

Abstract
I present a personal view of the beginning of two-dimensional gels and unsanctioned proteomics. I was still a young graduate student in the early 1970s when I developed methods for two-dimensional gel electrophoresis that became widely used. Though the method gave us the capacity to do things that had never been done, the value of global enumeration of proteins was not appreciated, and we were still two decades away from the invention of the term proteomics. I describe a period of exploration where, by exercising our new capability, we conducted the first proteomic type expression experiments, and made unforeseen contributions to advances in biology. Detection of single-amino acid substitutions validated genetic selections in cultured cells, and revealed a regulatory system that maintains the accuracy of protein synthesis by assuring an unbiased supply of its substrates. We documented biologic control with a dynamic range >108 fold, and, in a surprising turn, we identified an approach that provided a major breakthrough in recombinant DNA technology, the ability to express cloned sequences in Escherichia coli. The challenge then and now is to use a capability for global analysis to produce new insights into fundamental aspects of biology and to drive substantive technical advances.
Keywords: History, Missence errors, Mutation, Sensitivity, Two-dimensional gels
A few years ago, Angelika Goerg persuaded me to return to my roots, and to participate in the 4th Annual World Congress of the Human Proteome Organization by telling the story of the beginning of two-dimensional gels. Her biggest lever in convincing me was a flattering argument that, because proteomics has its roots in two-dimensional gel electrophoresis, the early history of its development would be of interest to those in the field. A positive reception of the presentation induced me to write this account of my involvement in the beginning of two-dimensional gel electrophoresis and the baby steps of the new field of proteomics.
I was not the person you might have expected to have developed two-dimensional gels, and the environment was not particularly oriented to technology. I was a young graduate student in the Department of Molecular Cellular, and Developmental Biology (MCDB), at the University of Colorado in Boulder. I was not working with anyone with a background in electrophoresis, and had no visions of starting a new field. However, like many novices in science, I had ambitious dreams, and, as a part of my thesis plan, I wanted to develop a new technique that would greatly increase our ability to resolve and detect the proteins being expressed in complex organisms.
I had had some early experience (1968) with electrophoresis as an undergraduate at McGill University, Montreal, where I was working in the laboratory of Barad Mukherjee. I was looking for possible histone changes during meiosis, and, thinking that electrophoresis would be the best way to resolve possible new histone variants, I introduced the technique to the laboratory. Based on literature descriptions and some help from the machine shop, I built a tube gel apparatus and identified procedures suitable for histone separations. This experience influenced my future work by removing any hesitancy to make whatever I needed when it was unavailable.
Upon moving to Boulder Colorado, I began graduate work on subjects distant from electrophoresis. I began by trying to purify a steroid binding protein, hoping that it would prove to be a metazoan transcription factor, which would have been novel at the time. However, the departure of my advisor, Joseph Daniel, initiated changes that would lead back to electrophoresis. A discussion with Jacques Pène introduced me to a possible new system for the study of developmental biology, and I joined his laboratory to initiate studies with the simple and beautiful colonial algae, Volvox (Fig. 1). The rest of Jacques’s lab was studying bacteriophage, but, nonetheless, there was an ideological connection. I set out with the goal of copying, at a much-expanded scale, an experimental approach that was being developed by the bacteriophage people to characterize the contributions of bacteriophage genes to the unfolding program of gene expression in the infectious cycle/life cycle.
Figure 1.

Images of the development of Volvox carteri. The asexual egg or gonidia (A) undergoes cleavage (B–D), producing large reproductive cells on the outside of a shell of numerous small flagellated somatic cells (E) and then a morphogenetic process called inversion (F), which I like to think of as a primitive version of gastrulation, inverts the organism to put its reproductive cells on the inside of the colony. This development normally occurs within a mother colony and (G) depicts mature colonies with developing young on the inside. Note that the adult is ball of about 4000 flagellated somatic cells organized so that flagellar beating of all of the cells produces a concerted force that rotates the colony and drives it forward toward a light source. The reproductive cells and their development occur inside this protective shell.
Shortly after I started graduate school, Laemmli’s modification of SDS gels was introduced [1]. This provided a simple elegant way to monitor expression of bacteriophage T4 genes. Because T4 shut off expression of E. coli host genes, the numerous bacterial proteins were not labeled by radioactive amino acids added after infection. Instead, only the proteins expressed from the simple bacteriophage genome were labeled. Following separation by SDS gel electrophoresis, these bacteriophage proteins were displayed as beautifully distinct bands that were detected by autoradiography of the gel once it had been dried down into a thin sheet [2]. Labeling at different times after infection showed bands emerging according to a stereotyped schedule representing the unfolding of the program of gene expression [3]. The roles that bacteriophage genes played in directing this choreography were revealed by examination of the patterns of proteins synthesized when bacteria were infected with bacteriophage compromised by specific mutations [2]. At the time, this was part of the grand field of molecular biology, but it could also be considered proteomics in miniature. The protein products of the full, albeit small, bacteriophage genome were detected, identified, and examined throughout the life cycle/infectious cycle. I was not working on bacteriophage, but I was impressed with what I saw. Analysis of gene expression by these one-dimensional gels was more powerful than had been achieved by other methods available at the time, and I thought it could go further.
Down the hallway, in Larry Gold’s laboratory, which was studying bacteriophage T4, SDS gels were just being introduced as a way to study protein expression. Because Patricia Zambryski/O’Farrell, my wife at that time, worked in Larry’s laboratory, I had a front seat view of events and I got involved in the technical aspects. There were no kits, indeed there were no suppliers providing the needed components. We designed and built the electrophoresis tanks, cut the glass plates, built gel driers, and developed practical methods for labeling proteins and for autoradiogaphic detection of the proteins labeled by incorporation of radioactive amino acids.
In this environment, I came up with the idea of analyzing the more complex life cycle of Volvox by first isolating mutants and then analyzing how these interrupted the developmental program of gene expression by examining the proteins expressed. To handle the much higher complexity of the organism I wanted to develop a two-dimensional separation system. I calculated that, if a two-dimensional approach achieved the product of the resolution of the individual methods, it would give me the ability to resolve thousands of proteins at a time. This would extend the capabilities of the protein separation approach to complex organisms, presumably allowing me to analyze my Volvox mutations much the way the bacteriophage mutants had been studied. I isolated numerous developmental mutants in Volvox based on simple morphological screens, but the technological advance of the two-dimensional gels was to steal the show, and that is what I focus on.
