

Abstract
Electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO) accepts electrons from electron transfer flavoprotein (ETF) and reduces ubiquinone from the ubiquinone pool. It contains one [4Fe-4S]2+,1+ and one FAD, which are diamagnetic in the isolated oxidized enzyme and can be reduced to paramagnetic forms by enzymatic donors or dithionite. In the porcine protein, threonine 367 is hydrogen bonded to N1 and O2 of the flavin ring of the FAD. The analogous site in Rhodobacter sphaeroides ETF-QO is asparagine 338. Mutations N338T and N338A were introduced into the R. sphaeroides protein by site-directed mutagenesis to determine the impact of hydrogen bonding at this site on redox potentials and activity. The mutations did not alter the optical spectra, EPR g-values, spin-lattice relaxation rates, or the [4Fe-4S]2+,1+ to FAD point-dipole interspin distances. The mutations had no impact on the reduction potential for the iron-sulfur cluster, which was monitored by changes in the continuous wave EPR signals of the [4Fe-4S]+ at 15 K. For the FAD semiquinone, significantly different potentials were obtained by monitoring the titration at 100 or 293 K. Based on spectra at 293 K the N338T mutation shifted the first and second midpoint potentials for the FAD from +47 and -30 mV for wild type to -11 and -19 mV, respectively. The N338A mutation decreased the potentials to -37 and -49 mV. Lowering the midpoint potentials resulted in a decrease in the quinone reductase activity and negligible impact on disproportionation of ETF1e- catalyzed by ETF-QO. These observations indicate that the FAD is involved in electron transfer to ubiquinone but not in electron transfer from ETF to ETF-QO. Therefore, the iron-sulfur cluster is the immediate acceptor from ETF.
Electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO)1 is responsible for linking electrons derived from the oxidation of fatty acids and some amino acids to the main mitochondrial respiratory chain (1). It is a monotopic membrane protein located on the inner mitochondria membrane facing into the mitochondrial matrix. ETF-QO has two redox-active cofactors, one [4Fe-4S]2+,1+ cluster and one flavin adenine dinucleotide (FAD). ETF-QO oxidizes electron transfer flavoprotein (ETF), a protein responsible for oxidizing nine flavoprotein dehydrogenases and two N-methyl dehydrogenases (2), and reduces ubiquinone (UQ). UQ then transfers the electrons to the cytochrome bc1 complex (complex III). Defects in ETF-QO, or its electron donor ETF, result in a metabolic disease known as multiple acyl-CoA dehydrogenase deficiency (MADD) or glutaric acidemia type II (GAII). The severity of this disease depends upon the particular mutation and can range from late-onset in the teenage years to death within the first days after birth (3, 4). It is important to understand better the physical properties of this enzyme because of its important role in oxidative metabolism and its link to GAII.
The locations of the redox centers in the crystal structure of porcine ETF-QO provide insights into the possible mechanism of electron transfer in this enzyme (5). The FAD molecule is closer to the UQ than the [4Fe-4S]2+,1+ cluster (9.9 Å as opposed to 18.8 Å), and the cluster is closer to the surface of the protein than the flavin ring of FAD (5) (Figure 1). This suggests that the [4Fe-4S]2+,1+ cluster is responsible for accepting electrons from ETF and that the FAD is responsible for reducing UQ. The closest approach between the [4Fe-4S]2+,1+ cluster and the FAD is 11.5 Å, which is consistent with electron transfer between the two cofactors (6). There is 98% sequence identity between porcine and human ETF-QO and 67% sequence identity between human and Rhodobacter sphaeroides ETF-QO (N. J. Watmough, F. E. Frerman, and J. N. Butt, 2008, unpublished results). This high sequence similarity predicts that the tertiary structures are closely related (7) and that the mechanism of electron transfer is likely similar in all three enzymes.
Figure 1.

Crystal structure of porcine ETF-QO (PDB data bank: 2GMH) highlighting the positions of the three redox centers, [4Fe-4S]2+,1+ cluster (red and yellow), FAD (pink), and ubiquinone (blue). On the right the ribbon structure has been removed for clarity, and distances of closest approach of the three redox centers are shown.
Recently, the mutations Y501F, T525A, and Y501F/T525A were introduced into R. sphaeroides ETF-QO (8). These residues were chosen for mutation because the same residues (although numbered differently) hydrogen bond to the cysteine Sγ atoms that are ligated to the [4Fe-4S]2+,1+ cluster of porcine ETF-QO. The mutations eliminate this hydrogen bonding and thus change the redox potential of the [4Fe-4S]2+,1+ cluster, which in turn affects the activity of the enzyme. The [4Fe-4S]2+,1+ midpoint potentials were lowered by about 100 mV for either single mutation and by about 165 mV for the double mutant. The lower redox potentials of these mutants decreased the quinone reductase activity and the rates of disproportionation of ETF1e-compared to the wild-type enzyme (8). Both single mutations had similar impacts on activities. These results demonstrate that reduction of [4Fe-4S]2+,1+ is required for proper enzyme function. As expected, these mutations caused no change in FAD midpoint potentials.
