

Abstract
Objective
The receptor tyrosine kinase Axl and its ligand Gas6 are involved in the development of renal diabetic disease. In vascular smooth muscle cells (VSMC) Axl is activated by reactive oxygen species and stimulates migration and cell survival, suggesting a role for Axl in the vascular complications of diabetes.
Methods and Results
We investigated the effect of varying glucose concentration on Axl signaling in VSMC. Glucose exerted powerful effects on Gas6-Axl signaling with greater activation of Akt and mTOR in low glucose, and greater activation of ERK1/2 in high glucose. Plasma membrane distribution and tyrosine phosphorylation of Axl were not affected by glucose. However, co-immunoprecipitation studies demonstrated that glucose changed the interaction of Axl with its binding partners. Specifically, binding of Axl to the p85 subunit of PI3-kinase was increased in low glucose, whereas binding to SHP-2 was increased in high glucose. Furthermore, Gas6-Axl induced migration was increased in high glucose, while Gas6-Axl mediated inhibition of apoptosis was greater in low glucose.
Conclusion
This study demonstrates a role for glucose in altering Axl signaling through coupling to binding partners, and suggests a mechanism by which Axl contributes to VSMC dysfunction in diabetes.
Keywords: Axl, glucose, vascular smooth muscle migration, signaling
Introduction
Hyperglycemia alters vascular smooth muscle cell (VSMC) signaling, which is a major factor in the development of vascular complications of diabetes such as atherosclerosis and hypertension 1-3. The receptor tyrosine kinase Axl is expressed in several cell types including VSMC 4, 5. Axl is activated by Growth Arrest Gene 6 (Gas 6), which is a homologue of Protein S 6, 7. This leads to stimulation of downstream signaling cascades including the PI3K-Akt pathway, and ERK1/2 4, 8-10. Gas6 activation of Axl in VSMC increases directed migration and inhibits apoptosis 11, 12. There are several reports that suggest a role for Axl in the pathogenesis of vascular and diabetic disease. Expression of Axl and Gas6 are increased in the glomerulus of diabetic rats. Furthermore, diabetic nephropathy is less severe in Gas6 knockout mice and when Gas6 activity is inhibited by warfarin treatment 13. Axl is highly upregulated in balloon injured carotid arteries with a time course paralleling that of neointima formation 14. Axl mRNA and protein level in cultured VSMC are also increased by the G-protein-coupled receptor agonists thrombin and angiotensin II 14. Axl is activated by H2O2, which is increased in vascular injury and hyperglycemia, in both VSMC and ex vivo vessels 15. In addition, neointima formation induced by injury is decreased in Axl knockout mice 15. These studies suggest Gas6-Axl represents an important pathogenic mechanism for cardiovascular and renal complications associated with diabetes.
Recently, we demonstrated that high glucose enhanced phosphorylation of Akt and ERK1/2 by angiotensin II through alterations in epidermal growth factor receptor (EGFR) N-glycosylation 16. Based on this study, we hypothesized that glucose would modulate Axl signaling and thereby VSMC function. Interestingly, while Gas6-stimulated ERK1/2 signaling was greater in high glucose than low glucose, the opposite was true for Akt. This suggests that glucose modulates the downstream coupling of Axl to its effectors. Here we show increased interaction between PI3K and Axl in low glucose and increased interaction between the protein tyrosine phosphatase SHP-2 and Axl in high glucose. Furthermore, Gas6-Axl signaling increased cell survival in low glucose and increased migration in high glucose. Our data demonstrate that glucose modulates Axl signaling via different cell signaling mechanisms and this may contribute to the vascular complications of diabetes.
Methods
Materials
Antibodies to Axl and ERK1/2 were from Santa Cruz Biotechnology; antibodies to phospho-ERK, phospho-Akt (Ser-473), Akt, mTOR, phospho-mTOR (Ser-2448); p85 PI3K antibody and Akt antibody were from Upstate, LiCor fluorescent secondary antibodies were from Molecular Probes. Sulfo-NHS-SS- biotin and streptavidin agarose were from Pierce. Gas6 was kindly provided by Brian Varnum (Amgen). All other reagents and chemicals were obtained from Sigma, unless specifically indicated.
Cell Culture
Cultured VSMC were obtained from rat aorta as described 17. VSMC were grown in Dulbecco's modified Eagle Medium supplemented with 25 mM NaHCO3, 10 mM HEPES, pH 7.4, 50 IU/ml penicillin, 50 μg/ml streptomycin, 10 % fetal bovine serum (FBS) containing 5.5 mM glucose (low glucose (LG)) in a 5 % CO2/95% O2 incubator at 37 °C. For high glucose (HG), cells were grown in 27.5 mM glucose and controls received 22.5 mM mannitol and 5 mM glucose (total).
