured in galactose medium (Fig. 7). MitoSOX Red colocalized with MitoTracker Green, indicating that the excess superoxide anion was concentrated in mitochondria (Fig. 7). These observations were confirmed by quantitative analysis of the fluorescent signal using a FACscan cell analyzer, which showed significant increases of MitoSox Red signal in P2 and P3 fibroblasts, but not in P1 cells compared to controls (Table 1). In cells incubated in glucose-rich media, MitoSOX Red showed a trend toward higher staining only in P3 cells, but levels of superoxides were lower in all cell lines than in fibroblasts cultured in galactose media (Table 1). The MitoSox Red signal was 7.8-fold higher in P2 vs. control cells in galactose medium, whereas the increase in flurorescence in P3 vs. control cells in glucose-rich media was 3.1-fold (Table 1).
Figure 7.

Assessment of superoxide generation. MitoSOX Red stain (top panels) revealed increased superoxide anion in COQ2 mutant fibroblasts (P2 and P3) but neither in PDSS2 mutant (P1) cells nor in all five control cell lines cultured in galactose medium (C is a representative control cell line). MitoSOX Red colocalized with MitoTracker Green (middle panel) in merged images (bottom panels), indicating that the excess superoxide anion was concentrated in mitochondria.
TABLE 1.
Flow cytometry quantitation of MitoSox Red fluorescence
| Cell culture medium | C | P1 | P2 | P3 |
|---|---|---|---|---|
| Galactose | 2.15 ± 0.65 | 1.14 ± 0.17 | 16.7 ± 0.06** | ND |
| Glucose-rich | 0.07 ± 0.03 | 0.02 ± 0.01 | 0.05 ± 0.01 | 0.22 ± 0.02* |
Data are means ± SD. C, control; ND, not determined; P1, P2, P3, patient 1, 2, 3, respectively.
P < 0.05
P < 0.01
In galactose medium, levels of lipid peroxides, MDA, and 4HDA were significantly higher than normal in P2 and P3 cells, but not in P1 fibroblasts (Fig. 8A). However, no differences between controls and patients were appreciated in lipid peroxidation when the cells were grown in glucose-rich medium (Fig. 8B).
Figure 8.

Quantitation of oxidative damage. A, B) Lipid peroxidation assessed by Bioxytech LPO-568 assay kit. In galactose medium, levels of lipid peroxides, malondialdehyde (MDA), and 4-hydroxyalkenals (4HDA) were significantly higher than normal in P2 and P3 cells but not in P1 fibroblasts. However, no differences between controls and patients were appreciated in lipid peroxidation when the cells were grown in glucose-rich medium. C, D) Protein oxidation estimated by OxyBlot. Quantitation of carbonyl groups in proteins revealed increases in all three patient cell lines in galactose medium but was clearly higher in P2 and P3 fibroblasts. By contrast, protein oxidation was virtually absent in both patient and control skin fibroblasts grown in high-glucose medium. Control data are expressed as the mean ± SD of 5 different samples in triplicate experiments. Patient data are expressed as the mean ± SD of triplicate experiments.*P < 0.05 vs. control; **P < 0.01 vs. control.
Protein oxidation, estimated by OxyBlot quantitation of carbonyl groups in proteins of cell homogenates, was increased in all three patient cell lines in galactose medium (Fig. 8C) but was clearly higher in P2 and P3 fibroblasts. By contrast, protein oxidation was virtually absent in both patient and control skin fibroblasts grown in high-glucose medium (Fig. 8D).
Antioxidant defenses
Assessment of the glutathione system showed a trend toward increased proportion of GSSG in P2 and P3 cells, but not in P1 fibroblasts relative to controls. This change was accompanied by slight increases in the activities of glutathione enzymes, GPx, and GRd, in P2 cells (Table 2).
TABLE 2.