In this study mutations were introduced to modulate the FAD redox potentials and investigate its importance for ETF-QO function. It has been shown that amino acids that form hydrogen-bonding interactions with the isoalloxazine headgroup of a FAD molecule can modulate its redox potentials (9). The X-ray crystal structure of porcine ETF-QO (5) shows that the location of threonine 367 is suitable for formation of hydrogen bonds with the N1 and O2 of the FAD. In R. sphaeroides ETF-QO asparagine 338 is at the position equivalent to threonine 367 of the porcine enzyme. Therefore, it is proposed that this residue is in position to form hydrogen bonds with N1 and O2 of the FAD (Figure 2). The mutations N338A and N338T were introduced to eliminate or lower the energy of the hydrogen bond to the FAD, respectively. The impact of these changes on FAD redox potential and enzyme activity was determined. Results from this study and the previous mutagenesis near the [4Fe-4S]2+,1+ cluster can be combined to give a description of the roles of the FAD and [4Fe-4S]2+,1+ cluster in ETF-QO electron transfer.
Figure 2.
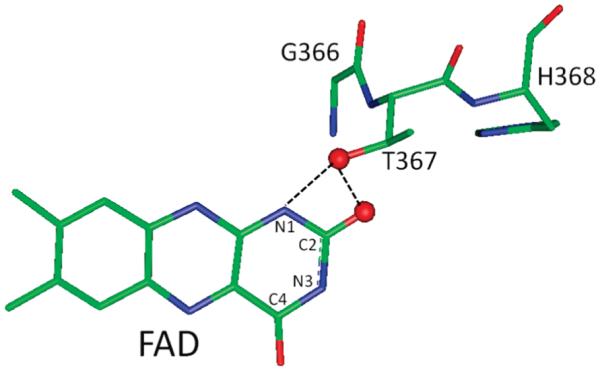
Position of threonine 367 and the C2 oxygen of the FAD isoalloxazine group from the porcine ETF-QO crystal structure (PDB data bank: 2GMH). The corresponding amino acid in R. sphaeroides ETF-QO is asparagine 338, which is in position to interact with the C2 oxygen and N1 of the isoalloxazine ring.
EXPERIMENTAL PROCEDURES
Mutagenesis, Expression, and Purification
The R. sphaeroides ETF-QO gene was cloned into the pET21a expression vector, and the wild-type protein was expressed in Escherichia coli C43 as previously described (8). Site-directed mutagenesis was carried out using the Stratagene QuikChangeII XL site-directed mutagenesis kit. Mutations were confirmed by sequencing. Mutant and wild-type plasmids were transformed into E. coli C43 cells (10). Cells were grown and protein purified as described previously (8). Ubiquinone was not present in the isolated protein. The iron and FAD contents of the enzyme were determined by the methods of Wu et al. (11) and Siegel et al. (12), respectively. The iron:FAD ratios are 4.0:1 for wild type, 3.6:1 for N338T, and 4.1:1 for N338A.
Enzyme Assays
ETF-QO quinone reductase activity was measured in a coupled reaction containing 10 mM Hepes(K+) buffer, pH 7.4, 2 μM medium-chain acyl-CoA dehydrogenase (MCAD), 2 μM porcine ETF, 100 μM octanoyl-CoA, 10 mM n-dodecyl β-d-maltopyranoside, and 55 μM Q1 (coenzyme Q1, Sigma Chemical Co.) at 25 °C (13). Q1 reduction was initiated by the addition of ETF-QO and was monitored by the decrease of absorbance at 275 nm (Δ∊ = 7.4 mM-1 cm-1).
Disproportionation of ETF1e- catalyzed by ETF-QO was assayed under anaerobic conditions as described by Beckmann and Frerman (8, 14), using 10 μM ETF as substrate. Reactions were initiated by the addition of ETF-QO, and the decrease in absorbance at 370 nm was monitored. Km of R. sphaeroides ETF-QO is 25 μM so it was impractical to use saturating concentrations of ETF in these assays. However, the assay conditions are satisfactory for comparative purposes because under nonsaturating conditions, the velocity is proportional to the second-order rate constant (15).
Spectrophotometric Titrations
Enzymatic reduction of ETF-QO was carried out as reported in ref 8. Reduction with dithionite was performed on 20 μM ETF-QO in 10 mM Hepes(K+), pH 7.4, 8 mM CHAPS, and 0.4 mM protocatechuate. The reaction mixtures were made anaerobic by alternate evacuation and purging with argon. Residual O2 was removed by addition of protocatechuate dioxygenase (16). The dioxygenase was a gift from Dr. David Ballou (University of Michigan).
Potentiometric Titrations
Potentiometric titrations of ETF-QO (∼40-100 μM enzyme, 20 mM Hepes (K+), 20% glycerol (v/v), pH 7.4) using dithionite as reductant were carried out in an anaerobic vessel under continuous N2(g) flow (8, 17).