Preparation of cell lysates and Immunoprecipitations
Cell monolayers were rinsed with ice-cold phosphate-buffered saline (PBS; 150 mM NaCl, 20 mM Na2PO4, pH 7.4) and then scraped in 1 ml of PBS. After a brief centrifugation, the cells were solubilized in 1 ml of cell lysis buffer (10 mM HEPES, pH 7.4, 50 mM Na Pyrophosphate, 50 mM NaF, 50 mM NaCl, 5 mM EDTA, 5 mM EGTA, 1 mM Na3VO4, 0.5 % Triton plus 1:1000 protease inhibitor cocktail). Cells were sonicated for 20 s, agitated on a rotating rocker at 4 °C for 30 min and centrifuged at 12,000 g for 30 min to remove insoluble cellular debris.
For immunoprecipitation studies, lysates were precleared for 1 hr with protein G agarose (Invitrogen) followed by incubation with anti-Axl antibody for 3 hr, and protein G agarose for a further 1 hr. Immunoprecipitates were then washed 4 times with 1 ml cell lysis buffer before the addition of Laemmli sample buffer. After heating at 95 °C for 3 min, proteins were resolved on SDS-PAGE and transferred to nitrocellulose membranes for Western analysis. Immunoreactive bands were detected with LiCor fluorescent secondary antibodies and the LiCor Odyssey infrared Imaging system. Analysis of blots was performed using the LiCor densitometry software.
Glycosidase Digestion
Peptide N-glycosidase F (PNGase-F, New England Biolabs) digestion was performed as described16.
Cell surface biotinylation
Cells were biotinylated as described 16. Cells were then lysed in 1 ml of N+ buffer (in mM: 60 HEPES, pH 7.4, 150 NaCl, 3 KCl, 5 EDTA trisodium, 3 EGTA, and 1% Triton X-100) and sonicated to clarity on ice with a Branson 450 probe sonicator. NHS-SS-biotin labeled proteins were precipitated using streptavidin agarose. The avidin-agarose beads were washed five times in N+ buffer, and bound proteins were solubilized in sample buffer yielding the surface fraction. Samples were separated on a 9% gel by SDS-PAGE, and Western analysis was then performed.
Subcellular Fractionation
Subcellular fractionation was performed as described 16 and an equal volume of each fraction was analyzed by SDS-PAGE.
Caspase-3-Enzyme Activity
Caspase-3-activity was detected by the color absorbance of the chromophore p-nitroanilide (pNA) obtained after proteolytic cleavage from the labeled substrate Asp-Glu-Val-Asp-pNA (DEVD-pNA) and pNA as standard using a spectrophotometer 405 nm (Clontech ApoAlert Caspase-3 assay kit) according to the manufacturers protocol.
Migration Assays
VSMC migration was measured using a Boyden chamber assay. Cells were incubated in LG or HG serum free media for 24 hr. Control medium (0% serum DMEM), Gas6 medium (0% serum DMEM with 200 ng/ml Gas6) or serum medium (10 % serum DMEM) was placed in the lower chamber. A collagen-coated polyvinylpyrrolidone-free polycarbonate membrane was placed on top, and 2 × 105 cells suspended in 50 μl 0% serum DMEM/0.1 % BSA were seeded in the upper chamber. After 6 hr incubation at 37 °C in 95 % air/5% CO2, cells attached to the lower side were fixed and stained using a Diff-Quik stain set (Dade Behring). The number of migrating cells was quantified using densitometry (NIH Image).
Statistical analysis
All experiments were carried out at least 3 times. The difference between LG and HG was assessed by analysis of variance.
Results
Glucose modulates Akt and ERK1/2 phosphorylation upon stimulation with Gas6
Previously, we found that that HG altered transactivation of the EGFR by angiotensin II; with increased ERK1/2 and Akt phosphorylation in HG as compared to LG 16. To explore the effect of glucose on Gas6-Axl signal transduction, we studied Akt and ERK1/2 activation. The phosphorylation of ERK1/2 by Gas6 was dramatically increased in HG as compared to LG (Figure 1A). In contrast, the phosphorylation of Akt by Gas6 was opposite, dramatically increased in LG as compared to HG (Figure 1B). (Please see Figure I supplement at www.ahajournals.org for quantification of A and B). This demonstrates that the ability of Axl to stimulate Akt and ERK1/2 is differentially regulated by glucose concentration.