Glutathione system
| Galactose medium |
Glucose-rich medium |
|||||||
|---|---|---|---|---|---|---|---|---|
| C | P1 | P2 | P3 | C | P1 | P2 | P3 | |
| Total GSH (nmol/mg protein) | 27.4 ± 7.52 | 21.6 ± 0.99 | 24.9 ± 1.88 | 30.0 ± 1.12 | 25.5 ± 1.11 | 27.3 ± 3.72 | 27.8 ± 2.6 | ND |
| % GSSG | 3.53 ± 0.57 | 2.60 ± 0.65 | 4.24 ± 0.36 | 4.51 ± 0.58 | 4.83 ± 2.94 | 4.89 ± 0.70 | 5.59 ± 0.34 | ND |
| GPx (nmol/min/mg protein) | 23.4 ± 7.22 | 17.6 ± 2.62 | 31.2 ± 8.65 | ND | 32.2 ± 13.1 | 24.7 ± 1.27 | 44.0 ± 13.2 | ND |
| GRd (nmol/min/mg protein) | 15.8 ± 4.08 | 11.9 ± 2.77 | 17.4 ± 2.02 | ND | 21.1 ± 5.0 | 19.4 ± 0.99 | 27.9 ± 8.1 | ND |
Data are means ± SD. n = 5. Experiments performed in triplicate. C, control; GPx, glutathione peroxidase; Grd, glutathione reductase; GSH, glutathione; GSSG, oxidized glutathione; ND, not determined; P1, P2, P3, patient 1, 2, 3, respectively.
Identification of CoQ4
In the standard electrochemical chromatograph, only CoQ9 and CoQ10 were identified in controls and patient fibroblasts. However, because some peaks had retention times similar to CoQ4, to screen for the presence of CoQ4, we radiolabeled all forms of ubiquinone by incubating the cultured cells with 14C-PHB for 48 h, for detection by scintillation counter. The results showed the presence of radioactivity with the retention time of CoQ9 and CoQ10 in controls and patient fibroblasts but not with the CoQ4 retention time (data not shown).
DISCUSSION
Given the multiple important roles of CoQ10 in mitochondria (32) and other cellular compartments (2), we assessed the consequences of different severe forms of primary CoQ10 deficiency on oxidative stress and mitochondrial bioenergetics using COQ2 and PDSS2 mutant skin fibroblasts. Our studies are based on a small number of cell lines; therefore, factors other than CoQ10 deficiency may be contributing to our results. Nevertheless, our observations suggest that defects in these two enzymes of the CoQ10 biosynthetic pathway produce different patterns of biochemical dysfunction. In PDSS2 mutant fibroblasts with severe CoQ10 deficiency, oxidative phosphorylation is impaired, whereas in COQ2 mutant cells with milder CoQ10 deficiency and less severe respiratory chain defects, overproduction of superoxides leads to significantly increased oxidative damage to proteins and lipids.
The functions of CoQ10 in the cells are mainly related to its lipophilic and redox properties (1). Because it is synthesized in the inner mitochondrial membrane, a high proportion of CoQ10 remains in this location, where its reduction potential (+45 mV) facilitates transfer of electrons from the dehydrogenases of complexes I and II to cytochrome b of complex III (33). As COQ2 and PDSS2 mutations reduce CoQ10 levels and decrease activities of ubiqui-none-dependent respiratory chain functions (17, 19), a priori, mutant cells would be expected to show decreased synthesis of ATP through oxidative phosphorylation. Our results verify this hypothesis and show a direct correlation between CoQ10 levels and ATP synthesis. In all three CoQ10-deficient fibroblast lines, respiratory chain activity is decreased; however, in P1 cells with 12% CoQ10 and 28% residual CII+III activity (17), the defect of ATP synthesis is more pronounced than in P2 and P3 cells, which have 30% CoQ10 and 48% residual CII+III activity (19) (Fig. 2). The reductions of mitochondrial respiratory chain activities and ATP synthesis in CoQ10-deficient cells are consistent with three prior reports. First, two fibroblast cell lines with severe deficiencies of CoQ10 (undetectable and<20% of normal) had 30 - 40% cell respiration relative to controls (23). Second, after hexane extraction of CoQ10, isolated mitochondria also showed reduced electron flow from complexes I and II to complex III (34). Third, acute in vitro pharmacological inhibition of CoQ10 biosynthesis, using p-aminobenzoate (1 mM for 4 days) to competitively inhibit COQ2 in cultured human myeloid leukemia HL-60 cells, reduced CoQ10 levels by ~50%, decreased CII+III activity by >75%, and increased superoxide production in ~35% of the treated cells compared to controls (35).