EPR Spectroscopy
EPR (electron paramagnetic resonance) spectroscopy, signal quantitation, and analysis of the data for [4Fe-4S]+ at 15 K and for FAD semiquinone (SQ-·) at 100 K were done by the methods of Fielding et al. (18) for wild-type ETF-QO and Usselman et al. (8) for ETF-QO mutants. EPR spectra of FAD SQ-· at 293 K were recorded in 1.0 mm i.d. capillaries on a Varian E9 spectrometer using a microwave power of 0.5 mW, a 2.0 G modulation amplitude, a time constant of 0.25 s, and averaging five 120 s scans. Spectra were quantitated by simulation (18) and compared with a tempol (4-hydroxy-2,2,6,6-tetramethylpiperidinooxy) standard.
Relaxation Enhancement Distance Measurements
Enhanced relaxation of the FAD SQ-· signal due to interaction with rapidly relaxing [4Fe-4S]+ was measured using inversion recovery EPR and was modeled using the locally written program MENOSR, which utilizes a modified version of the Bloembergen equation (19, 20). The inversion recovery measurements were performed on samples prepared in the potentiometric titration, selected for the maximum FAD SQ-· signal intensity. The inversion recovery curves are comprised of contributions from FAD SQ-· with neighboring diamagnetic [4Fe-4S]2+ and paramagnetic [4Fe-4S]+, but only the paramagnetic form of the iron-sulfur cluster enhances the FAD relaxation. In the MENOSR program the distance between the two centers and the fraction of iron-sulfur cluster in the diamagnetic state are both adjusted to give the best fit to the data.
The double mutation Y501F/T525A (8), which deletes two hydrogen-bonding interactions with the [4Fe-4S]2+,1+, was used to measure FAD SQ-· relaxation in the absence of enhancement. The [4Fe-4S]+ midpoint potential for Y501F/T525A ETF-QO is sufficiently low that at -15 mV all of the iron-sulfur cluster is in the diamagnetic +2 oxidation state and does not interact with the FAD SQ-·. In the previous study of ETF-QO by Fielding et al. (18) the FAD SQ-· in ETF was used to estimate the relaxation rate in the absence of relaxation enhancement. The relaxation rates that were measured for the SQ-· in reduced Y510F/T524A were in good agreement with values obtained previously for the SQ-· in ETF (8, 18).
Midpoint Potential Calculations
The relative concentrations of the paramagnetic [4Fe-4S]+ that are needed for the calculations of the midpoint potentials were based on peak-to-peak signal amplitudes (8). Double integration was not used because the signals for [4Fe-4S]+ and FAD SQ-· overlap. For the [4Fe-4S]+ titration curves the signal at gx value was selected for the calculations because it is relatively strong and does not overlap with the resonator background signal that was seen at lower field, overlapping with the gz value. The gy value was not used because of the close proximity of the FAD SQ-· signal. The midpoint potentials of the [4Fe-4S]2+,1+ cluster (Em) were calculated by nonlinear least-squares fitting to plots of [ox]/[red] versus E (mV) using the Nernst equation as described previously (8, 21). The Mathcad software package (PTC, Needham, MA) was used to fit the data by manually changing the midpoint potential (Em) to satisfy a least-squares error constraint. The scatter in the data points determines the uncertainty in Em, which is estimated as about ±5 mV. The first (Em1) and second (Em2) reduction potentials for the FAD were calculated by nonlinear least-squares fitting to plots of [SQ-·] versus E (mV) using the Nernst equation. The solver function in Microsoft Excel was used to vary the Em1 and Em2 values until a minimum sum of squares error between the data points and Nernst equation was found. The average of Em1 and Em2 can be determined with an uncertainty of about ±5 mV. The maximum values of the SQ-· concentrations measured at 293 K are 75% for wild-type, 39% for N338T, and 41% for N338A ETF-QO. These values are higher than those measured at 100 K, which are 53%, 32%, and 35% for wild type, N338T, and N338A, respectively. The separation between the two potentials, Em1 - Em2, is strongly dependent on the concentration of SQ-· observed at the peak of the titration curve, so the uncertainties in Em1 and Em2 are estimated as (15 mV.
Duplicate titrations of wild-type protein were carried out to determine the reproducibility of the experiment. At 293 K the Em1 values calculated from the two sets of data were 44 and 49 mV, respectively (an average of about 47 mV). The Em2 values were both found to be -30 mV. At 100 K the Em1 values were 8 and 3 mV (average of about 6 mV) and the Em2 values were -35 and -11 mV (average of -23 mV). A larger variation was observed in the Em2 values at 100 K, which is attributed to uncertainties in the calculated concentration of semiquinone at the peak of the titration curve.
