

Abstract
Background
Four voltage-gated potassium currents: Ito,f (KV4.2), Ito,s (KV1.4), IK,slow (KV1.5+KV2.1), and ISS (TASK1) govern murine ventricular repolarization. Although the accessory subunit, KChIP2, influences Ito,f expression, in preliminary experiments we found that action potential duration (APD) is maintained in KChIP2 knockout mice.
Objective
We tested the role of KChIP2 in regulating APD and studied the underlying ionic currents.
Methods
We used microelectrode techniques, whole-cell patch clamp studies, and real-time PCR amplification to characterize ventricular repolarization and its determinants in WT and KChIP2-/- mice.
Results
Despite comparable baseline action potentials, APD was more markedly prolonged by 4-aminopyridine (4-AP) in KChIP2-/- preparations. Peak K+ current densities were similar in WT and KChIP2-/- cells (mean±sem IP: 28.3±2 (n=27) vs. 29.2±2 pA/pF (n=24), respectively; P>0.05). Heteropodatoxin-2 (HpTx-2, 1 μM) had no effect on current amplitude in KChIP2-/- myocytes. The current fractions sensitive to 4-AP (50 μM and 1 mM) were larger in KChIP2-/- than WT (P<0.05). Real-time PCR demonstrated absence of KChIP2 and increased KV1.5 expression in KChIP2-/- ventricular myocardium.
Conclusion
KChIP2 deficiency eliminated HpTx-2-sensitive Ito,f, but had little impact on total APD, secondary to upregulation of 4-AP-sensitive IK,slow in association with increased KV1.5 expression. There is increased sensitivity to 4-AP-mediated APD prolongation in KChIP2-/-. Thus, KChIP2 appears important for murine repolarization in circumstances of reduced repolarization reserve.
Keywords: Potassium currents, action potentials, electrophysiology, animal models, repolarization
Introduction
Voltage-gated potassium (KV) currents are important determinants of mammalian myocardial repolarization that are differently regulated in a variety of cardiovascular pathologies.1-4 In mice, at least 4 pharmacologically, molecularly and kinetically distinct KV currents have been identified5, 6: Ito,f (encoded by KV4.2 and KV4.37), Ito,s (KV1.47), IK,slow (KV1.58 and KV2.19), and ISS (possibly involving the K2P channel TASK110). In contrast to larger animals, KV4.3 is not required for functional Ito,f in the mouse.11
The KV gene family encodes pore-forming α subunits, four of which comprise the functional channel. A number of accessory subunits contribute to the physiologically native current.12 Of the KV channel-interacting proteins (KChIPs) which primarily bind to the cytoplasmic N-terminal domain of the KV4 α subunits13, 14, only KChIP2 is expressed in heart.15-17 In mammalian cell lines, KChIP2 co-expression increases KV4-encoded current density, slows inactivation, and accelerates recovery from inactivation,15, 16 and also modulates KV1.5 expression.18
In human subjects, heart failure is associated with decreased Ito19, explained by decreased KV4.3 and KChIP2 mRNA and protein levels.20, 21 A mutation in the pore region22 or deletion of the pore-forming segment23 of KV4.2 produce dominant-negative subunits that reduce Ito,f and prolong action potential duration (APD) of murine myocytes. The pore deletion is associated with ventricular hypertrophy that progresses to heart failure, whereas the pore mutation does not lead to functional or structural dysfunction.22, 23 Deleting the entire KV4.2 gene completely abolishes Ito,f; however neither APD prolongation nor hypertrophy is observed.24
Although KChIP2 knockout reportedly leads to loss of Ito in murine cardiomyocytes25, our preliminary action potential recordings from intact ventricles were comparable to those in wild-type (WT) mice. Therefore, we designed the present studies to explore the KV-subtype specific consequences of KChIP2 deficiency. Our results demonstrate that while Ito,f is eliminated, total KV-current density remains unaltered. This is secondary to an upregulated 4-aminopyridine (4-AP) sensitive current, which translates to increased sensitivity to 4-AP-mediated APD prolongation.
Methods
Hearts were excised from anesthetized (0.1 mg/g ketamine, 0.01 mg/g xylazine, IP), adult (10-12 weeks), male C57BL6 WT control and KChIP2-/- mice. Genotyping was performed using DNA isolated from tail samples (Allele Biotech, CA) followed by PCR amplification of KChIP2 and neomycin. All experiments were approved by the Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health).
Transmembrane action-potential recordings
Multicellular left- (LV) or right- (RV) ventricular free wall preparations were superfused with oxygenated Tyrode's solution containing (mM): NaCl 131; NaHCO3 18; KCl 4; CaCl2 1.8; MgCl2 0.5; NaH2PO4 1.8; dextrose 5.5 (pH 7.4; T 37°C). High resistance (>30 MΩ) microelectrodes filled with 3M KCl were used to record (sub)epicardial action potentials from preparations paced at 500 ms cycle length via bipolar surface electrodes. After 2h equilibration, we recorded action potentials from 9-12 sites per preparation. Recordings were repeated 30 min after adding 1 mM 4-AP to the superfusate.