Because of the defect of respiratory chain activity induced by CoQ10 deficiency, we had expected to find reductions of steady-state levels of ATP both in the cells with COQ2 and with PDSS2 mutations. While, in fact, P2 and P3 cells grown in galactose media showed reductions in ATP level and ATP/ADP ratio, surprisingly P1 cells, despite their more severe CoQ10 deficiency, had less severe reduction of ATP/ADP and normal ATP levels (Fig. 4A-C). These apparently contradictory results can be explained by data obtained using 10% undialyzed FBS culture medium, which contains ~100 μg/ml glucose that can be utilized by cells to compensate for the deficit in mitochondrial ATP synthesis through increased glycolysis and enhanced transcription of genes encoding glycolytic enzymes (36, 37). Favoring this explanation are two observations: 1) the level of ATP and ATP/ADP ratio were reduced in P1 cells cultured in 10% dialyzed FBS culture medium (containing <0.5 μg/ml glucose); and 2) the activity of PFK was increased in P1 cells, indicating up-regulation of the rate-limiting step of glycolysis. In agreement with this concept, cultured cybrid cells with respiratory chain defects due to mitochondrial DNA mutations also showed compensatory up-regulation of glycolysis even in low glucose medium (38).
In addition to their critical function in energy production, mitochondria also appear to be a major source of ROS in cells (39, 40). Over the past three decades, studies have identified multiple mitochondrial sites that generate ROS under physiological and pathological conditions (33, 39, 41, 42). However, it has been unclear whether the block of electron transfer caused by partial deficiency of CoQ10 can increase ROS production. To address this issue, we cultured control and CoQ10-deficient cells under two different metabolic conditions: 1) high glucose media, where typically 60 - 65% of ATP is generated from glycolysis and the remainder from oxidative phosphorylation (37); and 2) galactose media, which forces energy production through oxidative phosphorylation (43). We observed a significant increase of mitochondrial superoxide production and concomitant increases of lipid and protein oxidation in P2 and P3 cells cultured in galactose media. P2 cells also showed slightly increased glutathione enzyme activities and increased proportion of oxidized to total glutathione; these changes in antioxidant defenses may have been induced by increased oxidative stress. Nevertheless, because the glutathione system is present in many cellular compartments, including cytoplasm, mitochondria, nucleus, and endoplasmic reticulum, selective changes in mitochondrial glutathione may have been masked by unchanged levels of glutathione in other compartments (44). Oxidative stress was not obvious in the cells cultured in glucose-rich medium probably because the respiratory chain is underutilized. Curiously, a subpopulation of P2 fibroblasts cultured in glucose media, but not in galactose media, showed decreased mitochondrial membrane potential, possibly reflecting reduced electron flow and proton pumping across the mitochondrial membrane in cells generating ATP predominantly through anaerobic glycolysis.
Our observation of signs of increased oxidative stress in P2 (COQ2 mutant) cells is also supported by the finding that Schizosaccharomyces pombe ppt-1 (COQ2 homologous)-deficient strains are unable to grow on minimal medium supplemented with glucose, require antioxidant supplementation to grow on minimal medium, and are vulnerable to H2O2 and Cu2+2exposure (45). Saccharomyces cerevisiae COQ2-null mutants are also unable to grow in nonfermentable carbon source (24).
