

Abstract
Protein-based vaccines have been explored as a safer alternative to traditional weakened or killed whole organism based vaccination strategies and have been investigated for their ability to activate the immune system against certain cancers. For optimal stimulation of T lymphocytes, protein-based vaccines should deliver protein antigens to antigen presenting cells in the context of appropriate immunostimulatory signals, thus mimicking actual pathogens. In this report, we describe the synthesis, characterization, and biological evaluation of immunostimulatory acid-degradable microparticles, which are suitable delivery vehicles for use in protein-based vaccines and cancer immunotherapy. Using a 3′ conjugation strategy, we optimized the attachment of immunostimulatory CpG DNA to our vaccine carriers and demonstrated that under acidic conditions similar to that found in endosomal compartments, these new particles were capable of simultaneously releasing a model protein antigen and a CpG DNA adjuvant. We found in an in vivo cytotoxicity assay that the co-encapsulation of ovalbumin, a model antigen, and immunostimulatory agent in the same particle led to superior cytotoxic T lymphocyte activity compared to particles co-administered with adjuvant in an unbound form. In addition, we investigated the ability of these acid-degradable particles to induce protective immunity in the MO5 murine melanoma model and found that they were effective until tumor escape, which appeared to result from a loss of antigen expression by the cancer cells due to in vivo selection pressure.
Keywords: Protein-based vaccines, subunit vaccines, acid-degradable materials, 3′ CpG, cancer immunotherapy, microparticle, hydrogel
INTRODUCTION
Traditional vaccination strategies utilizing live attenuated viruses or inactivated (killed) pathogens have been employed widely for the treatment and prevention of disease. Although these approaches have generated successful results for a large number of diseases, safety concerns have led to the development of vaccines based on pathogen-derived protein antigens (subunit vaccines).1 As protein antigens have been discovered for certain tumors, there has also been significant interest in the activation of the immune system against cancer cells expressing these antigens.2, 3 To date, the most promising cancer immunotherapy approaches have relied on the collection and ex vivo manipulation of dendritic cells (DCs),4 which are crucial for the initiation and orchestration of adaptive immune responses, or the drastic in vivo expansion of DCs using the systemic administration of Flt3 ligand, a bone marrow growth factor.5 Despite promising early clinical results, these strategies may ultimately be too laborious and expensive for widespread application. An ideal protein-based vaccine should target and activate DCs in vivo and effectively generate protective immunity while limiting the use of potentially hazardous immunostimulatory agents.
To meet these challenging demands, antigen delivery vehicles have been explored for use in subunit vaccines and cancer immunotherapy.6 For optimal performance, antigen delivery vehicles should closely mimic the composition and immunological processing of actual pathogens; they should actively or passively target antigen presenting cells (APCs) such as DCs, protect the antigenic protein from degradation until reaching these cells, direct the nature of the resulting immune response (i.e., cellular vs. humoral responses), and lastly, induce APC maturation by interacting with elements of the innate immune system such as Toll-like receptors (TLRs). To address some of these issues, several strategies have been reported in the literature such as directly conjugating TLR ligands to protein antigens7 or co-encapsulating immunostimulatory agents and proteins in liposomes8 or hydrophobic polymeric particles.9
We have explored an antigen delivery system capable of performing all of the above-mentioned functions, which is based on acid-degradable, acetal-crosslinked, hydrogel particles designed for uptake by APCs.10, 11 Compared to non-degradable systems, these microparticles greatly enhance the efficacy of major histocompatibility complex class I (MHC I) antigen presentation and the subsequent activation of CD8+ cytotoxic T lymphocytes (CTLs),12, 13 which are crucial in cancer immunotherapy and other applications demanding strong cellular immune responses. To effect APC maturation, we have recently reported a method for the incorporation of an immunostimulatory CpG oligonucleotide into the polymer backbone of the particles.14 Following phagocytosis by APCs, these particles were designed to degrade in the acidic environment of endosomal vesicles (Figure 1a–b) and release their protein payload as well as a CpG-polymer conjugate capable of binding TLR9, an intracellular receptor for unmethylated viral and bacterial DNA.15, 16 TLR9 ligation results in APC activation and maturation and leads to the subsequent migration of APCs to draining lymph nodes.17–19 Although these microparticles were effective in generating antigen specific immunity, they required a relatively high CpG content, which, we hypothesized, was due to a loss in activity of the CpG caused by its covalent linkage to the polymer scaffold.
Figure 1.

(a) General scheme for the preparation and acid-catalyzed degradation of microparticles loaded with protein and immunostimulatory DNA (CpG = 5′-TCCATGACGTTCCTGACGTT-3′). (b) Following internalization by APCs, acid-labile particles degrade to release their protein payload and a polymer-CpG conjugate, which can interact with TLR9. (c) Synthesis of macromonomer 8 from 3′ amine-functionalized oligonucleotide 6.
Building on our previous work, we describe an optimized attachment strategy of CpG DNA to acid-degradable protein-loaded microparticles, which shows no loss in immunostimulatory activity as a consequence of the covalent linkage. Ovalbumin (OVA) was encapsulated in these microparticles as a model antigen as there exist a number of immunological assays and disease models specific to this protein. We then studied the ability of this optimized system to induce a CTL response in vivo. In addition, we investigated the ability of these particles to generate protective immunity to a lethal tumor challenge in the MO5 murine melanoma model. In these studies, our aim was to analyze the synergistic effects arising from the delivery of a protein antigen and adjuvant to the same APC. We were specifically interested in determining if our particles would perform better than treatments relying on the potentially hazardous systemic administration of free immunostimulatory agents. Herein, we demonstrate that by incorporating both protein antigen and a TLR agonist, our delivery vehicles, in a single formulation, provide all the necessary components for the induction of robust cellular immune responses.