Patch-clamp recordings
Myocytes were disaggregated from the entire LV free wall by enzymatic dissociation and mechanical dispersion. Cells were maintained in enzyme-free solution at room temperature and used within 6h of isolation. Recording pipettes (1-2 MΩ) contained (mM): KCl 135; EGTA 10; HEPES 10; and glucose 5 (pH 7.2). The superfusate contained (mM): NaCl 136; KCl 4; MgCl2 2; CaCl2 1; CoCl2 5; tetrodotoxin (TTX; Sigma, St. Louis, MO) 0.01, HEPES 10; glucose 10 (pH 7.4; T 24°C). Cell capacitances were 184±10 pF (WT) versus 172±9 pF (KChIP2-/-; P>0.05). Series resistance was compensated electronically by 80-90%; time-constants of the compensated capacitance decay were comparable in WT and KChIP2-/- myocytes (393±26 versus 412±22 μs, respectively; P>0.05). Leak currents were <100 pA and were not corrected. 4-AP (50 μM and 1 mM5; Sigma) and heteropoda toxin 2 (HpTx-2; 1 μM5, 26; Sigma) were dissolved in water and added to the superfusate immediately before use.
Quantitative real-time PCR analysis
First strand cDNA was synthesized from 1 μg total mRNA isolated from WT and KChIP2-/- hearts. KV1.4, KV1.5, KV2.1, KV4.2, KV4.3, KChIP2, and Cyclophilin A expression was determined by quantitative real-time PCR (Roche, Basel, Switzerland). Amplicons were controlled by gel electrophoresis and sequencing. cDNA synthesis without reverse transcriptase served as negative control and showed no amplification. Results are presented as KChIP2-/- normalized to WT levels. Samples were tested in duplicate.
Data analysis
Peak outward currents (IP) were defined as maximal outward K+ current from 0 pA; IK1 amplitudes were measured at peak inward current and at the end of the 400 ms voltage step. The decay phase of the total KV current elicited by 4.5-s depolarization in LV free wall myocytes can be described by the sum of 2 exponential components, where the fast and slow τ are attributed to Ito,f and IK,slow, respectively.6, 24 Ito,s is found exclusively in cells from the interventricular septum. The steady-state current, ISS, does not inactivate and is described by a time-independent, constant current.7 Only fits with a correlation coefficient ≥98% were used. For each cell, current amplitudes are normalized to the whole-cell membrane capacitance, and current densities reported.
Data are presented as mean±SEM. Student's t test (when comparing 2 data sets) or 2-way ANOVA, followed by Bonferroni's test, when appropriate, were used to test differences statistically. P<0.05 was considered significant.
Results
Exaggerated APD prolongation by 4-AP in KChIP2-/- ventricles
Maximum diastolic potential, action potential amplitude and Vmax were comparable between WT and KChIP2-/- groups (Table). WT and KChIP2-/- APD were similar at control and prolonged significantly after exposure to 1 mM 4-AP (Table, Figure 1). However, 4-AP induced more marked APD prolongation in KChIP2-/- LV and RV preparations. In RV, 4-AP effects were greater in KChIP2-/- than WT (P<0.05; Table). Thus, the altered repolarizing currents in KChIP2-/- mice manifest as APD changes only in the presence of 4-AP; baseline APDs are equivalent to WT. This prompted us to characterize the voltage-gated potassium currents in disaggregated myocytes from WT and KChIP2-/- mice.
Table.
Action potential characteristics from intact LV and RV preparations.
| LV | n | APA, mV | MDP, mV | Vmax, V/s | APD50, ms | APD90, ms | ||
|---|---|---|---|---|---|---|---|---|
| WT | 4 | 73±3 | -69±2 | 110±27 | 11±1 | 75±10 | ||
| WT, 4-AP | 4 | 76±3 | -69±2 | 107±15 | 20±1* | 77% | 84±3* | 13% |
| KChIP2-/- | 3 | 70±3 | -67±2 | 87±11 | 10±1 | 70±4 | ||
| KChIP2-/-, 4-AP | 3 | 74±6 | -67±1 | 93±5 | 21±1* | 115% | 84±2* | 21% |
| RV | n | APA, mV | MDP, mV | Vmax, V/s | APD50, ms | APD90, ms | ||
| WT | 8 | 88±2 | -77±1 | 215±12 | 4.1±0.3 | 31±3 | ||
| WT, 4-AP | 8 | 87±3 | -73±1 | 196±15 | 8.9±0.8* | 113% | 41±4* | 28% |
| KChIP2-/- | 7 | 87±2 | -77±1 | 192±20 | 3.8±0.3 | 39±4 | ||
| KChIP2-/-, 4-AP | 7 | 93±2 | -74±2 | 160±13 | 13.6±0.9*† | 262% | 53±4*† | 41% |
APA, action potential amplitude; MDP, maximal diastolic potential; Vmax, maximal upstroke velocity; APD50 and APD90, action potential duration to 50 and 90% repolarization. Relative increases in APD induced by 4-AP are indicated in percent. Pacing-cycle length: 500 ms.