The idea of combining two techniques to produce a two-dimensional separation was not a great conceptual leap. Indeed, combining separations in two-dimensions had a long history in thin layer chromatography and in electrophoresis, and one combination of electrophoretic methods for the separation of proteins had already enjoyed wide success in resolving and identifying the ribosomal proteins of E. coli [4]. Nonetheless, development of a generally applicable method that achieves the full potential resolution of two-dimensional gels could prove to be a major advance, and this was my objective. I was blissfully unaware that anyone else might try to do the same thing I wanted to do. Of course, there were others [5–14]. To some extent, timing turned out to be on my side, but, rather than uniqueness or priority, it was exceptional resolution and generality that led to the impact of my efforts. The degree of my success was driven by a demanding standard and it required attention to many details. The result was a new capability whose influence was to grow slowly over two decades, eventually changing attitudes about how to study proteins [15, 16]. The “-omic” era that began in 1990’s with the focus on genomic sequencing, introduced a fascination with global analysis that led to the term proteomics and the elevation of a methodological approach to the status of a field. However, back in the 1970’s I was concerned with how to do experiments with the new global approach made possible by two-dimensional gels.
The story of the development of the method, in its details, would be long and dull. During the process, I ended up learning about chemical modifications of proteins, protein denaturation, detergents, and the pKa of an extraordinary range of compounds. However, it was a peculiar combination of optimism and stubbornness that was probably key to the success – no matter the disappointment at the end of a long experiment, I always came in the next day renewed, believing that I knew exactly what it would take to improve things.
Beyond technical details, a number of tactical decisions proved important. Chief among these was the choice of methods to combine to create the two-dimensions. Because any correlation in behavior in the two separations would cause the proteins to bunch together near a diagonal, to achieve maximal advantage of the two-dimensions, I needed two methods that separated proteins according to independent properties. As SDS gels separated proteins extremely well and did so according to molecular weight, I thought that I might do well by combining it with IEF. In IEF, proteins move to a position on pH gradient at which they are isoelectric at which point they stop. The pI and the final position depends on the balance of charged groups in the protein and is independent of size except that the proteins that have the biggest deviation from the mean pI tend to be small, an expected statistical effect due to the small sample of basic and acidic residues in small proteins. I realized that the pI of proteins tend to cluster near the pKa of the major charged aminoacyl side chains because it was at these pHs that a small change in pH would have the largest impact on the charge of a protein. Consequently, I spurned an objective that was common at the time, generation of linear pH gradients, in favor of conditions for isoelectric focusing that would give optimal dispersion of the proteins.
The method of detection was important for reasons that are easily overlooked. The proteins in biological samples differ enormously in abundance and there are only a very small number of proteins that are abundant. Consequently, to detect many proteins, I needed sensitivity sufficient to detect the more numerous low abundance proteins. Although improved protein staining techniques were to be introduced later, the autoradiographic detection of proteins labeled with 14C-amino acids or 35S-methionine, which I adopted from the bacteriophage people, gave an unsurpassed sensitivity. It often seems to me that the decreased popularity of using radioisotopes has led to an emphasis on inferior techniques, and that most modern comparisons of sensitivity do not justly credit the merits of autoradiography. Direct exposure of X-ray film by beta emissions (e.g. 35S or 14C decay) records intensity-differences over three orders of magnitude in a single exposure and with three exposures of a gel, I would typically cover six orders of magnitude. With pre-enrichment, we achieved a sensitivity of one part in 109, documented by reconstruction [17]. Indeed, while people often assume that only abundant proteins are visualized on gels, these detection levels are seldom matched even using modern approaches.
I was very impressed with the universal applicability of SDS gels, and I was hoping to devise a method that would work for all proteins and with essentially any type of sample. Given the extraordinary range of properties and behaviors of proteins, this was a major challenge. SDS, by denaturing proteins and binding to them, in abundance, solubilized virtually all proteins and wiped out most distinctions among proteins other than size. While this was advantageous for solubilization and compatible with size separation, the same reagent would disrupt separations, such as IEF, based on a different property, such as pI. I searched for alternative conditions that might achieve similar solubilization and still be compatible with IEF. In the end, the relatively obvious choice of using urea, nonionic detergents and a reducing agent to denature and solubilize proved effective for all but a few recalcitrant proteins. However, the use of these agents introduced their own problems such as sensitizing the sample to temperature-accelerated protein modification (carbamylation), destabilizing the pH gradient, and complicating the behavior of SDS (due to mixed micelle formation) in the transition to the second dimension. Once recognized, these problems were largely overcome by relatively subtle technical changes.
Finally, as resolution and sensitivity increases, the integrity of the proteins in the sample becomes increasingly important. Proteins are rather sensitive to spontaneous chemical modification. Hydrolytic deamidation of asparagine and glutamine will add negative charge to proteins, as will air oxidation of cysteine to the sulfinic or sulfonic acid derivatives. Furthermore, since there are many sites of possible modification within most proteins and addition of one charge to a tiny fraction of the protein can give a detectable new spot, the method is extraordinarily sensitive to modifications [18]. Indeed, it is so sensitive that even the biological background of misincorporation of amino acids contributes to satellite spots detected on a good gel [19, 20]. At the limits of translational fidelity the likelihood of amino acid misincorporation is about 10−4 per residue, and, at this level, a protein with 50 negatively charged residues will lack one of these negative charges ~0.5% of the time, a level that can be detected as a separate spot when resolution and sensitivity are high. Unfortunately, contaminants (probably nucleophiles) are present in many reagents, and I found that treatments that appeared innocuous could lead to accumulation of protein damage when concentrations of reagents were high. With experience, I grew very conservative in the treatment of biological samples, and one of my continuing disappointments is that a substantial fraction of the work appearing in the literature appears to be compromised by microheterogeneity that probably does not have a biological source.
I started experimenting with the introduction of denaturants in IEF gels in January of 1972 and by June, a month before my 23rd birthday, I had developed a credible two-dimensional separation of E. coli proteins (Fig. 2). A picture of this gel was used as a piece of supporting documentation with a grant application to the National Science Foundation (NSF) submitted by four faculty members at MCDB, Larry Gold, David Hirsh, David Prescott, and Ed McConkey. Herman Lewis, the administrative head of Genetic Biology at NSF, recognized the potential of the method. Even though the grant did not fare well, he invited an application for funds to develop the method. With Larry Gold’s encouragement, I wrote a letter outlining plans for technical improvements and development of computational analyses. As a result, Larry Gold and David Hirsh were given a small grant for the development of the method. At the time, I was working in Jacques Pène’s laboratory! These confusing inter-laboratory arrangements where resolved when Jacques left Boulder, and I moved to David Hirsh’s laboratory, rectifying the dichotomy between sponsorship and funding.