EXPERIMENTAL PROCEDURES
General Procedures and Materials
All reagents were purchased from commercial sources and used without further purification unless specified otherwise. Water for buffers was purified to a resistance of 18 M Ω using a NANOpure Diamond (Barnstead) purification system. Organic solvents were dried by passing through two columns of neutral alumina within a commercial solvent purification apparatus. Functionalized oligonucleotides (1 and 6) with phosphorothioate backbones were purchased from Integrated DNA Technologies (CpG-1826 sequence: 5′-TCCATGACGTTCCTGACGTT-3′). Particle samples were characterized by scanning electron microscopy (SEM) using an S-5000 microscope (Hitachi) after being sputter coated with a 2 nm layer of a palladium/gold alloy. Fluorescence measurements were obtained on a Spectra Max Gemini XS (Molecular Devices), absorbance measurements were obtained using a Lambda 35 spectrophotometer (Perkin Elmer) or, for microplate-based assays using a Spectra Max 190 (Molecular Devices). Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF) data was collected on a PerSeptive Biosystems Voyager-DE PRO instrument (Applied Biosystems) in positive ion mode; accelerating voltage: 25,000 V, grid: 95%, guide wire: 0.2%, delay time: 550 ns. MALDI samples were prepared in a matrix of a saturated solution of 3-hydroxypicolinic acid in a 1:1 mixture of acetonitrile/water. A 17-mer oligonucleotide with a known mass was used as an internal standard.
Acid-degradable Crosslinker 3 was synthesized as previously described.12
NHS Ester 7 was synthesized as previously reported20 and recrystallized from ethyl acetate/hexanes (1:3) prior to use.
3′-Methacrylamide-modified CpG Macromonomer 8 was prepared according to a procedure adapted from Hermanson.21 Briefly, a solution of NHS ester 7 (4.33 mg, 23.6 μmol) in DMF (250 μL) was added to a solution of 3′-amine-modified oligonucleotide 6 (1.29 mg, 0.197 μmol) dissolved in a 50 mM phosphate buffer (pH 8.0, 250 μL). The resulting mixture was vortexed gently then incubated at 37 °C for 4 h in the dark. The reaction mixture was diluted with distilled water (2 mL) and washed with CH2Cl2 (500 μL). The modified oligonucleotide was purified using size exclusion gel filtration (PD-10 column, GE Healthcare) and lyophilized. The purified DNA conjugate was quantified prior to use (1.22 mg, 95% yield) by an absorbance measurement at 260 nm. [M+H]+ m/z = 6612.4. Found MALDI-TOF: [M+H]+ m/z = 6609.8.
General Acid-degradable Particle Preparation
Acid-degradable particles containing both ovalbumin (OVA, grade VI, Sigma Aldrich) and a CpG macromonomer were prepared using an inverse-suspension free-radical polymerization as described previously.14 Briefly, OVA (7.0 mg) was dissolved in a 300 mM phosphate buffer (pH 8.0, 200 μL) followed by acrylamide (62.3 mg, 0.88 mmol), crosslinker 3 (62.9 mg, 0.12 mmol), and ammonium persulfate (6.6 mg, 0.029 mmol). The CpG macromonomer (either 1 or 8, amounts noted below) was added to the monomer solution in the same buffer (50 μL) and the combined solution was quickly added to an organic phase (2.5 mL) composed of 2.25% Span 80 (w/v) and 0.75% Tween 80 (w/v) in hexanes. This mixture was then sonicated for 30 s using a Branson Sonifier 450 with an output setting of 2, a duty cycle of 40%, and with a ½” flat tip probe. Polymerization was initiated by the addition of N, N, N′, N′-tetramethylethylenediamine (25 μL), and the resulting mixture was stirred at rt for 10 min. The particles were isolated by centrifugation (1400 × g), washed with hexanes (3 × 2 mL) and acetone (4 × 2 mL), and finally dried overnight under vacuum to yield a fine white powder (typical yields: 50–60 mg).
Particles with low, medium or high CpG content
To obtain particles with different CpG loadings, the amount of CpG macromonomer (1 or 8) used in the particle preparation was varied. Specifically, 0.19 mg, 0.58 mg, or 1.71 mg of the appropriate macromonomer was used to prepare particles containing a low, medium, or high quantity of CpG, respectively.
CpG-free particles
Particles containing only OVA were prepared in the same fashion except that the monomers and protein were dissolved in 250 μL buffer and the CpG macromonomer was omitted.
OVA-free particles
Particles containing only CpG were prepared as described above except that the OVA was omitted from the aqueous phase.
Empty particles
Particles containing neither OVA nor CpG were prepared as described above omitting both the OVA and CpG macromonomer from the aqueous phase.
Oligonucleotide and Protein Quantification
Particle samples were weighed out in triplicate and washed twice with a 300 mM phosphate buffer (pH 8.0). The washing procedure involved first suspending the particles in buffer by vortexing and sonication, collecting the particles by centrifugation (2500 × g), and removing the supernatant. The particles were then suspended in a 300 mM acetate buffer (pH 5.0) at a final concentration of 5 mg/mL to hydrolyze the acetal crosslinks. The resulting solutions were allowed to stand overnight at rt and were subsequently analyzed for protein content using the MicroBCA assay (Pierce) or, for oligonucleotide content, using the fluorescent probe OliGreen (Invitrogen) according to the manufacturers’ protocol. OVA-free particles and CpG-free particles were degraded in the same manner as described above and used to determine background values for the MicroBCA and OliGreen assays, respectively.
Animals
C57BL/6 mice 6 to 8 weeks of age were purchased from the Jackson Laboratory. All mice were housed in the Stanford and UC Berkeley Animal Facilities in accordance with NIH guidelines. All procedures were approved by the Stanford and UC Berkeley Animal Care and Use Committees.