, P<0.05 versus baseline.
, P<0.05 versus WT, 4-AP.
Figure 1.

APD prolongation by 4-AP is enhanced in KChIP2-/- ventricles. Representative action potentials recorded from intact WT and KChIP2-/- LV and RV in control and after addition of 1 mM 4-AP.
Comparable peak KV currents in WT and KChIP2-/- ventricular myocytes
Figure 2A shows representative K+ currents recorded from a WT and a KChIP2-/- myocyte. Mean peak current (IP) densities at +40 mV are 28.3±2.0 pA/pF in WT cells (n=27) versus 29.2±1.9 pA/pF in KChIP2-/- cells (n=24; P>0.05). No differences in IP density were seen using 4.5-s depolarizing steps between -40 and +50 mV (VH=-70 mV; Figure 2B). Activation kinetics, measured as the time from depolarization to peak current, were significantly slower in KChIP2-/- compared to WT, particularly at steps to positive voltages (Figure 2C).
Figure 2.
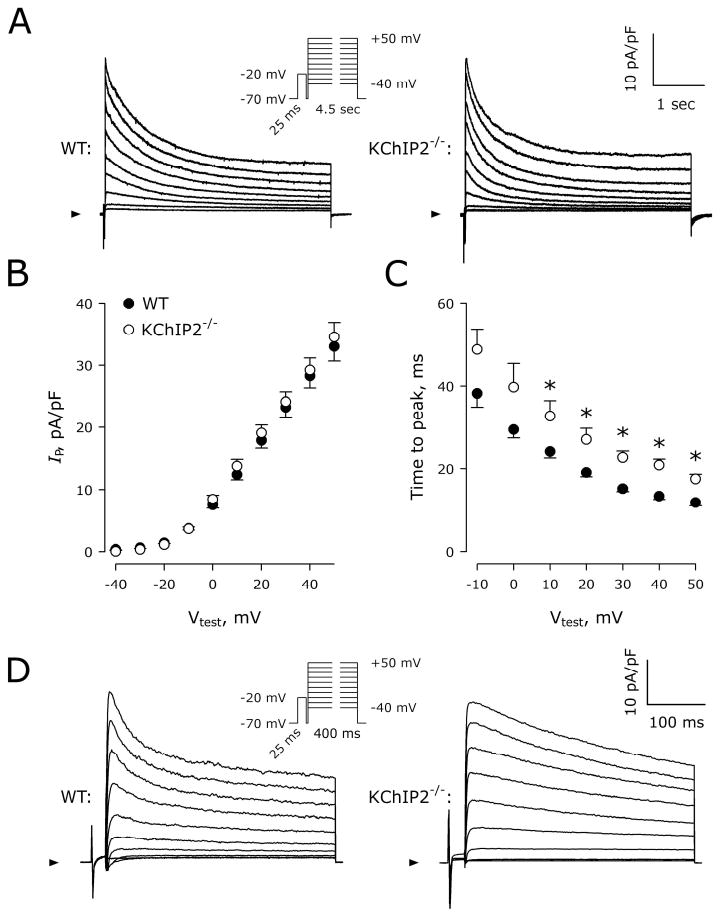
Comparison of KV currents in WT and KChIP2-/- myocytes. A, Representative current traces normalized to cell capacitance recorded from WT and KChIP2-/- cells using 4.5-s depolarizing steps from VH=-70 mV to Vstep=-40 to +50 mV (Inset). Each step was preceded by a 25 ms depolarization to -20 mV to eliminate contamination by inward Na+ current not completely blocked by TTX. Arrowheads indicate 0 pA. B, IP densities plotted as a function of Vtest in WT (n=27) and KChIP2-/- (n=24) myocytes. C, Time interval from cell depolarization to peak current as a function of Vtest. *, P<0.05 versus WT. D, Representative current traces normalized to cell capacitance recorded from a WT and a KChIP2-/- cell using 400-ms depolarizing steps. Although peak current amplitude is comparable, the WT cell displays faster current inactivation within the first ∼100 ms.
Significant differences were found in the double-exponential fit to the 4.5-s decay phase of the KV currents. At Vstep=+40 mV, the fast τ were 198±11 versus 226±19 ms in WT and KChIP2-/- myocytes, respectively (P<0.05). The slow τ were 1486±57 versus 1248±69 ms in WT and KChIP2-/- myocytes, respectively (P<0.05). The peak amplitudes of the fast component (9±2 versus 12±2 pA/pF, P>0.05), slow component (12±1 versus 12±1 pA/pF, P>0.05), and the constant component (6±1 versus 7±1 pA/pF, P>0.05), were all comparable between WT and KChIP2-/- myocytes, respectively. When 400-ms depolarizations were used, a fast inactivating component was clearly visible in WT cells but absent in KChIP2-/- myocytes (Figure 2D). This fast component had a time constant of inactivation of 38±2 ms, which is comparable to the previously reported τ for Ito,f.6
We measured peak and steady-state IK1 to address whether other K+ currents were affected by KChIP2-/- (Figure 3A). No measurable differences in the inwardly rectifying K+ current were observed in KChIP2-/- (n=7-10; Figure 3B).