Figure 2.

My first two-dimensional gel. Much of the early work in getting the gels working involved development of ways to run good IEF separations in presence of denaturants that would allow me to run whole cell preparation. After fussing with approaches for a few months this is what I obtained in my first attempt at adding the second dimension. The image is an auto-radiogram of E. coli proteins labeled by growth in the presence of 14C labeled amino acids. The isoelectric focusing dimension is on the horizontal axis with the basic side to the left, and the SDS separation displays high-molecular weight proteins toward the top and low toward the bottom. This is the orientation that I have always used, but others have chosen to change it creating a problem of standardization. The separation is compromised by clustering of the proteins in the center of the IEF dimension, by streaking, and by fuzzy spots. I was later able to improve these aspects.
While the switching from lab to lab might seem chaotic, I had excellent interactions with several faculties and with members of multiple laboratories. I thoroughly enjoyed my time both in Jacques’s laboratory, where this all began, and in David’s laboratory, where I completed my degree. Larry Gold and David Hirsh were extremely supportive. They introduced me to diverse scientists, and advocated the method that I developed. Later, they helped immensely in organizing a “Gel Course” that I taught to help disseminate the method.
At the time that I moved to David Hirsh’s laboratory in 1973, I had abandoned the computational effort that we had proposed to NSF. Using computers at that time had little in common with what people are familiar with today. It was extraordinarily cumbersome. Fortran was “THE computer language”. Programs were written using mammoth card punching machines in noisy stuffy rooms, and the resulting stack of cards was handed into a counter at the university computer facility. Two or three days later a ream of paper output would be returned to you detailing innumerable syntax errors in your program. If one ever succeeded in getting a program to run, there was no image output, only text. I did not enjoy the process at all, and I had no confidence that a useful computational process could be developed in a reasonable time frame. My interest was in doing experiments, and, as I turned my efforts in this direction, I realized that, with careful attention, the separations could be made extremely reproducible, that visual inspection reliably picked out meaningful differences, and that, with, a simple 14C standard, the autoradiographic exposures could be meaningfully quantified by visual comparison of spot intensities after different exposure times. It seemed that we could do without the cumbersome computers. With relief and enthusiasm, I launched into experimental analysis.
The method was pretty well developed at this time (Fig. 3), but I pursued experiments for another year during which I introduced additional refinements and developed a method for analysis of the basic proteins that were not included in the original gel system. Even though I had continued to isolate and analyze Volvox mutants, I never analyzed these on the gels. I felt that the testing grounds for the electrophoresis methods ought to be in a more well-defined experimental system, and, therefore, I focused my efforts on questions of gene regulation in E. coli.
Figure 3.
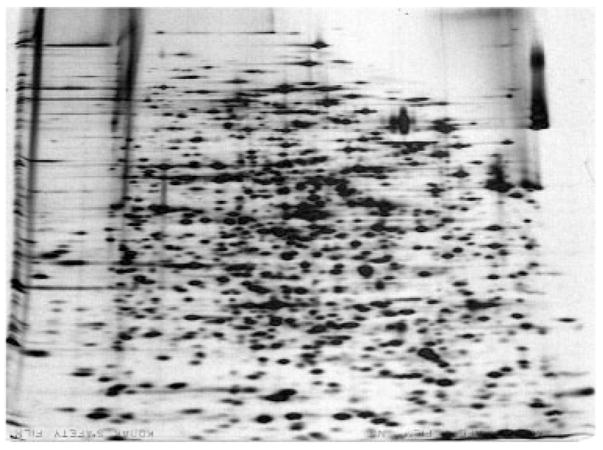
This gel was control (AMP added) from a large set of gels that I ran May 27, 1974 as part of the analysis of cyclic AMP responses in E. coli. The sample was wild-type E. coli K12 (strain 1100) grown in minimal glucose medium and labeled with 14C-amino acids. The gel was prepared and run as described [16] using a 10 to 14% exponential acrylamide gradient in the second dimension. A number of experiments carried out at this time gave reproducibly excellent results.
Identification of experimental questions well suited to the new methods involved new considerations. Everyone was familiar with looking at experimental outcomes in terms of one or at most a few genes, proteins or RNA. The idea of surveying the complete expression profile of an organism became widespread only with the onset of genomics and the ‘omics era two decades later. At the time, the common response upon showing someone an experiment was – how can you deal with, so many spots? While current attitudes make this response appear laughable, there is an underlying issue that is very serious and seldom given sufficient thought. While powerful ‘omics types of methods give a more thorough description of a biological response, I was hoping for more than description. I wanted to define specific experimental issues that could be uniquely addressed, by the new method.
Two-dimensional gels appeared to be particularly well suited to an analysis of a complex biological response were the ability to survey many changes might allow one to address new questions. My interests in developmental biology suggested many complex biological changes that might be analyzed, but I wanted a system compatible with rigorous experimental analysis. The most well known example of pleiotropic regulation at the time was the reconfiguration of the metabolism of E. coli in accord with the availability of its favorite source of energy, glucose. In the process, referred to as catabolite repression, genes involved in catabolism of numerous less favored nutrients are repressed in the presence of glucose. As was famously represented by regulation of the lactose operon, a cyclic AMP receptor protein, Crp, when bound to its activating ligand, cyclic AMP, stimulates transcription of these catabolic genes. Synthesis of cyclic AMP is down regulated by glucose leading to down regulation of “catabolite repressible” genes. Although intensive investigations had established an outline of the mechanism of cyclic AMP regulation of the Lac operon, the number of genes in the genome regulated by Crp was unknown nor was it known whether all catabolite repressible genes were similarly regulated.
Using two-dimensional gels to follow the level of expression of about 1000 proteins, I tested the effects of cyclic AMP, and explored the effects of mutants on this program of gene expression. I was able to show that cyclic AMP has no consequence in the absence of Crp. Thus, all the detected regulatory actions of cyclic AMP are mediated via this one receptor. Because the method surveyed a large representation of the genes in the organism, the observation suggested that all actions of cyclic AMP are likely to be mediated by this receptor protein. I also found that the expression of 10% of the proteins in E. coli is regulated by cyclic AMP/Crp, and that the effects on expression varied from protein to protein, ranging from mild repression to substantial induction along a continuum. This gave us the first view of the range of the response to this regulatory nucleotide. Finally, a mutant, crp−, in the Crp receptor, and a mutant, cya−, defective in cyclic AMP synthesis had almost identical expression patterns in the absence of added cyclic AMP. This identity in the two-dimensional gel patterns of the two mutants in the absence of cyclic AMP shows that Crp has no detectable effect on gene expression in the absence of its ligand. Apparently, all of the detected actions of Crp depend on cyclic AMP [21, 22].