Curiously, in both mutant fibroblasts lines, we observed small subpopulations of cells with decreased mitochondrial membrane potential that was more prominent in cells grown in glucose-rich than galactose medium and in COQ2 mutant than PDSS2 mutant cells. The significance of these findings is uncertain because it is not known whether the abnormalities are primary or secondary consequences CoQ10 deficiency.
In contrast to COQ2 mutant fibroblasts, PDSS1 mutant cells did not show any sign of oxidative stress, for which we propose three possible explanations, which are not mutually exclusive. First, in P1 cells, ATP is produced predominantly by anaerobic glycolysis, and electron flow through the respiratory chain is severely reduced, thus minimizing the uncontrolled electron leak, as has been proposed for cells harboring the mitochondrial ATP6 gene T8993G mutation (46). Second, the redox functions of CoQ10 are based on its ability to exchange two electrons in a redox cycle between the oxidized (ubiquinone) and the reduced (ubiquinol) forms, thus allowing CoQ10 to act as an antioxidant but also as a pro-oxidant through the semiquinone intermediate (47, 48). Therefore, in the setting of CoQ10 deficiency, increased ROS production could be the result of enhanced semiquinone generation from increased redox cycling of the limited CoQ10 pool. Third, the different subcellular locations of the biosynthetic blocks may influence ROS production; defects of extramitochondrial PDSS2 may produce a “balanced” respiratory chain, while the intramitochondrial COQ2 defect may interfere with assembly or stability of the respiratory chain enzymes and unbalanced oxidative phosphorylation with enhanced ROS production. These factors may also account for the lack of increased superoxide anion and lipid peroxides observed in CoQ10-deficient skin fibroblasts from a patient with defect of trans-prenyltransferase activity (23). We excluded the possibility that PDSS2 mutant cells synthesize a ubiquinol with a short oprenoid side chain (i.e., COQ4 derived from conjugation of geranylgeranyl pyrophosphate to para-hydroxybenozoate; Fig. 1), which might function as an antioxidant but not as an effective electron carrier in the mitochondrial respiratory chain. Although increased mitochondrial proton motive force has been associated with enhanced ROS production (49), COQ2 mutant cells did not show higher mitochondrial membrane potentials than PDSS2 mutant fibroblasts.
In conclusion, our observations, based on a small number of cell lines, reveal potentially important differences in the pathogenic mechanisms of CoQ10 deficiency due to different biosynthetic defects: mutations in COQ2 and PDSS2. These differences may explain the heterogeneous clinical phenotypes of patients and lead to more rational therapeutic strategies aimed at reducing oxidative stress, enhancing respiratory chain activity, or both. In fact, P1 presented with Leigh syndrome, a condition frequently lethal because of severe mitochondrial dysfunction due to a variety of genetic defects that impair ATP synthesis (50), and did not respond to CoQ10 supplementation, whereas patients with mutations in COQ2 have nephrotic syndrome, encephalopathies, or both that are responsive to CoQ10 (10, 51). Further investigation of the effects of CoQ10 deficiency is necessary to validate our findings and may provide additional insights into this emerging group of metabolic disorders.
Acknowledgments
The authors thank Estela Area and Beatriz Dorado for their technical support. This work was supported by U.S. National Institutes of Health (NIH) grants NS-11766 and HD-32062, by grants from the Muscular Dystrophy Association, and by the Marriott Mitochondrial Disorder Clinical Research Fund. L.C.L. is a postdoctoral fellow of the Ministerio de Educacion y Ciencia, Spain. C.M.Q. is supported by the Muscular Dystrophy Association.