Figure 3.
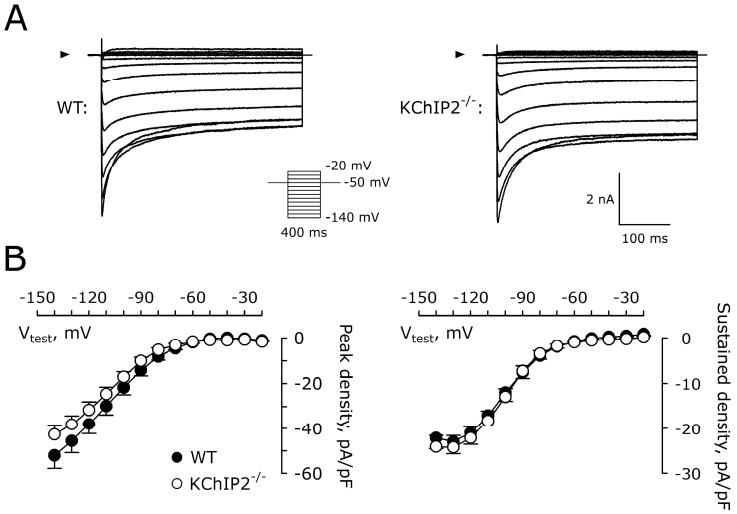
Similar inward rectifying K+ currents in WT and KChIP2-/- myocytes. A, Representative current traces recorded from a WT and KChIP2-/- myocyte. Arrowheads indicate 0 pA. Inset, voltage protocol. B, Peak inward IK1 (left) and steady state (right) IK1 in WT (n=7) and KChIP2-/- (n=10) myocytes.
No HpTx-2-sensitive current and larger 4-AP-sensitive currents in myocytes from KChIP2-/- mice
To evaluate the contribution of KChIP2 we used HpTx-2 (1 μM), which selectively blocks Ito,f without affecting other murine outward K+ currents.26 Figure 4A shows native KV currents in representative WT and KChIP2-/- myocytes. Addition of HpTx-2 abolished the fast-inactivating component of the KV-current decay in the WT cell and reduced IP (Figure 4B). HpTx-2 blocked 11±2% of the IP of the native KV current in WT myocytes (n=13; Figure 4C), but reduced the current by only 3±2% in KChIP2-/- myocytes (n=9; P<0.05 versus WT), suggesting KChIP2-/- mice lack the HpTx-2 sensitive Ito,f.
Figure 4.
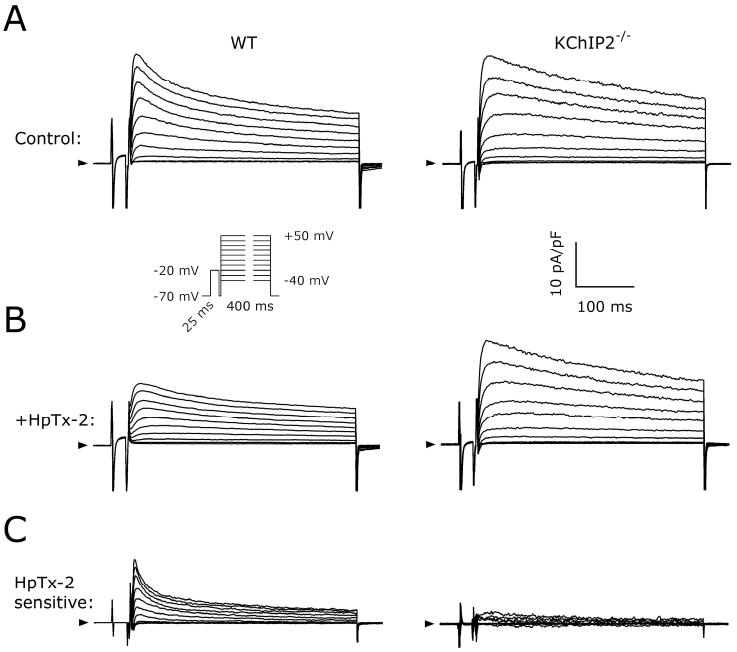
No effect of HpTx-2 on KV currents in KChIP2-/- myocytes. A, Representative baseline KV currents recorded using 400-ms pulses from VH=-70 mV to Vstep between -40 and +50 mV (inset) in myocytes from WT and KChIP2-/- mice. Arrowheads indicate 0 pA. B, Currents recorded in the same myocytes after addition of 1 μM HpTx-2 to the superfusion solution. C, The HpTx-2-sensitive currents obtained by digital offline subtraction of the current traces shown in A and B.