In probing the catabolite repression system, I addressed a new type of question. Of course, following the advent of full genome sequences and design of expression arrays, it became common to ask about the overall features of regulatory networks, but at the time, it was new. Indeed, I needed to develop a language to talk about the results. I called the fraction of the proteome that responded to a specific regulator the ‘domain of response’ for that regulator, and characterized the domain as having a heterogeneous response if different genes responded differently (direction or degree of response) [21, 22]. In many ways, the analyses foreshadowed considerations that are inherent to proteomic work.
These early experiments and the negative response of people to the complexity of the results obtained forced me to face the fact that the method, and proteomics in general, while powerful, are well suited to only specific types of questions. It might seem that two-dimensional gels are well suited to almost any problem based on the generalization that analysis of everything can be used to analyze anything; however, there usually are more efficient methods of assaying specific things. For example, the mechanisms of catabolite repression had been well established by study of lacZ expression. However, I was pleased with the analysis of the responses to cyclic AMP, because, despite the intensity of prior experimental dissection of this response, the two-dimensional gel analysis revealed new global features of the response to cyclic AMP and range of activities of Crp. As highlighted by these observations, two-dimensional gels best exhibit their merits when used to ask new questions that could not be addressed by traditional approaches, and much of the challenge was to define such questions. I believe that the definition of appropriate fundamental experimental goals remains an important challenge that proteomics must meet if it is to have a major influence on scientific developments of the future.
As a student, I also began to analyze the actions of another pleiotropic regulator in E. coli called, guanosine tetraphosphate (ppGpp). At the outset, it seemed that this analysis might be similar in approach and outcome to the analysis of cyclic AMP actions, but it led to a surprising realization and discovery that I am particularly proud of, and it illustrated another experimental strength of two-dimensional gels. When the availability of one amino acid is reduced, protein synthesis is forced to slow down, but I found that the manner in which E. coli achieves this slowdown depends on the activity of a regulatory system. In response to amino acid starvation, stringent strains (rel+) of E. coli synthesize ppGpp. This regulatory nucleotide was known to have a role in reducing the synthesis of ribosomal RNA, which could not be effectively assembled into ribosomes due to reduced synthesis of the protein components during starvation. However, ppGpp has another role. I found that rel− strains E. coli, which lack the ability to accumulate ppGpp in response to amino acid starvation, made errors in translation in which they substituted abundant amino acids in place of the one that was limiting [19]. Furthermore, such cells also showed a severe imbalance in the proteins that they produced: on a relative scale, they greatly over expressed proteins that lacked the amino acid for which they were starved. The translational errors showed up as heterogeneity in the isoelectric points of the proteins that were produced and the nature of the heterogeneity revealed the types of errors made in response to starvation for different amino acids. Amazingly, E. coli strains able to produce ppGpp showed neither the errors in translation nor the imbalance in expression when starved for different amino acids [19].
How could a regulator accommodate starvation for an amino acid and restore faithful translation? Although these analyses continued beyond my time in Boulder, I was able to show that ppGpp inhibited translation, apparently at all codons, while starvation alone resulted in inhibition only at the “hungry” codon. In the absence of ppGpp, the ribosomes would speed through, parts of a message where they were unencumbered by substrate-shortage and pile up at the hungry codon. Ribosomes at the hungry codon would consume the small amount of cognate substrate, thereby severely depleting the already limiting aminoacyl tRNA. In the absence of the correct aminoacyl tRNA, mass action promoted use of the much more abundant alternative aminoacyl tRNA resulting in misincorporation. In contrast, when ppGpp accumulated in proportion to the limitation for a particular amino acid, translation was reduced by generalized inhibition so that rates of protein synthesis would not exceed rates of supply of the limiting component. This arrangement protected translation from gross imbalance in the supply of its immediate substrates, the individual aminoacyl tRNA. Translation of a message can be viewed much like an assembly line where operation depends on the timely provision of all components, because slowing down one step will cause an intolerable pile up. A regulatory system that detects a deficiency in the rate of supply of any one part (amino acid) can provide a feedback signal to regulate the speed of the assembly line in order to avoid disastrous pileups. I was especially pleased, because it appeared that I had arrived at a mechanistic understanding of a fundamental aspect of biology, and it arose form the gels [19]. It was the abundance of information on the gels that revealed the misreading, the distortions in protein expression and their suppression by ppGpp. In this way, the gels identified the unrecognized problem that translation faced in coordinating its rate with substrate supply, and provided the means of identifying the mechanism by which it achieves coordination. It is interesting that amino acid starvation also can induce errors in translation in mammalian cells [20], but mammalian cells lack ppGpp and it is not yet known what measures might operate in higher systems to modulate the assembly line of protein synthesis.
During the course of my experiments with regulation in E. coli, I was also refining the method. The experiments provided a testing ground, since their success relied on the resolution and reproducibility of the separations. However, the experiments had also had a negative effect on dissemination of the method. I was so involved in the experiments, I needed some sort of push to finally decide to write them up. The thing that spurred me to finish and publish was the development of new plans for the future.
In 1973, Gordon Tomkins gave a seminar in Boulder and described work in which his lab had isolated mutants in tissue culture cells that interfered with the ability to respond to either a steroid hormone, glucocorticoid, or to cyclic AMP. I was excited about the possibility of extending the type of proteomics approach I was using in E. coli to mammalian cells. I talked to Gordon about joining his laboratory, and contacted a close friend Bob Ivarie, who had been a student in Jacques Pène’s laboratory and who had taken a postdoctoral position with Gordon. Things moved pretty fast after that. Gordon invited me to his laboratory and I began a long-distance collaboration with Bob Ivarie and a student in Gordon’s lab, Bob Steinberg. They sent me labeled tissue culture cells, uninduced or induced with the glucocorticoid, dexamethasone. In April of 1974, we had the first results showing the response of mammalian cells to hormones and I had working relationships with two collaborators who would influence the work I would do in San Francisco. But first, I needed to graduate.