Figure 1. Glucose modulates Akt and ERK1/2 phosphorylation upon stimulation of Axl with Gas6.
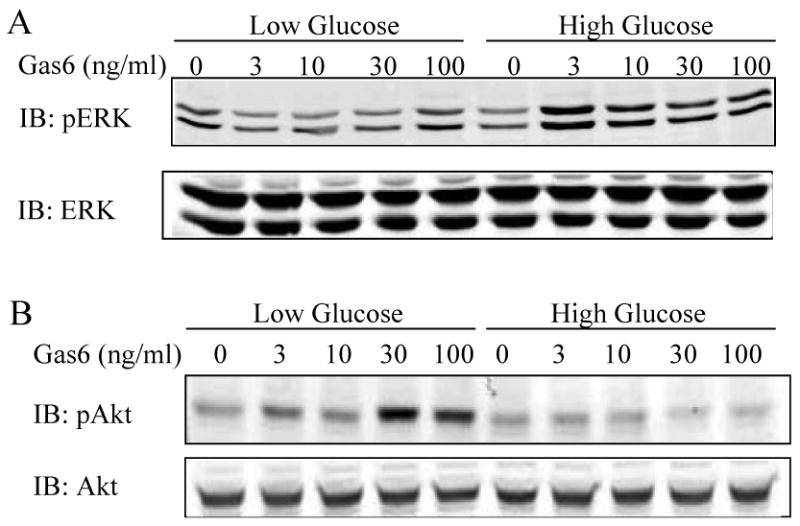
VSMC were cultured in serum free LG or HG DMEM for 24hr and treated with increasing Gas6 concentrations for 10 mins. Lysates were immunoblotted with: A. phosphospecific ERK1/2 antibody and reprobed with ERK1/2 antibody; B. phosphospecific Akt antibody and reprobed with Akt antibody.
Glucose does not modulate phosphorylation of Axl or cell surface expression of Axl
Previously, our lab demonstrated that the molecular mass of the EGFR varies depending on the glucose concentration due to N-glycosylation 16. We hypothesized that glucose might also affect Axl N-glycosylation and therefore studied the effect of LG vs. HG on Axl molecular mass. In HG Axl was present predominantly as a 140 kDa form (62.2 ± 1.7%), with a small proportion present as 120 kDa (16.1 ± 1.4%) and 114 kDa (21.7 ± 0.85%) forms (Figure 2A, B, C). In contrast, in LG the 114 kDa form of Axl was the major form (Figure 2A,B, C). To show that the Axl isoform change was due to glucose levels and not osmotic stress, the metabolically inactive sugar mannitol (22.5 mM) was added to LG media. This had no effect on the appearance of the 114 kD isoform in LG (data not shown).
Figure 2. Glucose does not alter Axl cell surface localization or phosphorylation by Gas6.
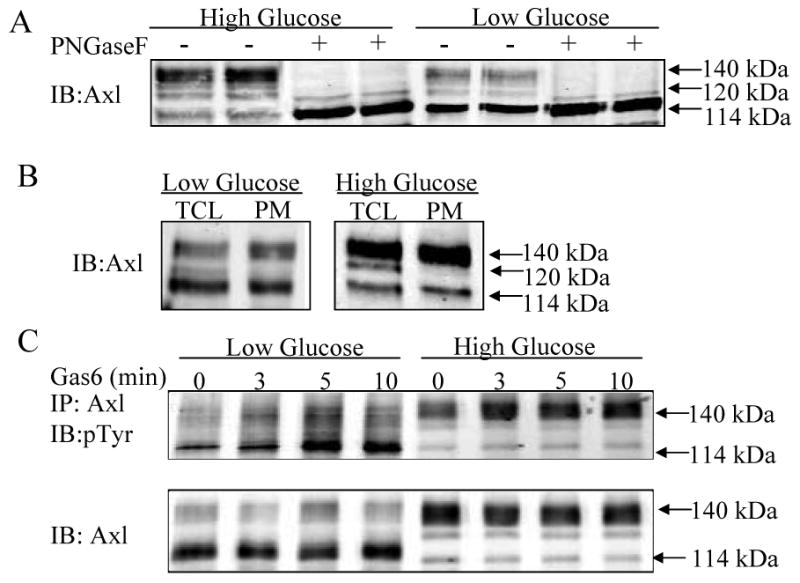
VSMC were cultured in serum free LG or HG DMEM for 24hr. A. Axl was immunoprecipitated, treated with or without PNGase-F and immunoblotted with Axl antibody. B. Proteins were biotinylated with NHS-SS-biotin and precipitated with streptavidin agarose. Total cell lysates (TCL) and plasma membrane fractions (PM) were immunoblotted with Axl antibody. C. Following Gas6 treatment Axl was immunoprecipitated. Immunoprecipitates were immunoblotted with phospho-tyrosine antibody (4G10). Results represent n=3.