Since total KV currents are comparable in WT and KChIP2-/- mice, but Ito,f is missing in KChIP2-/- myocytes, it is possible that a non-KV4 current is increased in KChIP2-/- mice. Therefore, we used a pharmacological approach to dissect the remaining currents in WT and KChIP2-/- myocytes. 4-AP ≥5 mM abolishes all outward K+ currents in mice. At lower concentrations (IC50 ≈ 50 μM), 4-AP most potently blocks IK,slow, whereas Ito,f is blocked at higher concentrations (IC50 ≈ 500 μM). ISS is blocked only in the mM range.7
4-AP concentration-dependently decreased peak-current densities in WT and KChIP2-/- myocytes (Figure 5). Figure 5A illustrates total KV current before and after adding 50 μM 4-AP to the superfusate in WT and KChIP2-/- myocytes. The 4-AP-sensitive current was obtained by digitally subtracting this current from the original KV current (Figure 5B). Peak 4-AP-sensitive current amplitude was significantly larger in KChIP2-/- myocytes than in WT myocytes at both 4-AP concentrations (Figure 5C). 4-AP-insensitive currents showed clear time-dependent inactivation in all WT myocytes (Figure 5A, left), whereas the 4-AP-insensitive current in KChIP2-/- myocytes showed very little inactivation over the depolarizing step (Figure 5A, right). When IP of 4-AP-insensitive (1 mM) currents were measured within the first 100 ms of the voltage step and compared to the mean current amplitude in the last 100 ms of the voltage step, 82±4% (n=12) of peak current remained in KChIP2-/- cells, compared to 58±2% in the WT myocytes (n=9; P<0.05). This suggests the absence of time-dependent current in KChIP2-/- myocytes exposed to 1 mM 4-AP.
Figure 5.

Larger fraction of KV current is sensitive to 4-AP in KChIP2-/- myocytes. A, Current traces normalized to cell capacitance recorded using 400-ms pulses from VH=-70 mV to Vstep=+40 mV from a WT and a KChIP2-/- cell, before and after addition of 50 μM 4-AP to the superfusate. Arrowheads indicate 0 pA. Note the prominent inactivation of the 4-AP resistant current in WT. B, The current fraction sensitive to 50 μM 4-AP obtained by digital offline subtraction of the current traces shown in A. C, 4-AP-sensitive current relative to the full IP in WT and KChIP2-/- cells. *, P<0.05.
Elevated KV1.5 transcription in KChIP2-/- mice
The altered pharmacological responsiveness combined with an unaltered native IP in KChIP2-/- myocytes suggest that currents contributing to overall repolarization may change subsequent to KChIP2 knockout. Therefore, we studied the molecular remodeling in KChIP2-/- LV, using quantitative real-time PCR with KV-subunit specific primers to quantify transcriptional activity in WT (n=6) and KChIP2-/- (n=6) tissue. We found significant increases in mRNA levels for KV4.2, KV4.3, and KV1.5 in KChIP2-/- ventricles versus WT (Figure 6). As expected, KChIP2 transcription was not detected in KChIP2-/- LV. KV2.1 and KV1.4 mRNA were comparable in WT and KChIP2-/- LV. Cyclophilin A levels (control) remained unaltered.
Figure 6.
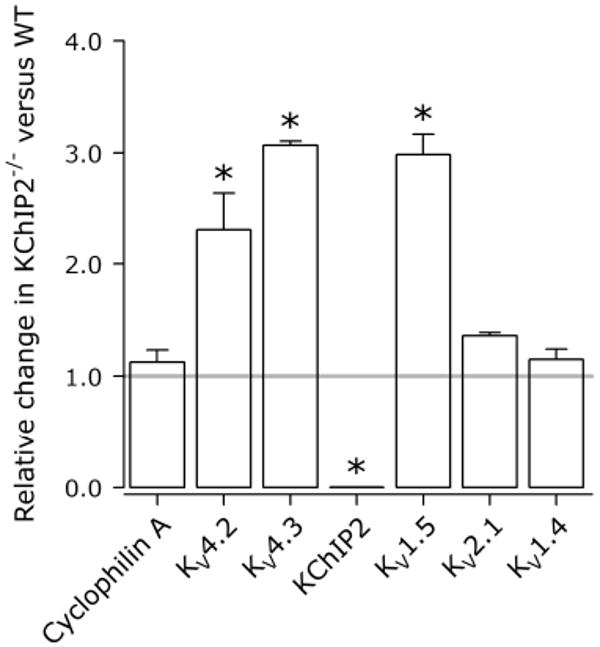
Compensatory remodeling potentially includes elevated KV1.5 transcription. Expression levels of the major ion channels underlying murine repolarization in KChIP2-/- (n=6) relative to WT (n=6) ventricles. Grey horizontal line indicates equal expression levels in WT and KChIP2-/- ventricles. KChIP2 was not detected in KChIP2-/- myocytes. *, P<0.05.
Discussion
The significant findings of this study are that myocytes from KChIP2-/- mice have no Ito,f, but show no change in IP density, versus WT myocytes. An increased 4-AP-sensitive current is likely responsible for the maintained IP in KChIP2-/- mice. Transmembrane action potentials recorded from intact ventricular preparations under control conditions are comparable in KChIP2-/- and WT; however, 4-AP-induced APD prolongation is intensified in KChIP2-/- preparations. Furthermore, the transcript level of the KV1.5 subunit is increased in KChIP2-/- ventricles. In total, this is consistent with an augmented IK,slow in KChIP2-/- myocytes, maintaining normal repolarization reserve.