RESULTS AND DISCUSSION
Characterization of Mutants
The optical spectrum of oxidized N338A ETF-QO is shown in Figure 3, trace 1. It is made up of overlapping contributions from the [4Fe-4S]2+,1+ and the FAD. The FAD contributes the dominant peaks at 380 and 430 nm, which are similar for the wild type and two mutants. Quantitation of FAD in wild-type ETF-QO and the two mutants demonstrated incorporation of the FAD, and the approximately 4:1 iron:FAD ratios confirm the presence of the [4Fe-4S]2+,1+ cluster.
Figure 3.

Anaerobic reductive titration of N338A ETF-QO. The solution contained 20 μM R. sphaeroides N338A ETF-QO, 1.5 μM human ETF, and 0.5 μM human MCAD in 10 mM Hepes(K+) at pH 7.4. The oxidized sample (spectrum 1) was titrated with octanyl-CoA. After each addition spectra were recorded every 5-10 min until less than 0.001 au change per minute in absorbance was observed. Spectra 2 and 3 correspond to mole ratios of 0.5:1 and 1:1, respectively, and spectrum 4 is the dithionite-reduced enzyme. The inset shows the Δε430nm vs mole ratio of titrant for (●) wild-type, (+) N338T, and (◇) N338A ETF-QOs.
The [4Fe-4S]2+,1+ cluster and the FAD are diamagnetic in the oxidized as-isolated enzyme but can be reduced to EPR-detectable forms. The continuous wave (CW) EPR spectra of the FAD SQ-· at 100 K in the two mutants are indistinguishable from those of wild-type protein (Figure 4A). The FAD signal was studied at 100 and 293 K because the relaxation rate of the [4Fe-4S]+ is so fast at these temperatures that it does not contribute to the CW signal (18). The FAD SQ-· signal becomes severely power-saturated at the lower temperatures and higher microwave powers that were used to monitor the [4Fe-4S]+ signal (18). Figure 4B shows the CW spectra of the [4Fe-4S]+ in the two mutants and wild-type ETF-QO at 15 K. As expected, the [4Fe-4S]+ line shapes are unchanged by the mutations near the FAD. CW EPR line widths and g-values (Table 1) are not changed by mutation.
Figure 4.

X-band CW EPR spectra of R. sphaeroides ETF-QO. (A) FAD semiquinone signal recorded at 100 K, 9.23 GHz with 0.01 mW power. A similar line shape, but with lower signal to noise, was observed at 293 K. (B) [4Fe-4S]+ signal recorded at 15 K, 9.35-9.42 GHz with 0.05 mW power. The saturated FAD semiquinone signal was deleted from (B) to aid in comparison (dashed lines). The anaerobic samples contained 20 mM Hepes (K+) and 20% glycerol (v/v) at pH 7.4. Spectra were recorded at their respective maximum semiquinone signals from the potentiometric titrations. The potentials were wild type (-10 mV), N338A (-75 mV), and N338T (-35 mV).
Table 1.
| sample | [4Fe-4S]+gz (line width) | [4Fe-4S]+gy (line width) | [4Fe-4S]+gx (line width) | SQ-·g |
|---|---|---|---|---|
| wild type | 2.089 (22) | 1.934 (15) | 1.875 (29) | 2.0036 |
| N338T | 2.084 (22.5) | 1.930 (17) | 1.870 (30.5) | 2.0037 |
| N338A | 2.084 (22.5) | 1.930 (17.5) | 1.870 (30.5) | 2.0039 |
The uncertainty of the iron-sulfur cluster g-values was ∼±0.003.
The uncertainty of the semiquinone g-values was ∼±0.0005.
Peak-to-peak first derivative line widths were obtained by computer simulation. Uncertainty of the line widths was ±2 G.
The temperature dependence of the spin-lattice relaxation rates, 1/T1, for [4Fe-4S]+ at the gy turning point in the spectrum was measured for the two mutants. Values are the same, within experimental uncertainty, as in the wild-type protein (Figure 5). Analysis of the temperature dependence of the relaxation in terms of contributions from Raman and Orbach process (18, 22) showed that the energy of the low-lying excited state, ΔE = 210 ± 5 K (146 cm-1), is unchanged by the mutations.
Figure 5.

Temperature dependence of spin-lattice relaxation rates for dithionite-reduced [4Fe-4S]+ in R. sphaeroides ETF-QO. The circles are the average values for wild type, N338T, and N338A mutants. The error bars reflect the standard deviations for the three proteins. The fit line is the sum of contributions from the Raman and Orbach processes. The Debye temperature was fixed at 100 K, and the energy of the low-lying excited state was ΔE = 210 K (146 cm-1).