REFERENCES
- 1.Turunen M, Olsson J, Dallner G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta. 2004;1660:171–199. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Bentinger M, Brismar K, Dallner G. The antioxidant role of coenzyme Q. Mitochondrion. 2007;7(Suppl):S41–S50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Aure K, Benoist JF, Ogier de Baulny H, Romero NB, Rigal O, Lombes A. Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology. 2004;63:727–729. doi: 10.1212/01.wnl.0000134607.76780.b2. [DOI] [PubMed] [Google Scholar]
- 4.Boitier E, Degoul F, Desguerre I, Charpentier C, François D, Ponsot G, Diry M, Rustin P, Marsac C. A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J. Neurol. Sci. 1998;156:41–46. doi: 10.1016/s0022-510x(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 5.Di Giovanni S, Mirabella M, Spinazzola A, Crociani P, Silvestri G, Broccolini A, Tonali P, Di Mauro S, Servidei S. Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology. 2001;57:515–518. doi: 10.1212/wnl.57.3.515. [DOI] [PubMed] [Google Scholar]
- 6.Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. U. S. A. 1989;86:2379–2382. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobreira C, Hirano M, Shanske S, Keller RK, Haller RG, Davidson E, Santorelli FM, Miranda AF, Bonilla E, Mojon DS, Barreira AA, King MP, DiMauro S. Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology. 1997;48:1238–1243. doi: 10.1212/wnl.48.5.1238. [DOI] [PubMed] [Google Scholar]
- 8.Rahman S, Hargreaves I, Clayton P, Heales S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatr. 2001;139:456–458. doi: 10.1067/mpd.2001.117575. [DOI] [PubMed] [Google Scholar]
- 9.Rötig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P, Lebideau M, Dallner G, Munnich A, Ernster L, Rustin P. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–395. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 10.Salviati L, Sacconi S, Murer L, Zaccello G, Franceschini L, Laverda AM, Basso G, Quinzii CM, Angelini C, Hirano M, Naini A, Navas P, DiMauro S, Montini G. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology. 2005;65:606–608. doi: 10.1212/01.wnl.0000172859.55579.a7. [DOI] [PubMed] [Google Scholar]
- 11.Artuch R, Brea-Calvo G, Briones P, Aracil A, Galvan M, Espinos C, Corral J, Volpini V, Ribes A, Andreu AL, Palau F, Sanchez-Alcazar JA, Navas P, Pineda M. Cerebellar ataxia with coenzyme Q(10) deficiency: Diagnosis and follow-up after coenzyme Q(10) supplementation. J. Neurol. Sci. 2006;246:153–158. doi: 10.1016/j.jns.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 12.Gironi M, Lamperti C, Nemni R, Moggio M, Comi G, Guerini FR, Ferrante P, Canal N, Naini A, Bresolin N, DiMauro S. Late-onset cerebellar ataxia with hypogonadism and muscle coenzyme Q10 deficiency. Neurology. 2004;62:818–820. doi: 10.1212/01.wnl.0000113719.67643.b7. [DOI] [PubMed] [Google Scholar]
- 13.Lamperti C, Naini A, Hirano M, De Vivo DC, Bertini E, Servidei S, Valeriani M, Lynch D, Banwell B, Berg M, Dubrovsky T, Chiriboga C, Angelini C, Pegoraro E, DiMauro S. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60:1206–1208. doi: 10.1212/01.wnl.0000055089.39373.fc. [DOI] [PubMed] [Google Scholar]
- 14.Musumeci O, Naini A, Slonim AE, Skavin N, Hadjigeorgiou GL, Krawiecki N, Weissman BM, Tsao CY, Mendell JR, Shanske S, De Vivo DC, Hirano M, DiMauro S. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56:849–855. doi: 10.1212/wnl.56.7.849. [DOI] [PubMed] [Google Scholar]
- 15.Horvath R, Schneiderat P, Schoser BG, Gempel K, Neuen-Jacob E, Ploger H, Muller-Hocker J, Pongratz DE, Naini A, DiMauro S, Lochmuller H. Coenzyme Q10 deficiency and isolated myopathy. Neurology. 2006;66:253–255. doi: 10.1212/01.wnl.0000194241.35115.7c. [DOI] [PubMed] [Google Scholar]