KChIP2 is responsible for maintenance of mouse ventricular Ito,f
Using 4.5s depolarizing steps, KV current decay was described by 2 exponential components in KChIP2-/- and WT myocytes, with only minor differences in time constants of inactivation. A very fast component of inactivation (τ<50 ms) was present in WT myocytes within the first 100 ms of depolarization, and by using 400-ms pulses, this rapid component was amplified relative to the total current decay (compare Figure 2A and 2D). This component was blocked by HpTx-2, which had no effect in KChIP2-/- myocytes (Figure 4). Thus, we found no evidence that KV4.2 or KV4.3 produce functional Ito,f in the absence of KChIP2 in the mouse, despite increased mRNA levels of these α subunits in the LV (Figure 6).
The unaltered IP and APD in the absence of KV antagonists in KChIP2-/- mice suggest upregulation of a secondary repolarizing K+ current that compensates for the absent Ito,f. Interestingly, 4-AP had a larger effect on KV current amplitude in KChIP2-/- myocytes, translating to exaggerated APD prolongation upon 4-AP-treatment. Previous studies suggest that KChIP2 associates with the KV1.5 α subunit in myocytes and that KChIP2 co-expression decreases KV1.5-encoded K+ current in cell lines, suggesting a multifunctional role of KChIP2 in regulating murine repolarizing currents.18 Thus, by relieving KChIP2-mediated suppression of KV1.5 in KChIP2-/- mice, an increased IK,slow would be expected. We observe a 3-fold increase in KV1.5 mRNA in KChIP2-/- ventricles (Figure 6) and an augmented 4-AP-sensitive IK,slow (Figure 5). The lack of an anti-KV1.5 antibody that can detect endogenous KV1.5 in cardiomyocytes reliably27 precluded analysis of KV1.5 protein expression. Furthermore, it is unclear whether direct relief of suppression contributes to the increased IK,slow in KChIP2-/- myocytes.
In vitro, KChIP2 increases KV4-encoded current amplitude, slows inactivation, and accelerates recovery from inactivation.15, 16 Hence, if a KV4-dependent current plays a considerable role in murine outward voltage-gated K+ currents, corollaries of KChIP2-/- in vivo should include decreased IP density, faster current decay, and slowed recovery of IP from inactivation. Despite transcription of KV4.2 and KV4.3, we found no residual Ito,f in KChIP2-/- mice. Thus, although a small, fast inactivating current is recorded in cell lines transfected with KV4.2 or KV4.3 in the absence of KChIP2, no such current is found in KChIP2-/- mice, suggesting an essential role for KChIP2 in translating KV4 protein into functional current in vivo.
Ito remodeling in KChIP2-/- mice reduces repolarization reserve
The KChIP2-/- mouse adds to the animal models and patient data in which a normal QT interval is excessively prolonged by a challenge (e.g. IKr block), reflecting a reduced repolarization reserve.29, 30 Many patients with congenital or drug-induced long-QT syndromes have normal QT intervals in the absence of provocation, referred to as genotype-phenotype discordance.31, 32 Furthermore, downregulation of Ito is prominent in heart failure1, 4, where IKr-blocking drugs can induce excess QT interval prolongation.33 Additionally, the significant cardiac plasticity in cardiac memory in dogs34, includes decreased Ito and downregulated KV4.3 and KChIP2 transcription in myocardium near ventricular pacing sites.35, 36 Here, IKr blockade causes more marked QT prolongation than in control dogs.37
In the present study, removal of Ito,f from the assembly of murine repolarizing currents caused no overt reduction in total current (Figure 2), due to IK,slow upregulation. The phenotypic consequence of this remodeling became evident only when action potentials were recorded in the presence of 4-AP (Figure 1). Thus, a reduced repolarization reserve with respect to Ito increases susceptibility to pharmacological prolongation of repolarization. Finally, the differences in action potential morphology and 4-AP response between LV and RV (Figure 1) likely reflect regional differences in repolarizing currents reported in cells isolated from the ventricles.5
Relationship to other studies on mouse pathologies
Kuo et al.25 reported decreased total Ito density and significantly prolonged APD in disaggregated RV myocytes from KChIP2-/- mice. In their study, INa and ICa,L were allowed to contribute to the current recordings.25, 38 In contrast, our study evaluated the relative contributions of the different voltage-dependent K+ currents to repolarization in KChIP2-/- mice. This intent prompted us to use solutions containing antagonists for INa (TTX) and ICa,L (Co2+).6 Using protocols and solutions identical to those of Kuo et al., we observed a smaller total outward current in KChIP2-/- myocytes (data not shown). The discrepancy in APD likely reflects the difference between the uncoupled disaggregated myocytes in the previous study25 and the multicellular preparations in the present study. However, our results complement the previous report of unaltered QT intervals or effective refractory periods in anesthetized, intact KChIP2-/- mice.25
In agreement with earlier reports on KChIP2-/- mice, we observed no evidence of cardiac hypertrophy or impaired function.25 This is paralleled by results from KV4.2-/- mice24 and mice expressing a KV4.2-pore mutant.22 In contrast, mice expressing a carboxy-terminal deletion of KV4.2 display marked hypertrophy and ventricular dilatation progressing to congestive heart failure.23 Whereas the two mutants display prolonged APD, KV4.2-/- mice have an upregulated Ito,s and increased transcription of KV1.4.24 In our experiments, we found neither Ito,s nor increased KV1.4 mRNA in KChIP2-/- myocytes. Thus, the compensatory mechanisms clearly differ between KV4.2-/- and KChIP2-/- mice and the signal transduction causing this discrepant remodeling is of interest. Furthermore, the absence of functional remodeling in the two knockout mice argues against a direct link between deficient Ito,f and cardiac hypertrophy and heart failure.