Interaction between the rapidly relaxing [4Fe-4S]+ and the more slowly relaxing FAD SQ-· enhances the electron spin relaxation rate for the SQ-·, which can be used to find the point-dipole distance between the two centers (18, 19). This method was used to determine a distance of 18.4 Å between the center of the iron-sulfur cluster and the weighted average position of the spin densities of FAD in R. sphaeroides ETF-QO (8, 18). The inversion recovery curves for the FAD SQ-· signal in the two mutants were modeled using the program MENOSR (20). The inversion recovery curves for wild type and N338T were very similar to that for N338A (Figure 6). For the N338T mutation the calculated distance between centers was 17.7 ± 0.9 Å, and the fraction of the cluster in the noninteracting, diamagnetic form was 28 ± 10%. Inversion recovery data for the N338A mutant were in good agreement with a calculated distance of 17.4 ± 0.9 Å and 30 ± 10% noninteracting [4Fe-4S]2+ cluster (Figure 6). The stated errors are standard deviations for results at temperatures between 31 and 50 K. Also shown in Figure 6 is the inversion recovery curve for the double mutant, Y501F/T525A, which lacks relaxation enhancement. The distances in the mutants are slightly smaller than the value of 18.4 Å previously calculated for wild-type ETF-QO. One explanation for this difference could be that the mutants alter the spin density of the FAD SQ-· so that the point dipole is slightly closer to the cluster. The similarities in line shapes of the FAD SQ-· signals (Figure 4A) indicate that any changes in spin density distributions are too small to have large impacts on major hyperfine couplings.
Figure 6.

Inversion recovery curves for semiquinone in the reduced N338A (upper) and Y501F/T525A (lower) samples of R. sphaeroides ETF-QO at 50 K. The samples were produced by reductive titration to the potentials that give the maximum SQ-· signal: N338A (-75 mV) and Y501F/T525A (-10 mV). The curves are the sums of contributions from semiquinone with neighboring diamagnetic [4Fe-4S]2+ and paramagnetic [4Fe-4S]+. The difference between the two curves reflects the impact of the paramagnetic [4Fe-4S]+: N338A (70-80%) and Y501F/T525A (0%). The dashed line is a simulated curve calculated with MENOSR. Simulated curves for both FAD site mutants give an average interspin distance of 17.6 ± 0.9 Å.
The similarity in CW EPR line shapes and g-values, the similarity in optical spectra, the fact that no detectable change in the [4Fe-4S]+ relaxation times was seen, the observed 4:1 iron to FAD ratios, and the similarity in the distances between the iron-sulfur cluster and the FAD SQ-· radical all suggest that the mutations did not cause major structural changes to the enzyme.
Spectrophotometric Titration
ETF-QO can be reduced either enzymatically using octanoyl-CoA as the electron donor or chemically with dithionite. The extent of reduction can be monitored by changes in the optical spectra (1, 8, 14, 21). Figure 3 shows the absorption spectra for oxidized N338A (trace 1), samples reduced with 0.5:1 and 1:1 mol ratios of octanoyl-CoA to ETF-QO (traces 2 and 3, respectively), and with excess dithionite (trace 4). For wild-type ETF-QO these ratios represent roughly 1 and 2 electronreduced ETF-QO because each mole of octanoyl-CoA can transfer two electrons to the enzyme. The inset in Figure 3 is a plot of the change in ε430 as a function of octanoyl-CoA added for wild-type and mutant ETF-QOs. The mutant proteins have higher limiting values for ε430 than wild type because octanoyl-CoA is not a strong enough reductant to fully reduce the FAD in the two mutants, with lower Em1 compared to wild-type ETF-QO.
Potentiometric Titration
CW EPR signals of FAD SQ-· and [4Fe-4S]+ were used to monitor the dithionite titrations (Figures 7 and 8). The FAD potentiometric titration curves for wild type and N338T and N338A mutants of R. sphaeroides ETF-QO are shown in Figure 7A (titration at 277 K and spectra recorded at 100 K) and Figure 7B (titration at 293 K and spectra recorded at 293 K). The solid lines are least-squares fits of the Nernst equation to the data. Calculated midpoint potentials are shown in Table 2.
Figure 7.

Potentiometric titration curves of the FAD semiquinone EPR signals recorded at 293 K (A) and 100 K (B) for R. sphaeroides wild-type (●, ▲), N338T (+), and N338A (◇) ETF-QOs. Solid lines are least-squares fitting of the Nernst equation to the data points.
Figure 8.

Potentiometric titration curves of [4Fe-4S]+ for R. sphaeroides wild-type (●), N338T (+), and N338A (◇) ETF-QOs. Data points were based on the X-band gx EPR signal intensities at 15 K. The midpoint potentials were fit using a single Nernst curve of n = 1 (solid lines).
Table 2.
Midpoint Potentials and Enzymatic Titration Δ∊430 Values for Wild-Type and Mutant ETF-QOsa
| sample | [4Fe-4S]2+,1+Em (mV) | Q/SQ-·Em1 (mV) | SQ-·/QH2Em2 (mV) | Q/QH2 (Em1 + Em2)/2 (mV) | reductive titration Δ∊430 |
|---|---|---|---|---|---|
| wild type | +37 | +47 | -30 | +8 (+9) | 12790 |
| N338T | +48 | -11 | -19 | -15 (-31) | 10560 |
| N338A | +42 | -37 | -49 | -43 (-78) | 9490 |
[4Fe-4S]2+,1+ midpoints were measured at 15 K. FAD midpoint potentials were measured at 293 K. Values at 100 K are given in parentheses.