As I was beginning to write my thesis and a paper describing the two-dimensional gels [15, 16], Larry Gold came up with the idea of organizing a course to teach people the method. As was his wont, he was thinking big and suggested that we invite people from all over the country and by teaching people from, various research centers, we would provide a means to disseminate the method to others in research centers all across the country. We started organizing this, but, realizing that local people would be offended if they were not given an opportunity to learn the procedure, we set up a second course to teach local people. I wrote my thesis and the paper, made 400 copies of preprints, organized and taught the two courses, and defended my thesis in the summer of 1974.
The courses were a huge job. Each ran for 4 days. In each, I took ten students through all aspects of the technique, and I ran two courses in 2 weeks. Thanks largely to Larry’s connections, I found myself, a 25-year-old graduate student, teaching a remarkably prestigious class. For example, Fotis Kafatos was one of my students. Fotis, who was already a well-known professor at Harvard, went on to have a brilliant and influential career including twelve years as Director-General of the European Molecular Biology Laboratory, during which time the laboratories flourished. Bruce Ames, a UC Berkeley faculty member and leader in demonstrating the relationship between the mutagenic and oncogenic potential of chemicals, was also one my attentive students. During these courses, we examined the proteins expressed by everything from clam eggs (donated by Fotis) to rabbit embryos (donated by John Van Blerkom).
During this marathon of activity, I defended my thesis. It-was as much a celebration as a defense, but there was one question that haunts me still. Why had I not written up all the other interesting things I had done? There was a huge level of interest in the electrophoresis method and its potential, and that is all that I wrote up. The findings about cyclic AMP regulation were published much later and only in an abstract and in a review chapter, and studies with Volvox still remain buried in my notebooks. In the rush to move on and probe new questions, I have committed the same act of negligence repeatedly. However, perhaps the cost is smaller than the gain of moving forward to new things. I packed up my Volvox mutants, sent them to one other person working on the topic, submitted the two-dimensional gel paper to the Journal of Biological Chemistry and moved to San Francisco.
Before I arrived in Gordon Tomkins’s laboratory, I had considered what would be needed to get the two-dimensional gels going at a level required to pursue the various projects and had ordered Plexiglas cut to sizes appropriate for gel “boxes”, as we called the apparatus for running the second dimensional slab gels. Keith Yamamoto, a postdoctoral fellow in Gordon’s lab when I arrived, still complains about how I collared not only my new collaborators but also half the lab into helping to build 20 gel boxes and to organize the “gel room”. I soon became familiar with postdoctoral fellows in laboratories throughout the Department of Biochemistry and enjoyed an engaged and vigorous scientific community with whom I shared numerous scientific adventures. However, before we got very far, I got some bad news. The Journal of Biological Chemistry had rejected, my submission of the two-dimensional gel paper [23]. In a total of five sentences of assessment, two reviewers dismissed the accomplishment, arguing that the technique was “extrapolated, in terms of usefulness, far beyond what the author has any reason to expect”. Now what?
I was somewhat taken aback. Two members of the editorial board of the J. Biol. Chem. had been among my students at the gel course. I had already sent out more than a hundred preprints and the J. Biol. Chem. seemed like the logical place to publish the work because the journal was widely available and the articles comprehensive. Fortunately, with some help from Bill Rutter, the department chair and an editorial board member, and Bruce Ames, one of my “students” who was also a board, member, the journal offices were convinced to submit the paper for re-review. A new round of reviews, while still critical, did support its publication, unfortunately only after I eliminated discussion of applications that presaged many useful approaches such as fingerprinting based on separation of partial proteolytic products [15], an approach later developed in detail and introduced by Don Cleveland for one-dimensional gels [24]. In any case, the paper finally came out in May of 1975 [16].
The tribulations of publication did little to disrupt the exciting introduction to the research environment at UCSF; however, tragedy was to mar later events. About 10 months after my arrival, Gordon Tomkins underwent surgery for an acoustic neuroma. Although the surgery involved a sophisticated procedure that would not compromise Gordon’s hearing, something that was especially important to Gordon who was a talented musician, neither the neuroma, nor the surgery was supposed to be life threatening. We were all greatly relieved by the first reports of the success of the surgery. However, a small amount of intracranial bleeding that was not detected during recovery caused brain damage and Gordon never recovered consciousness. It was a shock, and a loss of someone we held dear. It robbed us of his scintillating personality and his intellectual input. It also disrupted the scientific world of everyone in his laboratory. It was a large laboratory with, nearly 20 postdoctoral fellows. Fortunately, funding of the laboratory continued for a while, and, over the next year and half, everyone found new alternatives.
Since I had just begun several projects and established several new scientific interactions, I was interested in continuing, which is just what I did. About the time that most of Gordon’s laboratory had disbanded, I learned that the department had recruited Bruce Alberts from Princeton. I had heard a great deal about Bruce and had met him a few years before, when I was still in Boulder. I thought he would be a great advisor and was thrilled when he agreed to have me join his laboratory upon his arrival at UCSF. At first, the change had little impact on me, because Bruce’s lab moved in around me and I continued ongoing projects in the same place. However, Bruce was to prove the most influential scientist in my career. I found that I could approach him about almost anything and it would inevitably trigger a wide-ranging discussion with plenty of intellectually challenging ideas. He had a habit of listening to observations and responding by detailing a molecular model that always seemed to highlight the important questions that needed to be answered. Our interactions were to grow along with a lasting friendship. However, the work relevant to two-dimensional gels and proteomics preceded this. From late 1974 to 1977 I pursued analyses that I had initiated in graduate school, or new projects that were developed in collaboration with postdoctoral fellows several of whom had been associated with Gordon Tomkins’s laboratory. Fortunately, despite the turmoil that followed Gordon’s death, the interactive environment of the Department of Biochemistry, and the pool of talented postdoctoral fellows continued to provide a pleasant and scientifically fertile environment.