Glucose concentration has previously been shown to alter the level of membrane protein N-glycosylation 16, 18-20. Therefore, we used N-glycosidase F (PNGase F), which removes carbohydrates from glycopeptides at N-glycosylation sites, to determine whether the difference in Axl molecular mass was due to Axl N-glycosylation. PNGase-F treatment yielded a 114 kDa form of Axl from lysates of HG and LG treated cells demonstrating that the difference in molecular mass is due to N-glycosylation (Figure 2A). An important function for N-linked glycosylation is to target membrane proteins to the plasma membrane. Lack of proper glycosylation can cause proteins to be improperly folded and retained in the endoplasmic reticulum 21. We used cell surface biotinylation to determine whether the 114 kDa form of Axl was present in the plasma membrane at similar levels to the 140 kDa Axl. Membrane proteins were labeled with a membrane impermeant form of biotin, then after cell lysis the biotinylated proteins were extracted using streptavidin agarose. As shown in Figure 2B, in both LG and HG the same amounts of 114 kDa Axl and 140 kDa Axl were present in the plasma membrane demonstrating that glucose concentration does not alter cell surface levels of Axl. Finally, in response to Gas6 both 114 kDa and 140 kDa were tyrosine phosphorylated with similar time courses (Figure 2C), consistent with cell surface localization.
Glucose modulates the interaction between p85-PI3-kinase (p85-PI3K) and Axl
To determine if enhanced activation of Akt by Gas6 in LG was due to an increased interaction between Axl and p85-PI3K, co-immunoprecipitation of Axl with p85-PI3K was determined. Gas6 increased interaction between p85-PI3K and Axl, as previously reported 22. However, in LG, the interaction between Axl and p85-PI3K was significantly increased as compared to in HG (Figure 3A). This suggests that the increased phosphorylation of Akt in LG after Gas6 stimulation is due to increased binding to p85-PI3K.
Figure 3. Glucose modulates the interaction between Axl and PI3K and Axl and SHP-2.
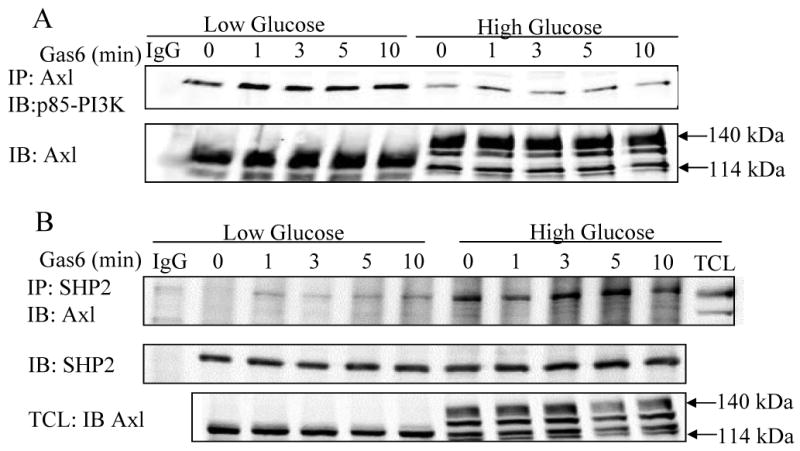
VSMC were cultured in serum free LG or HG DMEM for 24hr. A. Axl was immunoprecipitated from total cell protein and immunoblotted with p85 PI-3 kinase and Axl antibodies. B. SHP-2 was immunoprecipitated from total cell protein and immunoblotted with Axl and SHP-2 antibodies. Lower panel shows total cell lysates (TCL) immunoblotted with Axl antibody. Results represent n≥2.