The midpoint potentials calculated based on FAD SQ-· spectra recorded at 293 K differed significantly from those calculated from spectra at 100 K. The averages of Em1 and Em2 determined at 100 K were 16 to 45 mV more negative than at 293 K (Table 2). Differences in reduction potentials determined from EPR spectra at ambient and cryogenic temperatures have been reported previously for other proteins including Saccharomyces cereVisiae flavocytochrome b2 and E. coli sulfite reductase (23, 24). In the following paragraphs emphasis is placed on potentials determined by measurements at 293 K. At 293 K the N338T and N338A mutations lower Em1, the midpoint potential of the quinone/semiquinone (Q/SQ-·) couple from +47 mV for wild type to -11 and -37 mV, respectively. Mutations shift the Em2, the midpoint potential of the semiquinone/quinol (SQ-·/QH2) couple, to -19 and -49 mV for N338T and N338A compared to -30 mV for wild-type ETF-QO.
Figure 8 shows the [4Fe-4S]2+,1+ potentiometric titration curves for the two mutants and wild-type ETF-QO, calculated from EPR data at 15 K. The low temperature was used because the EPR signal becomes too broad to quantitate at higher temperatures. The Em of the [4Fe-4S]2+,1+ in the N338T (+48 mV) and N338A (+42 mV) mutants are within experimental uncertainty of the wild-type enzyme (+37 mV). These results show that mutations in the vicinity of the FAD do not impact the redox potentials of the [4Fe-4S]2+,1+. This is consistent with the observation that the [4Fe-4S]2+,1+ site mutations did not impact the redox potentials of the FAD (8).
Activity and Disproportionation Assays
The quinone reductase activity assay measures the rate at which electrons are shuttled through the enzyme from its biological electron donor, ETF, to its biological electron acceptor, ubiquinone. The relative quinone reductase activities for the N338T, N338A, Y501F, T525A, and Y501F/T525A mutated ETF-QOs are shown in Figure 9A. These values are reported as percents of wild-type activities to facilitate comparisons. Mutations in the vicinity of the [4Fe-4S]2+,1+, Y501F, T525A, and Y501F/T525A, lowered the relative activity to 37%, 35%, and 7% of wild type, respectively. Mutations in the vicinity of the FAD, N338T and N338A, lowered the relative activity to 58% and 35%, respectively (8). The differences can be explained by the fact that N338A mutation removes the hydrogen-bonding interaction with the FAD, whereas the N338T mutation only weakens the interaction. More generally it is observed that mutations at both centers decrease the quinone reductase activity. It can be concluded that both the [4Fe-4S]2+,1+ and the FAD are required for full biological function of the enzyme. It can also be concluded that the redox potentials of the two centers are carefully tuned in the wild-type enzyme and that small changes in potentials of one of the centers, such as in the N338T mutant, can cause a substantial decline in the enzyme’s activity.
Figure 9.

Activities of R. sphaeroides ETF-QO mutants in (A) quinone reduction and (B) disproportionation of ETF1e- compared to wild type. Error bars represent the standard deviation between multiple trials.
In the disproportionation assay, ETF-QO catalyzes the disproportionation of ETF1e-, where ETF1e- serves as both an electron acceptor and donor. Rate constants were measured for the disproportionation of human ETF1e- by mutant R. sphaeroides ETF-QOs and scaled to that for the wild-type enzyme (Figure 9B). The activity, expressed as an average turnover number for wild-type ETF-QO was 8 s-1, which is considerably lower with this heterologous coupled system than the 81.4 s-1 for the human ETF and ETF-QO homologue system (25). The turnover numbers found previously for the Y501F, T525A, and Y501F/T525A mutants near the iron-sulfur cluster decreased to 4.1, 5.1, and 0.66 s-1, respectively (8). These disproportionation rates correspond to roughly 51%, 64%, and 8% of wild-type activity. It is clear that mutations that remove a hydrogen-bonding interaction to the cysteine Sγ ligands of the [4Fe-4S]2+,1+ cluster reduce the rate at which ETF-QO can catalyze the disproportionation of ETF. This is not the case for the N338T and N338A mutations where a slight increase in disproportionation activity was seen. Relative to wild type, the N338A and N338T mutants had roughly 110% activity with rates of 8.8 and 8.9 s-1, respectively. The fact that changing the redox potential of the FAD has no negative effect on catalysis of the disproportionation of ETF SQ-· suggests that the FAD does not directly interact with the ETF. The increase in disproportionation rate in the N338A and N338T mutants could be caused by the lowering of the FAD Q/SQ-· redox potential. Lowering the Q/SQ-· redox potential makes the FAD harder to reduce. The point in the reductive titration curve where Δε corresponds to the same extent of FAD reduction for the wild-type and mutant proteins is at a lower potential for the mutants. At this potential there is a higher concentration of the [4Fe-4S]+ form of the cluster, which may enhance interaction with ETF.