Cell Lines and Culture
MO5 is a B16 melanoma cell line transfected with OVA cDNA,22 and was a kind gift from Dr. K. Rock (University of Massachusetts Medical Center, Worcester, MA). MO5 cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum, 4.5 g/L glucose, 2 mM L-glutamine (GlutaMAX-I), 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 0.055 mM 2-mercaptoethanol, 10 mM HEPES, and 2 mg/ml G418 (for antibiotic selection). B16-F10 cells were obtained from ATCC, and were maintained in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum, 4.5 g/L glucose, and 2 mM L-glutamine. All culture media components were from Invitrogen-Gibco with the exception of the serum, which was from Hyclone. Cells were incubated in a water-jacketed 37 °C/5% CO2 incubator. Bone marrow derived dendritic cells (BMDCs) were isolated from C57BL/6 mice and cultured as previously described.23
Detection of Intracellular Interleukin-12 Expression
BMDCs were plated in a 96-well plate at a density of 2×105 cells/well and incubated at 37 °C for 20 h with particle or control samples normalized to 300 ng CpG/well, then treated with Brefeldin A (1 mM final concentration) and incubated for another 4 h. Cells were washed, surface-stained with CD11c-PE (a DC specific surface marker) and CD86-Pacific Blue. The cells were then washed, fixed, and permeabilized with Cytofix/Cytoperm (Pharmingen), and stained with IL-12p40-APC. Relative expression of surface markers and intracellular IL-12 was then determined by flow cytometry.
Simultaneous Release of Protein and CpG
Particles containing both OVA and 3′-linked CpG were weighed out in triplicate and washed as described above. The particles were suspended in phosphate buffered saline (PBS, pH 7.4) at a concentration of 5 mg/mL and incubated at 37 °C under gentle agitation (300 rpm) using a Thermomixer R heating block (Eppendorf). After various time points, the samples were centrifuged (2500 × g), the supernatant collected and an equivalent volume of fresh PBS added. After time points beyond 6 h, the particles were suspended in 300 mM acetate buffer (pH 5.0) instead of PBS. The collected supernatants were analyzed for protein and single-stranded DNA content using the assays described above. Values for complete protein and DNA release were determined by incubating the samples overnight in pH 5.0 buffer at 37 °C. OVA-free particles and CpG-free particles were degraded in parallel and used to determine an appropriate background for the MicroBCA and OliGreen assays, respectively. Results represent the mean ± standard deviation of triplicate measurements.
In Vivo Cytotoxicity Assay
C57BL/6 mice (n=10) were vaccinated subcutaneously (s.c.) with PBS or each particle type normalized to 50 μg ovalbumin in 200 μL PBS. For mice receiving CpG-free particles, a corresponding low, medium or high amount of free CpG-1826 DNA (phosphorothioate backbone, Oligos Etc.), was co-injected in a final volume of 200 μL PBS. At day 7 after vaccination, mice were injected intravenously with 1 × 107 CFSE-labeled target cells consisting of 50% SIINFEKL(OVA258–265)-pulsed splenocytes labeled with 5 μM 5-(and 6)-carboxyfluorescein diacetate, succinimidyl ester (CFSE, Invitrogen) and 50% unpulsed splenocytes labeled with 0.5 μM CFSE. Mice were tail vein bled 8 or 18 h after adoptive transfer and the CFSE profile of transferred cells was determined by flow cytometry for analysis of OVA-specific cytotoxicity. Results are presented as means ± 95% confidence intervals. Statistical significance was analyzed using a two-tailed Student’s t-Test.
In Vivo Tumor Protection Experiment
The protection experiment was performed with age-matched female C57BL/6 mice. All immunizations were administered s.c. using 27.5 gauge needles. On day − 7, mice were immunized in the left flank with PBS or with 3′ CpG/OVA particles, OVA particles plus free CpG, or free OVA plus free CpG in 100 μL PBS. All treatments doses were normalized to 50 μg OVA and 10 μg CpG per mouse. On day 0, tumors were established by administering a s.c. injection of 6 × 105 MO5 cells in 100 μL PBS into the contralateral flank of each mouse. Tumor growth was measured using calipers and mice with tumors 1.5 cm in diameter were removed from the experiment and euthanized. Mice were also removed if they showed other signs of pain or distress such as immobility, a hunched posture or a lack of eating. Tumors from euthanized mice were removed and stored at − 20 °C in Allprotect Tissue Reagent (Qiagen) prior to analysis. Survival data was analyzed using MedCalc software (version 8.2.0.2) and statistical significance was determined using a logrank test.
Analysis of Tumor Tissue for OVA Content by ELISA
Tumor samples from each mouse (10–55 mg) were blotted dry and placed in 2 mL microcentrifuge tubes to which 400 uL of T-PER extraction reagent containing Halt Protease Inhibitor Cocktail and 5 mM EDTA (Pierce) was added. Samples were disrupted/homogenized at rt for 30 s at full speed (33,000 rpm) using a TissueRuptor with disposable probes (Qiagen), then centrifuged three times (10,000 × g, rt, 5 min each) to remove residual solids. Samples were stored at − 20 °C before analysis. B16 and MO5 cells from in vitro culture (approximately 8×106 cells each) were prepared in the same manner.
The wells of a 96-well microtiter plate (Falcon Pro-Bind, BD Labware) were coated with 100 uL of a 1:1000 dilution of goat anti-OVA antibody (IgG fraction, MP Biomedicals) overnight at 4 °C. After washing the wells once with PBS containing 0.2% Tween-20 (PBS-T), 200 μL of blocking buffer (2% fetal bovine serum in PBS) was added to each well and incubated at 37 °C for 1 h. After washing once with PBS-T, 100 μL of samples diluted in blocking buffer were added to the wells and incubated for 1 h at rt. The wells were washed three times with PBS-T, and 100 μL of a 1:4000 dilution of rabbit anti- OVA antibody (IgG fraction, MP Biomedicals) in blocking buffer was added to each well. After incubation at rt for 1 h, the wells were washed four times with PBS-T, and 100 μL of a 1:5000 dilution of AffiniPure HRP-conjugated donkey anti-rabbit IgG antibody (Jackson Immunoresearch) in blocking buffer was added to each well. After incubation at rt for 1 h, the wells were washed six times with PBS-T, and the plate was developed with 100 μL of 1-Step Ultra TMB-ELISA per well for 20 min at rt. The development was stopped by adding 100 μL of 2 M H2SO4, and absorbances were recorded at 450 nm. Dilutions of free OVA were used as a standard, and OVA content was normalized to total protein in each sample as determined by the Bradford method24 using the Coomassie Assay Protein Reagent (Pierce) according to the manufacturer’s instructions. Results represent the mean ± standard deviation of triplicate measurements.