Bob Ivarie and I pursued the analysis of the changes in protein expression in response to glucocorticoid hormone [25]. Using a system that had been developed in Gordon Tomkins’s Lab, we examined the response of a rat hepatoma cell line, HTC cells, to the administration of glucocorticoid hormone. Despite the widespread metabolic effects of glucocorticoids, we found that expression of less than 1% of the detected proteins was increased. Analysis of different cells lines revealed a similarly restricted domain of response. The response of mammalian cells to steroids was considerably less pleiotropic than the response of E. coli to cyclic AMP. Nonetheless, there were more complexities to take into account. The domain of response of mammalian cells to glucocorticoids was cell type specific. This cell type specificity exhibited itself in different ways. Most simply, if a particular cell type did not express a protein that was inducible in other cells, it would not be included in the domain of response of that cell. However, some proteins appeared inducible in some cell types and not in others, suggesting that inducibility could be modulated. It was difficult to describe this plastic domain of response. Our use of two-dimensional gels to define the domain of response of cells to a condition or treatment foreshadowed global gene expression studies that gained immense popularity decades later when genome sequences became available and arrays were developed [22, 25]. Although preceded by the prokaryotic work. [26], this analysis of glucocorticoid induction [25] was the first prominent publication of what would become a classical type of “proteomic study”.
In another series of analyses, we took advantage of the resolution of two-dimensional gels to look at mutations and protein modification. Bob Steinberg graduated, but stayed in San Francisco studying cyclic AMP response in tissue culture cells as a fellow with Philip Coffino. They were doing very interesting work isolating mutants defective in the response to cyclic AMP, but at that time, there were a number of uncertainties about what type of change was responsible for “mutations” selected in tissue culture. There was considerable skepticism that “real mutants” involving a change in the structure of the coding sequence would be isolated by selecting for defects in tissue culture cells. There were several reasons for this skepticism. For example, the cells are diploid, and mutation of both copies to give a homozygous defect seemed overwhelmingly improbable. Furthermore, because many cell lines exhibited plasticity in behavior, it was proposed that epigenetic changes might be common and that these could alter expression of genes to give rise to phenotypes. I had previously shown that the two-dimensional gels could detect a missense mutation in a T4 protein, the helix destabilizing protein p32 [15, 16] and a bacterial protein, MetE (unpublished). Hoping to directly detect mutant protein in mammalian cells, we used two-dimensional gels to examine the regulatory subunit of the cyclic AMP stimulated protein kinase (PKA-RS) from cells that were wild type or defective in their ability to respond to cyclic AMP. We first identified the spot corresponding to the wild-type protein and developed a one-step affinity chromatography procedure to enrich it before the two-dimensional gel analysis. We showed that the PKA-RS gave rise to two spots of similar size, where the more acidic spot was labeled with 32P showing that it is phosphorylated. Several of the mutants produced PKA-RS that was shifted in the isoelectric focusing dimension. The shifted spots came in both phosphorylated and unphosphorylated forms and were found in addition to the wild-type spots. We developed an approach based on chemical modification of proteins (carbamylation of lysine) to calibrate the amount of the shift caused by unitary changes in charge. In this way were able to show that the shift due to phosphorylation was compatible with a single phosphate (slightly more than single charge change because of the degree of ionization) and that the shift in some of the mutants was consistent with the gain of a partially ionized carboxyl group as would be expected for a missence change that introduced an aspartate or glutamate in place of a neutral amino acid. Another mutation shifted the charge toward the basic side as if it transformed an acidic residue to a basic one. This detailed analysis showed that the gel system is capable of resolving subtle distinctions in charge and provided compelling evidence that structural gene mutations were isolated by selecting for the failure to respond to cyclic AMP. The wild-type protein clearly persisted in the mutant lines. The dominance of the mutant form could be explained by reduced responsiveness to ligand, such that the mutant regulatory proteins continued to repress the kinase in the presence of cyclic AMP [26].
Early during the development of the two-dimensional gel electrophoresis method, I recognized that I was losing the very basic proteins due to instability of the basic end of the pH gradient. I was never able to fully solve this instability, but, while still in graduate school, I tried a simple approach to circumvent it. Using mammalian ribosomal proteins as a test sample enriched in basic proteins, I loaded the sample on the acidic rather the basic end so that the basic proteins would have to run all the way through the gels. I then ran the “isoelectric focusing” gels for different amounts of time. As long as I did not run the gels for too long the basic proteins were caught before running off the first dimensional gels and they ran well in the second dimension. While this was successful, I did not include a description of this method in the original paper in part because I did not think its development was complete. In addition, I thought the method would be viewed with suspicion, because it really was not IEF. While pragmatically successful, the parameters that might be involved in the migration of the proteins were likely to very complex. Indeed, I did not even know what to call it -electrophoresis, isotachophoresis or isoelectric focusing, I decided to defer a more thorough analysis. Once in San Francisco, my wife, Patricia Zambryski O’Farrell, who was then a postdoctoral fellow in the laboratory of Howard Goodman, grew concerned about detection of more basic proteins because some of the viral proteins she was studying appeared to be lost [27]. She undertook an optimization of the method I had tried. In the end, none of the modifications she tried were an improvement but, in the process, we characterized the method, which proved flexible, reproducible and appeared to encompass virtually all proteins in an extract. Additionally, the separations of the more acidic proteins could be nicely related to their positions on the standard two-dimensional gel. The method provided a pragmatic solution to the difficulty of including all proteins on an equilibrium IEF gel. We opted for a descriptive name for the procedure, nonequilibrium pH gradient gel electrophoresis (NEPHGE). The paper describing NEPHGE was rapidly accepted for publication in Cell without any hints of the criticisms directed at the original two-dimensional gel paper, perhaps reflecting the conservative nature of scientific review [28]. The introduction of immobilized pH gradients has now largely overcome the stability problem that compromised analysis of basic proteins, and the increased ability to manipulate the pH range of separation has improved resolution. Despite these advances, NEPHGE continued to offer a simple approach for broad-spectrum analyses.
Well after Gordon’s lab had disbanded, Bob Ivarie and I initiated another collaboration that really stretched the sensitivity with which proteins could be detected on two-dimensional gels. We were curious about the range of biological regulation. Many of the regulatory systems known in E. coli reduce expression of targeted genes about 100-fold when the gene is “shut off”. Regulation of the Lac operon has an unusually large dynamic range in that there is somewhat more than a 1000-fold difference between induced and repressed states. In mammals, cell types produce particular characteristic proteins appropriate for their specialized function, and background expression of these cell type specific proteins in inappropriate tissues is low – but how low? Is the range of regulation of gene expression in mammals different from that in E. coli? Often specialized cell types produce their differentiated gene products in abundance. For example, about 30% of the protein in the somatotrophs of the anterior pituitary gland can be growth hormone, and a pituitary-derived rat cell line, GH3, expresses about 1% of its protein as growth hormone. We compared this cell line to a rat hepatoma (liver) cell line, asking whether we could detect inappropriate growth hormone synthesis in the “liver” cells. We used 35S-methioinine to label the cell lines. To achieve high sensitivity we began with 10 000 times as much incorporated isotope as we were using in a more typical analysis. After preparing an extract, we enriched any potential growth hormone (>1000 purification estimated by reconstruction) by immunoprecipitation, and analyzed everything that appeared in the immunoprecipitate by two-dimensional gels, which we exposed, to film for a very long time. We could not detect any growth hormone expression in the liver cell line. Importantly, using a reconstruction experiment as well as standard curves, we demonstrated that we had enough sensitivity to detect growth hormone if it were expressed at a level of one part in 109 [17], We combined this two-dimensional gel analysis of protein levels with an analysis of RNA levels to conclude that cell type specific control of gene expression resulted in regulation over about eight orders of magnitude or more [17]. This measure suggests that developmental regulation, at least in this context, operates over a much larger range than known modes of suppression of gene expression in E. coli.