Electron Transfer Mechanism
The previous finding that the ubiquinone reductase and disproportionation activities decreased when the midpoint potential of the [4Fe-4S]+ decreased demonstrates that reduction of the cluster is required for activity. Based on this evidence and the crystal structure of porcine ETF-QO (26), it was suggested that electrons from ETF enter ETF-QO through the [4Fe-4S]2+,1+ (8). The mutations in the present study shifted the redox potentials of the FAD while leaving the redox potential of the [4Fe-4S]2+,1+ unchanged. The shifted FAD redox potentials cause a decrease in the ubiquinone reductase activity but no decrease in the disproportionation activity. The lower ubiquinone reductase activity suggests that the FAD is required to shuttle electrons through the protein and reduce UQ and is consistent with crystallographic data (5). The slight increase in the disproportionation activity suggests that the FAD is not directly involved in the electron transfer between ETF and ETF-QO. Both of these findings support the theory that the electrons are transferred from ETF to ETF-QO through the [4Fe-4S]2+,1+ and then to the FAD which transfers the electrons to UQ as shown in the scheme:
CONCLUSION
The asparagine residue at position 338 of R. sphaeroides ETF-QO is postulated to interact with the FAD through hydrogen-bonding interactions. Replacement of the asparagine by threonine lowered Em1 by 58 mV and raised Em2 by 11 mV. The alanine mutation lowered Em1 by 84 mV and Em2 by 19 mV compared to wild type. It is proposed that the hydrogen-bonding interaction of the asparagine is completely removed by the alanine mutation but only weakened by the threonine mutation. No change in the midpoint potential of the [4Fe-4S]2+,1+ was detected in these mutants. No evidence of the mutations causing changes in the structure of the enzyme was observed based on the visible and EPR spectra. Quinone reductase activity was lowered by both mutations, demonstrating that reduction of the FAD is required for proper enzyme function. Rates of disproportionation of ETF1e- were not lowered by the mutations, suggesting that FAD does not interact directly with ETF. These results combined with the previous results for the mutations near the [4Fe-4S]2+,1+ support the proposal that electrons are accepted from ETF through the [4Fe-4S]2+,1+ and are then transferred to FAD which reduces UQ.
Footnotes
This work was supported by the National Institutes of Health, Grant NIBIB EB002807 (G.R.E. and S.S.E.), and the Children’s Hospital Research Foundation, Denver, CO (F.E.F.).
- CW
- continuous wave
- EPR
- electron paramagnetic resonance
- ETF
- electron transfer flavoprotein
- ETF-QO
- electron transfer flavoprotein-ubiquinone oxidoreductase
- FAD
- flavin adenine dinucleotide
- GAII
- glutaric acidemia type II
- MADD
- multiple acyl-CoA dehydrogenase deficiency
- MCAD
- medium-chain acyl-CoA dehydrogenase
- Q
- quinone
- Q1
- coenzyme Q1
- QH2
- hydroquinone/quinol
- SQ-·
- anionic semiquinone
- UQ
- ubiquinone
REFERENCES
- 1.Ruzicka FJ, Beinert H. A new iron-sulfur protein of the respiratory chain: a component of the fatty acid oxidation pathway. J. Biol. Chem. 1977;252:8440–8445. [PubMed] [Google Scholar]
- 2.Toogood HS, Leys D, Scrutton NS. Dynamics driving function-new insights from electron transferring flavoproteins and partner complexes. FEBS J. 2007;274:5481–5504. doi: 10.1111/j.1742-4658.2007.06107.x. [DOI] [PubMed] [Google Scholar]
- 3.Goodman SI, Binard RJ, Woontner MR, Frerman FE. Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein ubiquinone oxidoreductase (ETF-QO) gene. Mol. Genet. Metab. 2002;77:86–90. doi: 10.1016/s1096-7192(02)00138-5. [DOI] [PubMed] [Google Scholar]
- 4.Beresford MW, Pourfarzam M, Turnbull DM, Davidson JE. So doctor, what exactly is wrong with my muscles? Glutaric aciduria type II presenting in a teenager. Neuromuscular Disord. 2006;16:269–273. doi: 10.1016/j.nmd.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Frerman FE, Kim J-J. Structure of electron transfer flavoprotein ubiquinone oxidoreductase and electron transfer to the mitochondrial ubuiquinone pool. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16212–16217. doi: 10.1073/pnas.0604567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principals of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 7.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Usselman RJ, Fielding AJ, Frerman FE, Eaton GR, Eaton SS. Impact of Mutations on the Midpoint Potential of the[4Fe-4S]+ Cluster and on the Catalytic Activity in Electron Transfer Flavoprotein-ubiquinone Oxidoreductase (ETF-QO) Biochemistry. 2008;47:92–100. doi: 10.1021/bi701859s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breinlinger E, Niemz A, Rotello VM. Model systems for flavoenzyme activity. Stabilization of the flavin radical anion through specific hydrogen bond interactions. J. Am. Chem. Soc. 1995;117:5379–5380. [Google Scholar]