Limitations
Our recordings of WT IP are smaller and Ito,f are reduced versus other reports6, despite identical mouse strains, patch-clamp solutions, and voltage protocols. Different cell-disaggregation protocols or storage conditions between myocytes disaggregation and patch-clamp experiments (plating on laminin-coated coverslips, stored in 5% CO2 incubator in up to 24 h)6 could contribute to the differences seen between laboratories. Nevertheless, we studied WT and KChIP2-/- mice under identical conditions and the differences reported are consequent to KChIP2 deficiency.
Although increased mRNA expression may not be identical to increased protein translation and –trafficking, our functional data of upregulated IK,slow density complements the increased KV1.5 mRNA levels.
Conclusions
Loss of KChIP2 has little impact on total murine KV current, and the genotype is reflected phenotypically only during pharmacological challenges to repolarization. Ito,f is absent in KChIP2-/- mice, whereas 4-AP-sensitive IK,slow is upregulated. This is associated with excess prolongation of APD in the presence of 4-AP and with increased expression of KV1.5. Thus, KChIP2 and Ito,f appear critical to action potential morphology in the mouse only in circumstances of decreased repolarization reserve.
Acknowledgments
This work was performed during Dr. Thomsen's tenure as Research Fellow of the Heart Rhythm Society. Technical assistance from Patricia McLaughlin is greatly appreciated. The authors wish to thank Dr. Masahiko Hoshijima (University of California, San Diego, CA) and Dr. Kenneth Chien (Massachusetts General Hospital, MA) for providing the KChIP2-/- mice. We gratefully acknowledge Dr. Geoffrey S. Pitt for helpful discussions. This work was supported by USPHS-NHLBI grants HL-28958 and HL-67101.
Referenced literature
- 1.Kaab S, Dixon J, Duc J, et al. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98(14):1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- 2.Lue WM, Boyden PA. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation. 1992;85(3):1175–1188. doi: 10.1161/01.cir.85.3.1175. [DOI] [PubMed] [Google Scholar]
- 3.Huang B, Qin D, El-Sherif N. Spatial alterations of Kv channels expression and K(+) currents in post-MI remodeled rat heart. Cardiovasc Res. 2001;52(2):246–254. doi: 10.1016/s0008-6363(01)00378-9. [DOI] [PubMed] [Google Scholar]
- 4.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42(2):270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 5.Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. J Gen Physiol. 1999;113(5):661–678. doi: 10.1085/jgp.113.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunet S, Aimond F, Li H, et al. Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J Physiol. 2004;559(Pt 1):103–120. doi: 10.1113/jphysiol.2004.063347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo W, Xu H, London B, et al. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J Physiol. 1999;521(Pt 3):587–599. doi: 10.1111/j.1469-7793.1999.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.London B, Guo W, Pan X, et al. Targeted replacement of KV1.5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of I(K,slow) and resistance to drug-induced qt prolongation. Circ Res. 2001;88(9):940–946. doi: 10.1161/hh0901.090929. [DOI] [PubMed] [Google Scholar]
- 9.Xu H, Barry DM, Li H, et al. Attenuation of the slow component of delayed rectification, action potential prolongation, and triggered activity in mice expressing a dominant-negative Kv2 alpha subunit. Circ Res. 1999;85(7):623–633. doi: 10.1161/01.res.85.7.623. [DOI] [PubMed] [Google Scholar]
- 10.Marionneau C, Aimond F, Brunet S, et al. PPARalpha-mediated remodeling of repolarizing voltage-gated K+ (Kv) channels in a mouse model of metabolic cardiomyopathy. J Mol Cell Cardiol. 2008;44(6):1002–1015. doi: 10.1016/j.yjmcc.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niwa N, Wang W, Sha Q, et al. Kv4.3 is not required for the generation of functional Ito,f channels in adult mouse ventricles. J Mol Cell Cardiol. 2008;44(1):95–104. doi: 10.1016/j.yjmcc.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85(4):1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Yan Y, Liu Q, et al. Structural basis for modulation of Kv4 K+ channels by auxiliary KChIP subunits. Nat Neurosci. 2007;10(1):32–39. doi: 10.1038/nn1822. [DOI] [PubMed] [Google Scholar]
- 14.Callsen B, Isbrandt D, Sauter K, et al. Contribution of N- and C-terminal Kv4.2 channel domains to KChIP interaction [corrected] J Physiol. 2005;568(Pt 2):397–412. doi: 10.1113/jphysiol.2005.094359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.An WF, Bowlby MR, Betty M, et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403(6769):553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 16.Deschenes I, DiSilvestre D, Juang GJ, et al. Regulation of Kv4.3 current by KChIP2 splice variants: a component of native cardiac I(to)? Circulation. 2002;106(4):423–429. doi: 10.1161/01.cir.0000025417.65658.b6. [DOI] [PubMed] [Google Scholar]