While I was doing these experiments, a revolution was happening around me. UCSF was a major home of the beginning of recombinant DNA technology and the laboratories of several faculty members, Herb Boyer, the founder of Genentech, Bill Rutter, the founder of Chiron, and Howard Goodman, vigorously pursued the technological advances and their application. The atmosphere was thrilling. Like those around me, I began thinking in new directions. The first contribution I made to this new field was somewhat by chance, but it turned out to be major. It was directly due to the resolution of two-dimensional gels. I had been communicating with three other postdoctoral fellows, David Gelfand who become famous for his contributions to the development of PCR, Barry Polisky, and Bob Bishop, about the possibility of expressing cloned DNA as proteins in E. coli, and I got involved in the work when they wanted to detect products. The first step had been to clone the gene for E. coli β-galactosidase, lacZ, into a. plasmid. Tests for β-galactosidase activity suggested that they had succeeded, but in the absence of the now abundant sequence information, it was not clear exactly what had been cloned. The β-galactosidase produced by the plasmid baring cells appeared normal on one dimensional SDS gels, but when I examined the protein produced by the clone on two-dimensional gels, I found it was very slightly different from wild-type β-galactosidase. Based on some available peptide sequences and bit of deduction from the gels, we determined that the EcoR1 site that was used in the cloning of the gene lay 16 amino acids from the end of the gene so that the cloned gene was slightly truncated and the proteins produced where consequently fusion proteins reading into whatever sequence was cloned to the downstream side of the truncated lacZ. By chance, in the original clone, addition of a few residues encoded by downstream plasmid sequences produced a protein with an SDS gel mobility matching that of normal full-length β-galactosidase. We showed that insertion of new sequences into the EcoR1 site near the end of lacZ led to their expression as a fusion with the near full-length β-galactosidase, each new fusion giving distinctive products often much larger than β-galactosidase [29]. With the easily obtained sequencing information available today, this analysis might not seem so significant, but at the time, no one had ever built transgenes for expression and DNA sequencing was still a developing and very limited art for the especially skilled. The expression system that we defined provided the first expression of foreign DNA in E. coli and Genentech researchers adopted this approach in a hallmark study in which they produced somatostatin in E. coli [30–32].
As summarized briefly in this account, my colleagues and I explored many of the applications of two-dimensional gels in San Francisco during the seventies, but this period was also one where I learned many new things that drew me in directions unrelated to gels and proteomics. I started with an interest in developmental biology and I was still looking for technologies that take me in a direction that might uncover the mechanisms by which embryos assemble pattern. The rapidly advancing recombinant DNA techniques looked as if they might provide such an avenue, particularly if an approach could be found to identify and clone developmental regulators. It is hard to think, back to days when no developmental regulators were known, but that was where we were in late 1970s. It was the introduction of reverse genetics and the cloning of what were, at the time, obscure mutations in Drosophila that was to lead to the explosive identification of genes guiding development, and that is what I turned to. I was offered a position at UCSF and began my laboratory by embarking on the cloning of homeotic genes of Drosophila. I was inexperienced in the new endeavors, but since these were new approaches, I was not much behind anyone else. It proved practical and productive [33–35], and the 1980s saw remarkable advances in the understanding of embryonic development. From there, I have gone onto yet other new things, but I always look back with fondness on my days as a scientific youngster, when, with a little boldness and a lot of persistence I changed the standard for what could be achieved by electrophoresis of proteins.
Acknowledgments
I would like to acknowledge the numerous faculties in Boulder Colorado and at UCSF for encouraging the open research environment that fertilized numerous collaborations that drove this work.
Abbreviations
- AMP
adenosine monophosphate
- MCDB
Department of Molecular Cellular and Developmental Biology
- NSF
Nation Science Foundation
- ppGpp
guanosine tetraphosphate
- UC
University of California
- UCSF
University of California at San Francisco
Biography

Biographical material: Pat O’Farrell has been on the faculty at UCSF since 1979. After the described work on the development and early applications of two-dimensional gel electrophoresis, he investigated embryonic pattern formation in Drosophila, the developmental control of cell cycle, and, more recently, biological roles of nitric oxide in innate immunity and hypoxia.