Analysis of Tumor Tissue for OVA-encoding DNA
Tumor samples from each mouse (5–20 mg) were blotted dry and placed in 2 mL microcentrifuge tubes. Samples were disrupted/homogenized at rt for 30 s at full speed (33,000 rpm) using a TissueRuptor with disposable probes (Qiagen). Genomic DNA was purified with the AllPrep DNA/RNA/Protein Mini Kit (Qiagen) according to the manufacturer’s instructions. Genomic DNA was similarly purified from B16 and MO5 cells from in vitro culture (approximately 6×105 cells each).
Genomic DNA was amplified using the HotStarTaq Master Mix Kit (Qiagen), according to the manufacturer’s instructions. OVA transcripts were amplified using the forward primer 5′-(GGCTCCATCGGCGCAGCAAG)-3′ and reverse primer 5′-(GGGGAAACACATCTGCCAAA)-3′.25 GAPDH was amplified using QuantiTect Primer Assay validated primers (Qiagen). Each PCR reaction mixture (50 μl) contained 0.25 μg of template DNA and 25 pmol of each primer. PCR products were analyzed by electrophoresis on a 1% agarose gel (120 V) using TAE buffer (pH 8.0, 40 mM Tris, 20 mM acetic acid, 1 mM EDTA). The gel was stained with ethidium bromide and imaged under UV irradiation.
RESULTS AND DISCUSSION
Synthesis of CpG-loaded acid-degradable particles
We recently described the incorporation of a CpG oligonucleotide into acid-degradable polyacrylamide particles to facilitate co-delivery of a model protein antigen, OVA, and an immunostimulatory agent to the same APC (Figure 1a–b).14 CpG DNA was chosen as an adjuvant because its receptor, TLR9, is located in endosomal compartments, the same location in which our particles are designed to degrade.17, 19 Incorporation of the immunostimulatory DNA was achieved by copolymerizing a methacrylamide-modified CpG oligonucleotide, macromonomer 1, with acrylamide (2) and an acid-degradable crosslinker (3). In initial work, the CpG was covalently linked to the particle’s polymer scaffold through its 5′ terminus.26 Although these particles were found to generate antigen specific immunity in a number of assays, we hypothesized that the covalent attachment of the CpG oligonucleotide to the high molecular weight polymer in conjugate 4 may diminish its activity and lead to suboptimal DC activation. This hypothesis was supported by in vitro experiments in which bone marrow derived dendritic cells (BMDCs) were activated by free CpG oligonucleotides to a greater extent than with particles containing an equivalent amount of covalently bound oligonucleotide (see below).
In contrast to the 5′ end, modifications involving the 3′ terminus of CpG oligonucleotides are generally better tolerated and cause significantly less, if any, reduction in immunostimulatory activity.27, 28 Therefore, particles were prepared using a 20-mer of single-stranded CpG DNA that was modified on its 3′ terminus with a methacrylamide group (monomer 8). Polymerizable CpG monomer 8 was prepared in one step from N-hydroxysuccinimidyl ester 7 and amine functionalized oligonucleotide 6 with a good recovery of the product (Figure 1c). Following purification by size exclusion chromatography, macromonomer 8 was characterized using MALDI-TOF mass spectrometry and found to have a molecular weight within 0.04% of the predicted value. High-performance liquid chromatography and polyacrylamide gel electrophoresis were used to analyze the purity of CpG conjugate 8 (Figures S1 and S2). Both methods revealed the presence a single species, indicating that no significant degradation or polymerization reactions occurred during the synthesis and purification of 8.
Macromonomer 8 was used to prepare acid-degradable particles containing 3′-linked CpG DNA. Characterization of the particles by SEM (Figure 2) showed that they were similar in size, shape, and morphology to previously prepared particles with 5′-linked CpG.14 Based on SEM data, the particles were approximately 100–800 nm in diameter in the dry state, which we have previously found to be a suitable size for use in protein-based vaccines.29 To quantify the amount of CpG DNA incorporated, the particles were degraded in acidic buffer and the resulting solutions analyzed via a fluorescence-based assay utilizing the commercially available OliGreen reagent. Both CpG macromonomers were incorporated equally well into the particles with efficiencies of 60–70% based on the masses of starting monomers. Particles with loadings between 1 and 9 μg CpG/mg particles (see below) were easily obtained by varying the initial feed of monomers 1 or 8. Analysis of hydrolyzed particles by gel permeation chromatography was used to confirm the successful copolymerization of both CpG macromonomers into the polyacrylamide backbone of the carriers (see Figure S3).
Figure 2.

Representative SEM image of acid-degradable particles containing 3′-linked CpG.
3′-linked CpG particles are more immunostimulatory than 5′-linked CpG particles
To assess the relative immunostimulatory activity of acid-degradable particles containing 1 and 8, sets of particles were prepared containing no protein antigen and a low, medium or high loading of either 3′ or 5′-linked CpG (approximately 1, 3, and 9 μg CpG oligonucleotide per mg particles, respectively). These particles were then evaluated for their capacity to induce activation of immature BMDCs in vitro. The percentage of DCs producing interleukin-12 (IL-12),30 a pro-inflammatory cytokine, after overnight culture with CpG-containing particles was used to compare the stimulatory capacity of 3′-and 5′-linked CpG. For this experiment, particle concentrations were normalized such that all DCs were pulsed with the equivalent of 300 ng of CpG. As the particle formulations were almost entirely polyacrylamide by mass, CpG-free particles were used as a negative control to ensure that the carriers were not inherently immunostimulatory. As demonstrated in Figure 3, the 3′-linked CpG particles induced a consistently higher percentage of BMDCs to produce IL-12 compared to 5′-linked particles. In contrast to the 5′-linked CpG particles, DC activation levels correlated well with the total amount of CpG used in the 3′-linked particles, except for the particles with the lowest loading, suggesting that there might be a minimum CpG/particle ratio in order to achieve optimal DC activation. For all concentrations tested, the medium and high loaded 3′ CpG particles were as stimulatory, if not more so, than an equivalent amount of soluble CpG, indicating no loss in activity of the particle-associated CpG (data not shown). These findings are therefore in agreement with the structure-activity studies discussed above.