Glucose modulates the interaction between SHP-2 and Axl
The protein tyrosine phosphatase SHP-2 associates with tyrosine kinase receptors including Axl 23 and positively regulates ERK activation 24-26. Therefore we determined whether glucose concentration modulated SHP-2 interaction with Axl. SHP-2 co-immunoprecipitated with Axl in HG. However, an interaction between SHP-2 and Axl could not be detected in LG (Figure 3B). Therefore increased phosphorylation of ERK1/2 in HG may be due in part to increased association of SHP-2 with Axl. Gas6 induces Axl activation in LG leads to Akt activation via interaction of Axl with p85-PI3K (Figs 1 and 3). To characterize further the anti-apoptotic mechanism of Axl in LG conditions, modulation of downstream signaling molecules was determined. The PI3K-Akt pathway antagonizes apoptosis in VSMC as well as other cell types, in part via activation of mTOR. Because mTOR is expressed in VSMC 27, we tested the hypothesis that mTOR activation by Gas6 is stimulated in LG. There was a slight increase in Gas6 induced phosphorylation of mTOR at Ser 2448 in LG. However, the baseline level of mTOR phosphorylation was much less in HG and upon stimulation with Gas6 there was a significant time dependent decrease in phospho-mTOR (please see Figure II supplement at www.ahajournals.org).
To determine whether the altered interactions with p85-PI3K and SHP-2 were due to altered distribution of Axl within the plasma membrane in LG vs. HG, by subcellular fractionation sucrose gradient density centrifugation was performed (Figure 4). This demonstrated that Axl was predominantly present in caveolin enriched fractions both basally (Figure 4A) and after Gas6 (Figure 4B) stimulation, and this was not altered by glucose concentration (Figure 4).
Figure 4. Subcellular fractionation of Axl on a sucrose gradient is not altered by glucose.
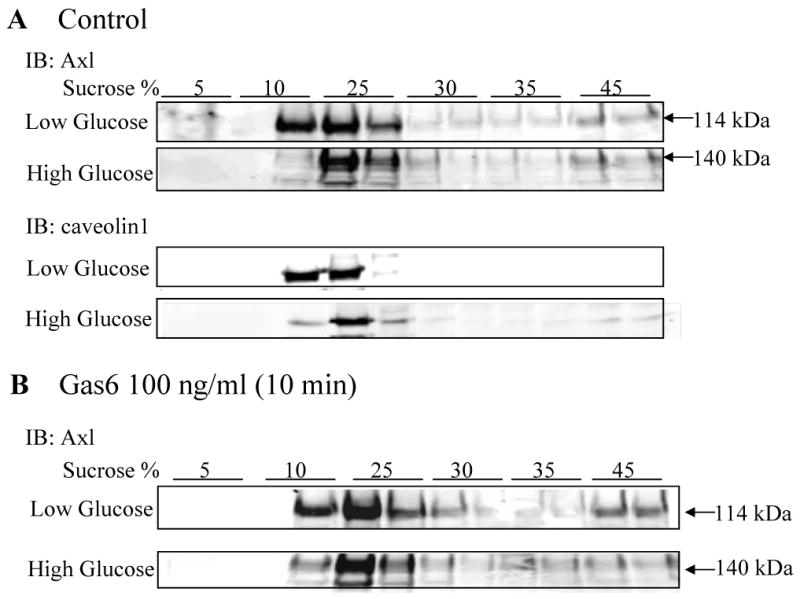
VSMC were cultured in serum free LG or HG DMEM for 24hr. Post-nuclear supernatant was prepared and loaded on a 5-45% sucrose density gradient. Following centrifugation, fractions were collected. 30 μl of each fraction was subjected to SDS-PAGE and blotted with the indicated antibodies. Results represent n=3. Cofractionation under (A) basal conditions and (B) after stimulation with Gas6.
The anti-apoptotic effect of Axl is enhanced in LG conditions
To determine whether the Gas6-Axl mediated increase in Akt signaling in LG has physiological consequences, the ability of Axl to protect VSMC from apoptosis was determined by measuring caspase-3 activity. VSMC were incubated in 0% serum media containing LG or HG for 24 hr. Apoptosis was induced by serum deprivation alone or serum deprivation and TNFα (10 ng/ml) for 24 hr in LG or HG media. Caspase-3 activity was increased by 22-fold vs. control in LG+0% serum in contrast to a 1.6-fold increase vs. control in HG+0% serum. After TNFα treatment, caspase-3 activity was 35-fold higher than control in LG in contrast to 2.4-fold higher than control in HG (Figure 5A). This is consistent with previous studies, which demonstrate that high glucose is anti-apoptotic 28-30. After treatment with Gas6, caspase-3 activity in LG+0% serum was reduced from 22-fold to 11-fold, but was unchanged in HG+0% serum (1.6-fold vs. 1.5-fold; Figure 5A). After TNFα treatment in LG, caspase-3 activity was reduced from 35-fold to 17-fold, but was unchanged in HG (2.4-fold vs. 3.4-fold; Figure 5A). Because the baseline level of apoptosis in HG was so low it would be difficult to achieve significant differences, but the fact that the trend shows no change toward less apoptosis is consistent with the Akt data in Fig. 1. The 114 kDa Axl is the Axl isoform that predominates in the signaling of this anti-apoptotic effect based on the data in Figure 2. These results are consistent with the greater Akt activation observed in LG (Figure 1B-C).