- 17.Rosati B, Pan Z, Lypen S, et al. Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol. 2001;533(Pt 1):119–125. doi: 10.1111/j.1469-7793.2001.0119b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Guo W, Mellor RL, et al. KChIP2 modulates the cell surface expression of Kv 1.5-encoded K(+) channels. J Mol Cell Cardiol. 2005;39(1):121–132. doi: 10.1016/j.yjmcc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Nabauer M, Beuckelmann DJ, Erdmann E. Characteristics of transient outward current in human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73(2):386–394. doi: 10.1161/01.res.73.2.386. [DOI] [PubMed] [Google Scholar]
- 20.Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73(2):379–385. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- 21.Radicke S, Cotella D, Graf EM, et al. Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc Res. 2006;71(4):695–703. doi: 10.1016/j.cardiores.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 22.Barry DM, Xu H, Schuessler RB, et al. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circ Res. 1998;83(5):560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- 23.Wickenden AD, Lee P, Sah R, et al. Targeted expression of a dominant-negative K(v)4.2 K(+) channel subunit in the mouse heart. Circ Res. 1999;85(11):1067–1076. doi: 10.1161/01.res.85.11.1067. [DOI] [PubMed] [Google Scholar]
- 24.Guo W, Jung WE, Marionneau C, et al. Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res. 2005;97(12):1342–1350. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- 25.Kuo HC, Cheng CF, Clark RB, et al. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107(6):801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 26.Sanguinetti MC, Johnson JH, Hammerland LG, et al. Heteropodatoxins: peptides isolated from spider venom that block Kv4.2 potassium channels. Mol Pharmacol. 1997;51(3):491–498. [PubMed] [Google Scholar]
- 27.Marionneau C, Brunet S, Flagg TP, et al. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res. 2008;102(11):1406–1415. doi: 10.1161/CIRCRESAHA.107.170050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deschenes I, Armoundas AA, Jones SP, et al. Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J Mol Cell Cardiol. 2008;45(3):336–346. doi: 10.1016/j.yjmcc.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21(5):1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 30.Thomsen MB, Matz J, Volders PG, et al. Assessing the proarrhythmic potential of drugs: current status of models and surrogate parameters of torsades de pointes arrhythmias. Pharmacol Ther. 2006;112(1):150–170. doi: 10.1016/j.pharmthera.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Kaab S, Hinterseer M, Nabauer M, et al. Sotalol testing unmasks altered repolarization in patients with suspected acquired long-QT-syndrome--a case-control pilot study using i.v. sotalol. Eur Heart J. 2003;24(7):649–657. doi: 10.1016/s0195-668x(02)00806-0. [DOI] [PubMed] [Google Scholar]
- 32.Vincent GM, Timothy KW, Leppert M, et al. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med. 1992;327(12):846–852. doi: 10.1056/NEJM199209173271204. [DOI] [PubMed] [Google Scholar]
- 33.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350(10):1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 34.Rosenbaum MB, Blanco HH, Elizari MV, et al. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol. 1982;50(2):213–222. doi: 10.1016/0002-9149(82)90169-2. [DOI] [PubMed] [Google Scholar]
- 35.Patberg KW, Obreztchikova MN, Giardina SF, et al. The cAMP response element binding protein modulates expression of the transient outward current: implications for cardiac memory. Cardiovasc Res. 2005;68(2):259–267. doi: 10.1016/j.cardiores.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 36.Yu H, McKinnon D, Dixon JE, et al. Transient outward current, Ito1, is altered in cardiac memory. Circulation. 1999;99(14):1898–1905. doi: 10.1161/01.cir.99.14.1898. [DOI] [PubMed] [Google Scholar]
- 37.Plotnikov AN, Shvilkin A, Xiong W, et al. Interactions between antiarrhythmic drugs and cardiac memory. Cardiovasc Res. 2001;50(2):335–344. doi: 10.1016/s0008-6363(01)00233-4. [DOI] [PubMed] [Google Scholar]
- 38.Fiset C, Clark RB, Larsen TS, et al. A rapidly activating sustained K+ current modulates repolarization and excitation-contraction coupling in adult mouse ventricle. J Physiol. 1997;504(Pt 3):557–563. doi: 10.1111/j.1469-7793.1997.557bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]