Footnotes
The author has declared no conflict of interest
References
- 1.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 2.O’Farrell PZ, Gold LM, Huang WM. The identification of prereplicative bacteriophage T4 proteins. J Biol Chem. 1973;248:5499–5501. [PubMed] [Google Scholar]
- 3.O’Farrell PZ, Gold LM. Transcription and translation of prereplicative bacteriophage T4 genes. in vitro J Biol Chem. 1973;248:5512–5519. [PubMed] [Google Scholar]
- 4.Kaltschmidt E, Wittmann HG. Ribosomal proteins. VII. Two-dimensional polyacrylamide gel electrophoresis for fingerprinting of ribosomal proteins. Anal Biochem. 1970;36:401–412. doi: 10.1016/0003-2697(70)90376-3. [DOI] [PubMed] [Google Scholar]
- 5.Raymond S, Aurell B. Two-dimensional gel electrophoresis. Science. 1962;138:152–153. doi: 10.1126/science.138.3537.152. [DOI] [PubMed] [Google Scholar]
- 6.Dale G, Latner AL. Isoelectric focusing of serum proteins in acrylamide gels followed by electrophoresis. Clin Chim Acta; Int J Clin Chem. 1969;24:61–68. doi: 10.1016/0009-8981(69)90141-7. [DOI] [PubMed] [Google Scholar]
- 7.Macko V, Stegemann H. Mapping of potato proteins by combined electrofocusing and electrophoresis identification of varieties. Hoppe-Seyler’s Zeitschrift fur physiologische Chemie. 1969;350:917–919. [PubMed] [Google Scholar]
- 8.Wright GL., Jr Separation of proteins in concentrated cerebrospinal fluid by two-dimensional electrophoresis in acrylamide gel. Clin Chem. 1971;17:430–432. [PubMed] [Google Scholar]
- 9.Orrick LR, Olson MO, Busch H. Comparison of nucleolar proteins of normal rat liver and Novikoff hepatoma ascites cells by two-dimensional polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1973;70:1316–1320. doi: 10.1073/pnas.70.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howard GA, Traut RR. A modified two-dimensional gel system for the separation and radioautography of microgram amounts of ribosomal proteins. Methods Enzymol. 1974;30:526–539. doi: 10.1016/0076-6879(74)30052-3. [DOI] [PubMed] [Google Scholar]
- 11.MacGillivray AJ, Rickwood D. The heterogeneity of mouse-chromatin nonhistone proteins as evidenced by two-dimensional polyacrylamide-gel electrophoresis and ion-exchange chromatography. Eur J Biochem/FEBS. 1974;41:181–190. doi: 10.1111/j.1432-1033.1974.tb03258.x. [DOI] [PubMed] [Google Scholar]
- 12.Scheele GA. Two-dimensional gel analysis of soluble proteins. Charaterization of guinea pig exocrine pancreatic proteins. J Biol Chem. 1975;250:5375–5385. [PubMed] [Google Scholar]
- 13.Klose J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Human-genetik. 1975;26:231–243. doi: 10.1007/BF00281458. [DOI] [PubMed] [Google Scholar]
- 14.Anderson L, Anderson NG. High resolution two-dimensional electrophoresis of human plasma proteins. Proc Natl Acad Sci USA. 1977;74:5421–5425. doi: 10.1073/pnas.74.12.5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Farrell PH. High resolution two-dimensional electrophoresis. Thesis. 1974:68. [Google Scholar]
- 16.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 17.Ivarie RD, Schacter BS, O’Farrell PH. The level of expression of the rat growth hormone gene in liver tumor cells is at least eight orders of magnitude less than that in anterior pituitary cells. MoL Ceil Bioi. 1983;8:1460–1467. doi: 10.1128/mcb.3.8.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Farrell PH, O’Farrell PZ. Two-dimensional polyacrylamide gel electrophoretic fractionation. Methods Cell Biol. 1977;16:407–420. doi: 10.1016/s0091-679x(08)60116-8. [DOI] [PubMed] [Google Scholar]
- 19.O’Farrell PH. The suppression of defective translation by ppGpp and its role in the stringent response. Cell. 1978;14:545–557. doi: 10.1016/0092-8674(78)90241-6. [DOI] [PubMed] [Google Scholar]
- 20.Parker J, Pollard JW, Friesen JD, Stanners CP. Stuttering: high-level mistranslation in animal and bacterial cells. Proc Natl Acad Sci USA. 1978;75:1091–1095. doi: 10.1073/pnas.75.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Farrell PH. Proceedings: Catabolite repression as a pleiotropic response. J Biochem. 1976;79:33P. [PubMed] [Google Scholar]
- 22.O’Farrell PH, Ivarie RD. The glucocorticoid domain of response: measurement of pleiotropic cellular responses by two-dimensional gel electrophoresis. Monographs Endocrinol. 1979;12:189–201. doi: 10.1007/978-3-642-81265-1_10. [DOI] [PubMed] [Google Scholar]
- 23.O’Farrell PH. This weeks citation classic. Current contents. 1982;51:18. [Google Scholar]
- 24.Cleveland DW, Fischer SG, Kirschner MW, Laemmli UK. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J Biol Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 25.Ivarie RD, O’Farrell PH. The glucocorticoid domain: steroid-mediated changes in the rate of synthesis of rat hepatoma proteins. Cell. 1978;13:41–55. doi: 10.1016/0092-8674(78)90136-8. [DOI] [PubMed] [Google Scholar]
- 26.Steinberg RA, O’Farrell PH, Friedrich U, Coffino P. Mutations causing charge alterations in regulatory subunits of the cAMP-dependent protein kinase of cultured S49 lymphoma cells. Cell. 1977;10:381–391. doi: 10.1016/0092-8674(77)90025-3. [DOI] [PubMed] [Google Scholar]
- 27.Crawford LV, O’Farrell PZ. Effect of alkylation on the physical properties of simian virus 40 T-antigen species. J Virol. 1979;29:587–596. doi: 10.1128/jvi.29.2.587-596.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Farrell PZ, Goodman HM, O’Farrell PH. High resolution two-dimensional electrophoresis of basic as well as acidic proteins. Cell. 1977;12:1133–1141. doi: 10.1016/0092-8674(77)90176-3. [DOI] [PubMed] [Google Scholar]
- 29.O’Farrell P, Polisky B, Gelfand DH. Regulated expression by readthrough translation from a plasmid-encoded beta-galactosidase. J Bacteriol. 1978;134:645–654. doi: 10.1128/jb.134.2.645-654.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itakura K, Hirose T, Crea R, Riggs AD, et al. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science. 1977;198:1056–1063. doi: 10.1126/science.412251. [DOI] [PubMed] [Google Scholar]
- 31.Andrews EL. The New York Times. 1988. Biotechnology dispute is rekindled; p. 2. [Google Scholar]
- 32.Rich, IN RE PATRICK H. O’FARRELL, BARRY A. POLISKY and DAVID H. GELFAND. No. 87–1486: Decision of the UNITED STATES COURT OF APPEALS FOR THE FEDERAL CIRCUIT Fed. Cir. 1988, 853 F.2d 894, 1–9.
- 33.Kuner JM, Nakanishi M, Ali Z, Drees B, et al. Molecular cloning of engrailed: a gene involved in the development of pattern in Drosophila melanogaster. Cell. 1985;42:309–316. doi: 10.1016/s0092-8674(85)80126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desplan C, Theis J, O’Farrell PH. The Drosophila developmental gene, engrailed, encodes a sequence-specific DNA binding activity. Nature. 1985;318:630–635. doi: 10.1038/318630a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiNardo S, Kuner JM, Theis J, O’Farrell PH. Development of embryonic pattern in D. melanogaster as revealed by accumulation of the nuclear engrailed protein. Cell. 1985;43:59–69. doi: 10.1016/0092-8674(85)90012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]