Figure 3.

IL-12 production by CD11c+ BMDCs pulsed with 3′- or 5′-linked CpG particles. Numbers in histograms represent percentages of intracellular IL-12 positive DCs after overnight culture with particles. The CpG concentration in each sample was normalized to 300 ng/mL. Data are representative of three experiments with similar results.
In addition, the expression levels of CD86 on BMDCs treated with the CpG-loaded particles described above were determined. CD86 provides a necessary co-stimulatory signal to naïve T cells during priming via CD86–CD28 interaction.31 As was the case for the IL-12 assay, the 3′-linked CpG particles were superior at inducing high levels of CD86 expression in BMDCs compared to 5′-linked particles (see Figure S4 for representative flow cytometry histograms). These data demonstrate that by carefully controlling the direction of conjugation, CpG DNA can be covalently linked to particulate vaccine carriers without compromising the overall immunostimulatory activity of the oligonucleotide. Based on these results, all subsequently used CpG-loaded particles involved attachment exclusively via a 3′-linkage.
Co-encapsulation of CpG and protein antigen leads to superior in vivo CTL responses
For the generation of optimal immune responses, APCs need to receive antigens in the context of appropriate maturation signals. In the natural processing of microbes by APCs, these signals are provided by elements of the innate immune system which are capable of recognizing a diverse array of pathogen-associated molecular patterns (PAMPs) such as unmethylated CpG dinucleotide motifs, lipopolysaccharides, heat shock proteins, flagellin, and double-stranded RNA.32, 33 Recognition of one or more PAMPs induces APC maturation, which typically includes the secretion of cytokines, upregulation of co-stimulatory molecule expression and increased antigen presentation. In comparison, APC processing of protein antigens without concomitant immunostimulation may lead to T cell anergy and immunological tolerance rather than the desired immune response.34
Having determined the optimal CpG attachment chemistry, OVA was next encapsulated in the acid-degradable, CpG-loaded particles to provide a vehicle capable of delivering both a model antigen and an immunostimulatory agent to the same APC. As was done for CpG quantification, protein-loaded particles were degraded in acidic buffer and the resulting solutions were analyzed for OVA content using a bicinchoninic acid-based assay. The protein loading was found to be approximately 40 μg OVA/mg particles, an incorporation efficiency of about 70% based the mass of starting materials, and this loading was used in all particles tested in this report.
To investigate the ability of the particles to simultaneously deliver an antigen and an immunostimulatory agent, a release experiment was performed to model the behavior of particles prior to and after uptake by APCs (Figure 4). Particles containing both 3′-linked CpG and OVA (3′ CpG/OVA particles) were incubated at 37 °C in PBS (pH 7.4), modeling the neutral environment prior to uptake by APCs. After 6 hours, the PBS was replaced with an acidic buffer to simulate phagocytosis and particle degradation by APCs. At various time points throughout the experiment, the particle solutions were analyzed for the release of OVA and CpG using the assays described above. After 6 hours under neutral conditions the particles had released no more than 10% of their encapsulated OVA or CpG. In contrast, nearly all of the OVA and CpG was released within 2 hours after introduction of the acidic buffer. These bulk release data suggest that our particles are indeed capable of simultaneously delivering antigen and immunostimulatory CpG DNA, which should be beneficial in the generation of robust immune responses.
Figure 4.
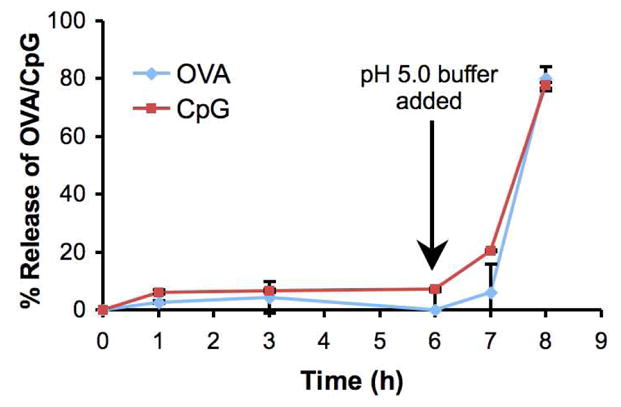
Release of OVA and 3′-linked CpG from acid-degradable particles in a bulk model phagocytosis experiment. 3′-CpG/OVA particles were first incubated at 37 °C at physiological pH (7.4). After 6 hours, the buffer was replaced with acidic buffer (pH 5). The release of OVA and CpG from the particles was quantified at various time points before and after acidification. Total release was determined after overnight incubation at pH 5 and 37 °C.
To investigate the synergistic effects arising from the co-delivery of antigen and immunostimulatory agent to the same APC in vivo, three types of 3′ CpG/OVA particles were prepared, each with a constant protein loading (approximately 40 μg OVA/mg particles) and a low, medium or high loading of CpG (approximately 1, 3 or 9 μg CpG per mg particles, respectively). These particles were then tested for their capacity to induce OVA-specific CTL activity compared to OVA-loaded particles co-administered with a corresponding amount of free CpG (Figure 5). In this experiment, mice were vaccinated subcutaneously (s.c.) with the particle samples described above normalized to a constant dose of OVA (50 μg). Seven days later, an equal mixture of two populations of CFSE labeled splenocytes was injected intravenously. A target cell population was pulsed with SIINFEKL (OVA258–265), the immunodominant OVA-derived CD8+ T cell epitope in C57BL/6 mice. As a control, a second population was not pulsed with SIINFEKL and was stained with a lower CFSE concentration. To determine the extent of antigen-specific cytotoxicity, the relative amount of each population remaining after 8 and 18 hours was quantified by flow cytometric analysis (see Figure S5 for representative histograms). The percentage of target cell-specific lysis was found to be highly dependent on the absolute quantity of CpG administered at both time points tested. In addition, for all CpG particle loadings and for both time points studied, the 3′ CpG/OVA combination particles induced a significantly higher level of specific lysis (p < 0.01) of target cells compared to the co-injected mixture of OVA particles plus an equivalent amount of free CpG. The largest differences (approximately 2-fold) in induced cytotoxicity between these two samples were observed at the lower CpG doses, and after 18 hours these differences tended to decrease as the amount of CpG increased. Beyond suggesting co-delivery related synergistic effects, our data indicate that, at least in the case of our optimized particles, the magnitude of these effects increases as stimulant doses are lowered. This is a significant finding because, for vaccine purposes, it may be desirable to avoid the use of free adjuvants or at least use the lowest possible dose in order to minimize the likelihood of developing autoimmune disorders or provoking other side effects.35
Figure 5.