- 10.Miroux B, Walker JE. Overproduction of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 11.Wu G, Mansey SS, Wu S, Suresus KK, Foster MW, Cowan JA. Characterization of an iron-sulfur cluster assembly protein (ISU1) from Schizosaccharomyces pombe. Biochemistry. 2002;41:5024–5032. doi: 10.1021/bi016073s. [DOI] [PubMed] [Google Scholar]
- 12.Siegel LM. Quantitative determination of noncovalently bound flavins: types and methods of analysis. Methods Enzymol. 1978;53:419–429. doi: 10.1016/s0076-6879(78)53046-2. [DOI] [PubMed] [Google Scholar]
- 13.Ramsay RR, Steenkamp DJ, Husain M. Reactions of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase. Biochem. J. 1987;241:883–892. doi: 10.1042/bj2410883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beckmann JD, Frerman FE. Electron transfer flavoprotein-ubiquinone oxidoreductase from pig liver: purification and molecular, redox and catalytic properties. Biochemistry. 1985;24:3913–3921. doi: 10.1021/bi00336a016. [DOI] [PubMed] [Google Scholar]
- 15.Fersht A. Structure and Mechanism in Protein Science. W. H. Freeman and Co.; New York: 1988. pp. 104–105. [Google Scholar]
- 16.Patil PV, Ballou DP. The use of protocatechuate dioxygenase for maintaining anaerobic conditions in biochemical experiments. Anal. Biochem. 2000;286:187–192. doi: 10.1006/abio.2000.4802. [DOI] [PubMed] [Google Scholar]
- 17.Dutton PL. Redox potentiomery: determination of midpoint potentials of oxidation reduction components of biological electron transfer systems. Methods Enzymol. 1978;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- 18.Fielding AJ, Usselman RJ, Watmough N, Simkovic M, Frerman FE, Eaton GR, Eaton GR. Electron Paramagnetic Resonance Characterization and Interspin Distance Measurement of Electron Transfer Flavoprotein Ubiquinone Oxidoreductase (ETF-QO) J. Magn. Reson. 2008;190:222–232. doi: 10.1016/j.jmr.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eaton SS, Eaton GR. Determination of distances based on T1 and Tm effects. Biol. Magn. Reson. 2000;19:347–381. [Google Scholar]
- 20.Rakowsky MH, More KM, Kulikov AV, Eaton GR, Eaton SS. Time-Domain Electron Paramagnetic Resonance as a Probe of Electron-Electron Spin-Spin Interaction in Spin-Labeled Low-Spin Iron Porphyrins. J. Am. Chem. Soc. 1995;117:2049–2057. [Google Scholar]
- 21.Paulsen KE, Orville AM, Frerman FE, Lipscomb JD, Stankovich MT. Redox properties of electron-transfer flavoprotein ubiquinone oxidoreductase as determined by EPR-spectroelectrochemistry. Biochemistry. 1992;31:11755–11761. doi: 10.1021/bi00162a012. [DOI] [PubMed] [Google Scholar]
- 22.Eaton SS, Eaton GR. Relaxation times of organic radicals and transition metal ions. Biol. Magn. Reson. 2000;19:29–154. [Google Scholar]
- 23.Tegoni M, Silvestrini MC, Guigliarelli B, Asso M, Brunori M, Bertrand P. Temperature-jump and potentiometric studies on recombinant wild type and Y143F and Y254F mutants of Saccharomyces cereVisiae flavocytochrome b2: Role of the driving force in intramolecular electron transfer kinetics. Biochemistry. 1998;37:12761–12771. doi: 10.1021/bi980192z. [DOI] [PubMed] [Google Scholar]
- 24.Coves J, Zeghouf M, Macherel D, Guigliarelli B, Asso M, Fontecave M. Flavin mononucleotide-binding domain of the flavoprotein component of the sulfite reductase from Escherichia coli. Biochemistry. 1997;36:5921–5928. doi: 10.1021/bi9623744. [DOI] [PubMed] [Google Scholar]
- 25.Simkovic M, Degala GD, Eaton SS, Frerman FE. Expression of human electron transfer flavoprotein-ubiquinone oxidoreductase from a baculovirus vector: kinetic and spectral characterization of the human protein. Biochem. J. 2002;364:659–667. doi: 10.1042/BJ20020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Frerman FE, Kim J-J. Structure of electron transfer flavoprotein ubiquinone oxidoreductase and electron transfer to the mitochondrial ubuiquinone pool. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16212–16217. doi: 10.1073/pnas.0604567103. [DOI] [PMC free article] [PubMed] [Google Scholar]