Figure 5. The anti-apoptotic effect of Gas6-Axl is increased in low glucose and the pro-migratory effect of Gas6-Axl is increased in high glucose.

VSMC were cultured in serum free LG or HG DMEM for 24hr. A. Apoptosis was induced using 0%-serum or 0%-serum + TNF-α with and without Gas6 for 24hr. In LG caspase-3 activity was significantly less than control; *=p<0.05, n=4. B. 0%-serum, Gas6 or serum was placed in the lower Boyden chamber. VSMC were seeded in the upper chamber for 6hr. In response to Gas6 in HG VSMC migration was significantly greater compared to LG; *=p<0.005, n=7.
Migration is enhanced in response to Gas 6 in HG
Since Gas6 stimulation of Axl increases VSMC migration 12, we determined the effect of LG and HG on Gas6 stimulated VSMC migration. VSMC were incubated in 0% serum DMEM containing LG or HG. Baseline migration in LG and HG was similar as was migration in response to 10% serum (Figure 5B). However, in response to 200 ng/ml Gas6, migration in HG was 27 ± 2% higher than in LG (p<0.005;Figure 5B). This demonstrates that the pro-migratory effect of Axl is increased in HG and suggests that the 140 kDa Axl plays a more significant role in migration than the 114 kDa Axl.
Discussion
In this study we investigated the effect of glucose concentration on Axl signal transduction in VSMC. The major finding is that glucose alters Gas6-Axl signaling by modulating interaction of Axl with specific signaling proteins. In HG, Gas6-Axl stimulation increased ERK1/2 activation while in LG Gas6-Axl stimulated PI3K-Akt-mTOR. It appears that the 140kDa Axl is primarily responsible for ERK 1/2 activation while the 114kDa Axl is responsible for Akt activation. Increased interaction of Axl with SHP-2 in HG and increased interaction with PI3-K in LG correlated with these alterations in Axl downstream signals. Functionally, this resulted in increased migration in response to Gas6 in HG and increased protection against apoptosis in LG.
Glucose has previously been shown to modulate signaling in VSMC. HG activates protein kinase C and this inhibits VSMC apoptosis 30. Increased activation of ERK1/2 and p38 also occurs in HG 31, 32. Recently, we demonstrated that transactivation of the EGFR by angiotensin II was increased in HG, which increased both Akt and ERK1/2 phosphorylation 16. Interestingly, the activation of Akt by Axl was higher in LG than in HG and in contrast, Axl activated ERK1/2 more in HG as compared to LG. Axl is anti-apoptotic and pro-migratory in VSMC 11, 12. Previous studies have shown that the anti-apoptotic effect is predominantly through PI3K-Akt signaling 11. The present study demonstrates that in LG Axl predominantly activates Akt signaling and therefore mediates cell survival, via a mechanism that includes caspase 3 inhibition. The anti-apoptotic effect of Gas6 stimulation in LG is further supported by the activation of signals downstream of Akt, including mTOR. The effects of Akt and mTOR interaction have been shown to decrease apoptosis in endothelial cells 33 and the Gas6-Axl and Akt/mTOR pathways play a crucial role in mesangial and glomerular hypertrophy 13. Thus we propose a similar signaling mechanism in VSMC. We also demonstrated increased migration in HG as compared to LG in response to Gas6, suggesting a key role for ERK1/2 signaling in Gas6-Axl mediated migration in HG.