Specific lysis of SIINFEKL-pulsed target cells in mice immunized with PBS, 3′ CpG/OVA particles, or OVA particles plus free CpG after 8 and 18 hours using 3 different CpG doses. High, medium and low CpG doses correspond to 9, 3, and 1 μg CpG per mg particles (or an equivalent amount of free CpG) respectively. Specific lysis was determined using flow cytometry by comparing the remaining population of stained target cells to a population of unpulsed control cells (see Figure S5). In all cases, vaccination with 3′ CpG/OVA combination particles produced a superior immune response compared to the co-injection of OVA particles plus free CpG (* p < 0.01, n = 10, mean ± 95% confidence intervals).
3′ CpG/OVA particles enhance survival in a tumor protection experiment
Having demonstrated their promising in vivo activity, the ability of the 3′ CpG/OVA particles to induce protective immunity and prevent the growth of an OVA expressing tumor using a murine melanoma model system was investigated. For this study, different groups of C57BL/6 mice (n = 10) were vaccinated (on day −7) s.c in their left flank with PBS, OVA-containing particles plus free CpG, 3′ CpG/OVA particles, or free CpG plus free OVA. In the case of the treatment groups, each mouse received the equivalent of 50 μg of OVA and 10 μg of CpG. Seven days later (day 0), the mice were challenged with a s.c. injection in the contralateral flank of 6×105 MO5 cells, a derivative of the B16 melanoma cell line transfected with the OVA gene,22 and the subsequent tumor growth was monitored for the next 64 days. The survival of each treatment group as a function of time is shown in Figure 6 (see Figure S6 for plots of tumor volume vs. time). The mice in the PBS group developed tumors first (9 of 10 mice had visible tumors by day 9) and all mice from this group were removed from the study due to large tumor burdens within 28 days. In contrast, tumors were not visible in those mice receiving the 3′ CpG/OVA particles until much later, with only two mice developing visible tumors by day 23. Throughout the study, mice in this group maintained the highest probability of survival compared to all other groups. The probability of survival for the OVA-particle plus free CpG group remained higher than the free OVA plus free CpG group until approximately day 33 at which point they became nearly identical. Importantly, the 3′ CpG/OVA combination particles performed at least as well, if not better, than two other treatments employing the potentially hazardous administration of unbound immunostimulatory agents. Based on the results obtained in the in vivo cytotoxicity experiment described above, it is possible that more pronounced differences between the treatment groups may have been observed if the CpG dose were lowered.
Figure 6.

Kaplan-Meier survival plot from in vivo tumor protection experiment. The different groups of C57BL/6 mice (n = 10) were immunized on day −7 s.c in their left flank with PBS, OVA-containing particles plus free CpG, 3′ CpG/OVA particles, or free CpG plus free OVA. In the case of the treatment groups, each mouse received the equivalent of 50 μg of OVA and 10 μg of CpG. Seven days later (day 0), the mice were challenged with a s.c. injection in the right flank of 6×105 MO5 cells, a derivative of the B16 melanoma cell line transfected with the OVA gene, and the subsequent tumor growth was monitored for the next 64 days. The survival of each treatment group as a function of time is plotted.
Tumor escape is caused by a loss of OVA expression in vivo
The co-encapsulation of antigen and immunostimulatory agent in the same acid-degradable vehicle appears to have produced a robust immune response, however complete tumor rejection was not achieved even in this experimental group. We hypothesized that the late onset of tumors in the treatment groups may have been related to the loss of OVA expression by the MO5 cells in vivo. In addition to the absence of antibiotics, which are used to select for OVA-expressing cells in culture, the selection pressure of a strong in vivo CTL response may have led to the appearance of tumors with low or even no OVA expression, a phenomenon referred to as immunoediting.36 To explore this hypothesis, we excised and saved tumor tissue from each mouse as it was removed from the experiment. After the final mouse was removed from the study on day 54, the tumor tissue samples were analyzed using an ELISA to look for the presence of OVA (Figure 7a). Consistent with our hypothesis, we only observed OVA protein above background levels in tumor tissue removed from mice in the PBS group. Additionally, the OVA content from tumors in this group decreased the longer the mice remained in the study. To further explore our hypothesis, the tumor tissue was analyzed for the presence of OVA-encoding DNA using PCR. As shown in Figure 7b, only mice in the PBS group were positive for this gene, and, consistent with our ELISA data, the intensity of the corresponding band decreased the longer the mice remained in the study. In contrast, the tumors from mice in all of the treatment groups were negative for the OVA-encoding gene. Based on these data, the MO5 cells used in our study likely lost their expression of OVA after approximately 25 days in vivo. This result agrees well with a study by Goldberger et al. in which mice with greater than 90% OVA-specific CTLs developed tumors after approximately the same time following a challenge of MO5 cancer cells.25 Although it is clear that the treatment groups generated a significantly better immune response compared to PBS (p < 0.0001), we found no significant differences between the 3′ CpG/OVA particles and treatment with free antigen and CpG (p = 0.38) or treatment with OVA particles plus free CpG (p = 0.21). It must be noted however, that the loss of OVA expression by the tumor cells complicates the interpretation of these data.