- 16.Lalani SR, Vladutiu GD, Plunkett K, Lotze TE, Adesina AM, Scaglia F. Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch. Neurol. 2005;62:317–320. doi: 10.1001/archneur.62.2.317. [DOI] [PubMed] [Google Scholar]
- 17.Lopez LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006;79:1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, Bacq D, de Lonlay P, Munnich A, Rotig A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Invest. 2007;117:765–772. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, DiMauro S, Hirano M. A mutation in parahydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006;78:345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Ber I, Dubourg O, Benoist JF, Jardel C, Mochel F, Koenig M, Brice A, Lombes A, Durr A. Muscle coenzyme Q10 deficiencies in ataxia with oculomotor apraxia 1. Neurology. 2007;68:295–297. doi: 10.1212/01.wnl.0000252366.10731.43. [DOI] [PubMed] [Google Scholar]
- 21.Quinzii CM, Kattah AG, Naini A, Akman HO, Mootha VK, DiMauro S, Hirano M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology. 2005;64:539–541. doi: 10.1212/01.WNL.0000150588.75281.58. [DOI] [PubMed] [Google Scholar]
- 22.DiMauro S, Quinzii CM, Hirano M. Mutations in coenzyme Q10 biosynthetic genes. J. Clin. Invest. 2007;117:587–589. doi: 10.1172/JCI31423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geromel V, Kadhom N, Ceballos-Picot I, Chretien D, Munnich A, Rötig A, Rustin P. Human cultured skin fibroblasts survive profound inherited ubiquinone depletion. Free Radic. Res. 2001;35:11–21. doi: 10.1080/10715760100300551. [DOI] [PubMed] [Google Scholar]
- 24.Lopez-Martin JM, Salviati L, Trevisson E, Montini G, Dimauro S, Quinzii C, Hirano M, Rodriguez-Hernandez A, Cordero MD, Sanchez-Alcazar JA, Santos-Ocana C, Navas P. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007;16:1091–1097. doi: 10.1093/hmg/ddm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manfredi G, Yang L, Gajewski CD, Mattiazzi M. Measurements of ATP in mammalian cells. Methods. 2002;26:317–326. doi: 10.1016/S1046-2023(02)00037-3. [DOI] [PubMed] [Google Scholar]
- 26.Ferraro P, Nicolosi L, Bernardi P, Reichard P, Bianchi V. Mitochondrial deoxynucleotide pool sizes in mouse liver and evidence for a transport mechanism for thymidine monophosphate. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18586–18591. doi: 10.1073/pnas.0609020103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iuso A, Scacco S, Piccoli C, Bellomo F, Petruzzella V, Trentadue R, Minuto M, Ripoli M, Capitanio N, Zeviani M, Papa S. Dysfunctions of cellular oxidative metabolism in patients with mutations in the NDUFS1 and NDUFS4 genes of complex I. J. Biol. Chem. 2006;281:10374–10380. doi: 10.1074/jbc.M513387200. [DOI] [PubMed] [Google Scholar]
- 28.Solans A, Zambrano A, Rodriguez M, Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum. Mol. Genet. 2006;15:3063–3081. doi: 10.1093/hmg/ddl248. [DOI] [PubMed] [Google Scholar]
- 29.Mattiazzi M, Vijayvergiya C, Gajewski CD, DeVivo DC, Lenaz G, Wiedmann M, Manfredi G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 2004;13:869–879. doi: 10.1093/hmg/ddh103. [DOI] [PubMed] [Google Scholar]
- 30.Esterbauer H, Cheeseman KH. Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 1990;186:407–421. doi: 10.1016/0076-6879(90)86134-h. [DOI] [PubMed] [Google Scholar]
- 31.Floreani M, Napoli E, Martinuzzi A, Pantano G, De Riva V, Trevisan R, Bisetto E, Valente L, Carelli V, Dabbeni-Sala F. Antioxidant defences in cybrids harboring mtDNA mutations associated with Leber's hereditary optic neuropathy. FEBS J. 2005;272:1124–1135. doi: 10.1111/j.1742-4658.2004.04542.x. [DOI] [PubMed] [Google Scholar]