We propose a model for the downstream cell signaling mechanisms of Gas6 mediated activation of Axl in HG and LG (please see Figure III supplement at www.ahajournals.org). The mechanism by which glucose modifies Axl signaling appears to be altering Axl binding with signaling molecules. Activation of p85-PI3K and its downstream target Akt by Gas6-Axl requires interaction between PI3K and Axl 9, 34, 35. This study suggests increased association of Axl with p85-PI3K mediates increased activation of Akt and mTOR in LG. Previous studies demonstrate that the protein tyrosine kinase SHP-2 is required for ERK1/2 activation by growth factors 24-26. Gas6 activates SHP-2 and increases SHP-2 association with Axl 23. We demonstrated that there is increased association of SHP-2 with Axl under HG conditions and this may mediate the increased ERK1/2 activation. The mechanism by which PI3K associates with 140kDa Axl and SHP2 with 114kDa Axl selectively remains to be determined.
In the case of the EGFR, N-glycosylation appears to change the distribution of EGFR within plasma membrane microdomains thereby modulating angiotensin II transactivation of the EGFR 16. However, this cannot explain the effect of glucose on Axl since plasma membrane distribution was predominantly in the caveolae fraction in both LG and HG conditions. N-glycosylation of the EGFR also alters ligand binding properties and receptor dimerization, thus modifying EGFR downstream signaling 36-38. This seems unlikely for Gas6-Axl since signaling to ERK1/2 and Akt occurred at high levels. A likely possibility is that cellular distribution of p85-PI3K and SHP-2 is altered depending on glucose concentration. Further studies will elucidate the mechanism by which glucose concentration and N-glycosylation state of Axl regulate interaction of Axl with signaling molecules.
This novel finding that Gas6-Axl-mediated Akt and ERK1/2 activation differs depending on glucose concentration has important implications for Axl function in vivo. Several studies point to an important role for Axl in the pathogenesis of diabetic cardiovascular disease. Expression of Axl is increased in injured VSMC, in diabetic mesangial cells and in mesangial cells stimulated with high glucose 13, 14, 39. Diabetic nephropathy is less severe in Gas6 knockout mice and after treatment of rats with the Gas6 inhibitor warfarin 13. Axl is also activated by H2O2 in VSMC 15. Therefore under conditions of oxidative stress, as occurs in diabetes, Axl is likely to be activated. In rat vasculature, Axl is present as 140 kDa, and 114 kDa forms in approximately equivalent ratio 14, 15. Studies in cultured VSMC suggest that activation of the 140 kDa Axl in HG will result in increased ERK1/2 signaling and VSMC migration. VSMC migration is increased in patients with diabetes and diabetes accelerates the accumulation of VSMC in atherosclerotic lesions 40, 41. Therefore, this study suggests that altered Gas6-Axl signaling will contribute to vascular dysfunction in diabetes.
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health Grant HL68286 (to B.C.B.) and by an American Heart Association SDG grant (Award No. 05535197N, to M.E.C.)
References
- 1.Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. Jama. 2002;287:2570–2581. doi: 10.1001/jama.287.19.2570. [DOI] [PubMed] [Google Scholar]
- 2.Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108:1527–1532. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 3.King GL, Kunisaki M, Nishio Y, Inoguchi T, Shiba T, Xia P. Biochemical and molecular mechanisms in the development of diabetic vascular complications. Diabetes. 1996;45 3:S105–108. doi: 10.2337/diab.45.3.s105. [DOI] [PubMed] [Google Scholar]
- 4.Melaragno MG, Fridell YW, Berk BC. The Gas6/Axl system: a novel regulator of vascular cell function. Trends Cardiovasc Med. 1999;9:250–253. doi: 10.1016/s1050-1738(00)00027-x. [DOI] [PubMed] [Google Scholar]
- 5.Berk BC. Angiotensin II signal transduction in vascular smooth muscle: pathways activated by specific tyrosine kinases. J Am Soc Nephrol. 1999;10 11:S62–68. [PubMed] [Google Scholar]