Figure 7.

Time dependent loss of OVA expression in tumors. (a) Loss of OVA expression in tumors from treatment groups, but not PBS group, as analyzed by ELISA. The concentration of OVA in tumor tissue from the first, middle and last mouse from each group to be removed from the study was analyzed and compared to MO5 and B16 cells prepared from cultures (positive and negative controls, respectively). (b) Loss of OVA gene in tumors from treatment groups, but not PBS group as analyzed by PCR. The presence of the OVA gene (1.1 kb) in the first, middle and last mouse from each group to be removed from the study was analyzed.
Despite the tumor escape, these tumor rejection and other results are encouraging and provide validation for our proof-of-concept acrylamide-based system. In future immunotherapy experiments using more biocompatible particle platforms,37 we plan to explore the incorporation of multiple natural cancer antigens in the same carrier, which may make immune system evasion by the tumor cells more difficult.
CONCLUSION
This study demonstrates the validity of the design of a model delivery vehicle for protein-based vaccines that is capable of simultaneously delivering protein antigens and immunostimulatory CpG DNA. This design is based on an understanding of the uptake and processing of pathogenic species, which consist of discrete units composed of antigens and immunostimulatory moieties. A clear advantage of the CpG conjugation strategy used is that it avoids the potentially hazardous use of unbound immunostimulatory agents. In an in vivo CTL activity assay, we found that for all time points and stimulant concentrations tested, packaging CpG DNA and protein antigen in a single particle formulation induced significantly stronger immune responses compared to controls in which these components were administered separately. In a preliminary cancer immunotherapy experiment, we found that our microparticles were capable of inducing protective immunity in a mouse model until the tumors lost antigen expression, presumably due to selection pressure by the immune system. In general, this study validates the concept of covalently attaching immunostimulatory CpG DNA to delivery vehicles for use in antigen-based vaccine formulations.
Supplementary Material
Acknowledgments
The authors thank the National Institutes of Health for funding of this research (NBIB grant R01-EB005824). We thank Ann Fischer and Michelle Yasukawa for help with cell culture studies and Lianjun Shen (University of Massachusetts Medical Center, Worcester, MA) for advice on MO5 culture and ELISA assay development and Professors Ellman and Bertozzi (UC Berkeley) for access to equipment.
Footnotes
Supporting Information Available: Figures S1–S6, characterization of macromonomer 8 and CpG-polymer conjugates, representative flow cytometry histograms, and plots of tumor volume vs. time. This material is available free of charge at http://pubs.acs.org.
Contributor Information
Tristan T. Beaudette, College of Chemistry, University of California, Berkeley, California 94720-1460
Eric M. Bachelder, College of Chemistry, University of California, Berkeley, California 94720-1460
Joel A. Cohen, College of Chemistry, University of California, Berkeley, California 94720-1460
Allie C. Obermeyer, College of Chemistry, University of California, Berkeley, California 94720-1460
Kyle E. Broaders, College of Chemistry, University of California, Berkeley, California 94720-1460
Jean M. J. Fréchet, College of Chemistry, University of California, Berkeley, California 94720-1460.
Eun-Suk Kang, Department of Pathology, Stanford University School of Medicine, Palo Alto, California, 94304..
Ines Mende, Department of Pathology, Stanford University School of Medicine, Palo Alto, California, 94304..
William W. Tseng, Department of Pathology, Stanford University School of Medicine, Palo Alto, California, 94304.
Matthew G. Davidson, Department of Pathology, Stanford University School of Medicine, Palo Alto, California, 94304.
Edgar G. Engleman, Department of Pathology, Stanford University School of Medicine, Palo Alto, California, 94304..
LITERATURE CITED
- 1.Byrd W, de Lorimier A, Zheng ZR, Cassels FJ. Microencapsulated subunit vaccine approach to enterotoxigenic Escherichia coli and other mucosal pathogens. Adv Drug Delivery Rev. 2005;57(9):1362–1380. doi: 10.1016/j.addr.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 2.van der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, Chapiro J, Van den Eynde BTJ, Brasseur F, Boon T. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev. 2002;188:51–64. doi: 10.1034/j.1600-065x.2002.18806.x. [DOI] [PubMed] [Google Scholar]
- 3.Sheng KC, Pietersz GA, Wright MD, Apostolopoulos V. Dendritic cells: Activation and maturation - Applications for cancer immunotherapy. Curr Med Chem. 2005;12(15):1783–1800. doi: 10.2174/0929867054367248. [DOI] [PubMed] [Google Scholar]
- 4.Tacken PJ, de Vries IJM, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7(10):790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 5.Merad M, Sugie T, Engleman EG, Fong L. In vivo manipulation of dendritic cells to induce therapeutic immunity. Blood. 2002;99(5):1676–1682. doi: 10.1182/blood.v99.5.1676. [DOI] [PubMed] [Google Scholar]
- 6.Reddy ST, Swartz MA, Hubbell JA. Targeting dendritic cells with biomaterials: developing the next generation of vaccines. Trends Immunol. 2006;27(12):573–579. doi: 10.1016/j.it.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Datta SK, Cho HJ, Takabayashi K, Horner AA, Raz E. Antigen-immunostimulatory oligonucleotide conjugates: mechanisms and applications. Immunol Rev. 2004;199(1):217–226. doi: 10.1111/j.0105-2896.2004.00149.x. [DOI] [PubMed] [Google Scholar]
- 8.Jerome V, Graser A, Muller R, Kontermann RE, Konur A. Cytotoxic T lymphocytes responding to low dose TRP2 antigen are induced against B16 melanoma by liposome-encapsulated TRP2 peptide and CpG DNA adjuvant. J Immunother. 2006;29(3):294–305. doi: 10.1097/01.cji.0000199195.97845.18. [DOI] [PubMed] [Google Scholar]