- 32.Lenaz G, Fato R, Formiggini G, Genova ML. The role of coenzyme Q in mitochondrial electron transport. Mitochondrion. 2007;7(Suppl):S8–S33. doi: 10.1016/j.mito.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Turrens JF. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasquali P, Landi L, Cabrini L, Lenaz, G. Effect of ubiquinone extraction on ubiquinol-1 oxidase activity in beef heart mitochondria. J. Bioenerg. Biomembr. 1981;13:141–148. doi: 10.1007/BF00763836. [DOI] [PubMed] [Google Scholar]
- 35.González-Aragón D, Burón MI, López-Lluch G, Herman MD, Gómez-Diáz C, Navas P, Villalba JM. Coenzyme Q and the regulation of intracellular steady-state levels of superoxide in HL-60 cells. Biofactors. 2005;25:31–41. doi: 10.1002/biof.5520250105. [DOI] [PubMed] [Google Scholar]
- 36.Heddi A, Stepien G, Benke PJ, Wallace DC. Coordinate induction of energy gene expression in tissues of mitochondrial disease patients. J. Biol. Chem. 1999;274:22968–22976. doi: 10.1074/jbc.274.33.22968. [DOI] [PubMed] [Google Scholar]
- 37.Soderberg K, Nissinen E, Bakay B, Scheffler IE. The energy charge in wild-type and respiration-deficient Chinese hamster cell mutants. J. Cell. Physiol. 1980;103:169–172. doi: 10.1002/jcp.1041030121. [DOI] [PubMed] [Google Scholar]
- 38.Pallotti F, Baracca A, Hernandez-Rosa E, Walker WF, Solaini G, Lenaz G, Melzi D'Eril GV, Dimauro S, Schon EA, Davidson MM. Biochemical analysis of respiratory function in cybrid cell lines harbouring mitochondrial DNA mutations. Biochem. J. 2004;384:287–293. doi: 10.1042/BJ20040561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 40.Nohl H, Gille L. Lysosomal ROS formation. Redox. Rep. 2005;10:199–205. doi: 10.1179/135100005X70170. [DOI] [PubMed] [Google Scholar]
- 41.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 42.Miwa S, St-Pierre J, Partridge L, Brand MD. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic. Biol. Med. 2003;35:938–948. doi: 10.1016/s0891-5849(03)00464-7. [DOI] [PubMed] [Google Scholar]
- 43.Robinson BH, Petrova-Benedict R, Buncic JR, Wallace DC. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibro-blasts. Biochem. Med. Metab. Biol. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- 44.Smith CV, Jones DP, Guenthner TM, Lash LH, Lauterburg BH. Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicol. Appl. Pharmacol. 1996;140:1–12. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 45.Uchida N, Suzuki K, Saiki R, Kainou T, Tanaka K, Matsuda H, Kawamukai M. Phenotypes of fission yeast defective in ubiquinone production due to disruption of the gene for p-hydroxybenzoate polyprenyl diphosphate transferase. J. Bacteriol. 2000;182:6933–6939. doi: 10.1128/jb.182.24.6933-6939.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baracca A, Sgarbi G, Mattiazzi M, Casalena G, Pagnotta E, Valentino ML, Moggio M, Lenaz G, Carelli V, Solaini G. Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim. Biophys. Acta. 2007;1767:913–919. doi: 10.1016/j.bbabio.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 47.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 48.Navas P, Villalba JM, de Cabo R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion. 2007;7(Suppl):S34–S40. doi: 10.1016/j.mito.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 49.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 50.Hirano M, Kaufmann P, De Vivo DC, Tanji K. Mitochondrial neurology I: encephalopathies. In: DiMauro S, Hirano M, Schon EA, editors. Mitochondrial Medicine. Informa Healthcare; London: 2006. pp. 27–44. [Google Scholar]
- 51.Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer L, Barisoni L, Pastore A, Muda AO, Valente ML, Bertini E, Emma F. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]