- 9.Kaiser-Schulz G, Heit A, Quintanilla-Martinez L, Hammerschmidt F, Hess S, Jennen L, Rezaei H, Wagner H, Schatzl HM. Polylactide-coglycolide microspheres coencapsulating recombinant tandem prion protein with CpG-oligonucleotide break self-tolerance to prion protein in wild-type mice and induce CD4 and CD8 T cell responses. J Immunol. 2007;179(5):2797–2807. doi: 10.4049/jimmunol.179.5.2797. [DOI] [PubMed] [Google Scholar]
- 10.Murthy N, Thng YX, Schuck S, Xu MC, Frechet JMJ. A novel strategy for encapsulation and release of proteins: Hydrogels and microgels with acid-labile acetal cross-linkers. J Am Chem Soc. 2002;124(42):12398–12399. doi: 10.1021/ja026925r. [DOI] [PubMed] [Google Scholar]
- 11.Murthy N, Xu MC, Schuck S, Kunisawa J, Shastri N, Frechet JMJ. A macromolecular delivery vehicle for protein-based vaccines: Acid-degradable protein-loaded microgels. Proc Natl Acad Sci U S A. 2003;100(9):4995–5000. doi: 10.1073/pnas.0930644100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Standley SM, Kwon YJ, Murthy N, Kunisawa J, Shastri N, Guillaudeu SJ, Lau L, Frechet JMJ. Acid-degradable particles for protein-based vaccines: Enhanced survival rate for tumor-challenged mice using ovalbumin model. Bioconjugate Chem. 2004;15(6):1281–1288. doi: 10.1021/bc049956f. [DOI] [PubMed] [Google Scholar]
- 13.Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Frechet JMJ. Acetalated Dextran is a Chemically and Biologically Tunable Material for Particulate Immunotherapy. Proc Natl Acad Sci U S A. 2009;106(14):5497–5502. doi: 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Standley SM, Mende I, Goh SL, Kwon YJ, Beaudette TT, Engleman EG, Frechet JMJ. Incorporation of CpG oligonucleotide ligand into protein-loaded particle vaccines promotes antigen-specific CD8 T-cell immunity. Bioconjugate Chem. 2007;18(1):77–83. doi: 10.1021/bc060165i. [DOI] [PubMed] [Google Scholar]
- 15.Krieg aM, Yi aK, Matson S, Waldschmidt TJ, Bishop Ga, Teasdale R, Koretzky Ga, Klinman DM. Cpg Motifs in Bacterial-DNA Trigger Direct B-Cell Activation. Nature. 1995;374(6522):546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 16.Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4(4):248–257. doi: 10.1038/nri1329. [DOI] [PubMed] [Google Scholar]
- 17.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7(1):49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 18.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discovery. 2006;5(6):471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 19.Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Delivery Rev. 2008;60(7):795–804. doi: 10.1016/j.addr.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Mammen M, Dahmann G, Whitesides GM. Effective Inhibitors of Hemagglutination by Influenza-Virus Synthesized from Polymers Having Active Ester Groups - Insight into Mechanism of Inhibition. J Med Chem. 1995;38(21):4179–4190. doi: 10.1021/jm00021a007. [DOI] [PubMed] [Google Scholar]
- 21.Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego: 1996. [Google Scholar]
- 22.Falo LD, Kovacsovicsbankowski M, Thompson K, Rock KL. Targeting Antigen into the Phagocytic Pathway in-Vivo Induces Protective Tumor-Immunity. Nat Med. 1995;1(7):649–653. doi: 10.1038/nm0795-649. [DOI] [PubMed] [Google Scholar]
- 23.Bachelder EM, Beaudette TT, Broaders KE, Paramonov SE, Dashe J, Frechet JMJ. Acid-degradable polyurethane particles for protein-based vaccines: Biological evaluation and in vitro analysis of particle degradation products. Mol Pharmaceutics. 2008;5(5):876–884. doi: 10.1021/mp800068x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradford MM. Rapid and Sensitive Method for Quantitation of Microgram Quantities of Protein Utilizing Principle of Protein-Dye Binding. Anal Biochem. 1976;72(1–2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Goldberger O, Volovitz B, Machlenkin A, Vadai E, Tzehoval E, Eisenbach L. Exuberated numbers of tumor-specific T cells result in tumor escape. Cancer Res. 2008;68(9):3450–3457. doi: 10.1158/0008-5472.CAN-07-5006. [DOI] [PubMed] [Google Scholar]
- 26.Macromonomer 1 was used in initial work due to synthetic convenience. As opposed to macromonomer 8, this molecule can be prepared entirely using solid phase DNA synthesis.
- 27.Kandimalla ER, Bhagat L, Yu D, Cong YP, Tang J, Agrawal S. Conjugation of ligands at the 5 ′-end of CpG DNA affects immunostimulatory activity. Bioconjugate Chem. 2002;13(5):966–974. doi: 10.1021/bc0200374. [DOI] [PubMed] [Google Scholar]
- 28.Lenert P, Goeken AJ, Ashman RF. Extended sequence preferences for oligodeoxyribonucleotide activity. Immunology. 2006;117(4):474–481. doi: 10.1111/j.1365-2567.2006.02320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen JA, Beaudette TT, Tseng WW, Bachelder EM, Mende I, Engleman EG, Frechet JMJ. T-Cell Activation by Antigen-Loaded pH-Sensitive Hydrogel Particles in Vivo: The Effect of Particle Size. Bioconjugate Chem. 2009;20(1):111–119. doi: 10.1021/bc800338n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3(2):133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 31.Sharpe AH, Freeman GJ. The B7–CD28 superfamily. Nat Rev Immunol. 2002;2(2):116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 32.Medzhitov R, Janeway C. The Toll receptor family and microbial recognition. Trends Microbiol. 2000;8(10):452–456. doi: 10.1016/s0966-842x(00)01845-x. [DOI] [PubMed] [Google Scholar]
- 33.Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 35.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6(11):823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 37.Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Frechet JMJ. Acetal-derivatized dextran: An acid-responsive biodegradable material for therapeutic applications. J Am Chem Soc. 2008;130(32):10494–10495. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.