- 6.Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, Mattsson K, Fisher J, Gies DR, Jones PF, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80:661–670. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 7.Varnum BC, Young C, Elliott G, Garcia A, Bartley TD, Fridell YW, Hunt RW, Trail G, Clogston C, Toso RJ, Yanagihara D, Bennett L, Sylber M, Merewether LA, Tseng A, Escobar E, Liu T, Yamane HK. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature. 1995;373:623–626. doi: 10.1038/373623a0. [DOI] [PubMed] [Google Scholar]
- 8.Fridell YW, Jin Y, Quilliam LA, Burchert A, McCloskey P, Spizz G, Varnum B, Der C, Liu ET. Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol Cell Biol. 1996;16:135–145. doi: 10.1128/mcb.16.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goruppi S, Ruaro E, Varnum B, Schneider C. Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol Cell Biol. 1997;17:4442–4453. doi: 10.1128/mcb.17.8.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goruppi S, Ruaro E, Varnum B, Schneider C. Gas6-mediated survival in NIH3T3 cells activates stress signalling cascade and is independent of Ras. Oncogene. 1999;18:4224–4236. doi: 10.1038/sj.onc.1202788. [DOI] [PubMed] [Google Scholar]
- 11.Melaragno M, Cavet M, Yan C, Tai L, Jin Z, Haendeler J, Berk B. Gas6 inhibits apoptosis in vascular smooth muscle: Role of Axl kinase and Akt. Journal of Molecular and Cellular Cardiology. 2004 doi: 10.1016/j.yjmcc.2004.06.018. In Press. [DOI] [PubMed] [Google Scholar]
- 12.Fridell YW, Villa J, Jr, Attar EC, Liu ET. GAS6 induces Axl-mediated chemotaxis of vascular smooth muscle cells. J Biol Chem. 1998;273:7123–7126. doi: 10.1074/jbc.273.12.7123. [DOI] [PubMed] [Google Scholar]
- 13.Nagai K, Arai H, Yanagita M, Matsubara T, Kanamori H, Nakano T, Iehara N, Fukatsu A, Kita T, Doi T. Growth arrest-specific gene 6 is involved in glomerular hypertrophy in the early stage of diabetic nephropathy. J Biol Chem. 2003;278:18229–18234. doi: 10.1074/jbc.M213266200. [DOI] [PubMed] [Google Scholar]
- 14.Melaragno MG, Wuthrich DA, Poppa V, Gill D, Lindner V, Berk BC, Corson MA. Increased expression of Axl tyrosine kinase after vascular injury and regulation by G protein-coupled receptor agonists in rats. Circ Res. 1998;83:697–704. doi: 10.1161/01.res.83.7.697. [DOI] [PubMed] [Google Scholar]
- 15.Konishi A, Aizawa T, Mohan A, Korshunov VA, Berk BC. Hydrogen peroxide activates the gas6-axl pathway in vascular smooth muscle cells. J Biol Chem. 2004;279:28766–28770. doi: 10.1074/jbc.M401977200. [DOI] [PubMed] [Google Scholar]
- 16.Konishi A, Berk BC. Epidermal growth factor receptor transactivation is regulated by glucose in vascular smooth muscle cells. J Biol Chem. 2003;278:35049–35056. doi: 10.1074/jbc.M304913200. [DOI] [PubMed] [Google Scholar]
- 17.Ishida T, Ishida M, Suero J, Takahashi M, Berk BC. Agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in vascular smooth muscle cells require c-Src. J Clin Invest. 1999;103:789–797. doi: 10.1172/JCI4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang JB, Frost SC. Effect of alternative glycosylation on insulin receptor processing. J Biol Chem. 1999;274:22813–22820. doi: 10.1074/jbc.274.32.22813. [DOI] [PubMed] [Google Scholar]
- 19.Kitzman HH, Jr, McMahon RJ, Williams MG, Frost SC. Effect of glucose deprivation of GLUT 1 expression in 3T3-L1 adipocytes. J Biol Chem. 1993;268:1320–1325. [PubMed] [Google Scholar]
- 20.Zheng Z, Cummings RD, Pummill PE, Kincade PW. Growth as a solid tumor or reduced glucose concentrations in culture reversibly induce CD44-mediated hyaluronan recognition by Chinese hamster ovary cells. J Clin Invest. 1997;100:1217–1229. doi: 10.1172/JCI119635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parodi AJ. Role of N-oligosaccharide endoplasmic reticulum processing reactions in glycoprotein folding and degradation. Biochem J. 2000;348(Pt 1):1–13. [PMC free article] [PubMed] [Google Scholar]
- 22.Goruppi S, Ruaro E, Schneider C. Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3T3 fibroblasts. Oncogene. 1996;12:471–480. [PubMed] [Google Scholar]
- 23.Gallicchio M, Mitola S, Valdembri D, Fantozzi R, Varnum B, Avanzi GC, Bussolino F. Inhibition of vascular endothelial growth factor receptor 2-mediated endothelial cell activation by Axl tyrosine kinase receptor. Blood. 2005;105:1970–1976. doi: 10.1182/blood-2004-04-1469. [DOI] [PubMed] [Google Scholar]
- 24.Deb TB, Wong L, Salomon DS, Zhou G, Dixon JE, Gutkind JS, Thompson SA, Johnson GR. A common requirement for the catalytic activity and both SH2 domains of SHP-2 in mitogen-activated protein (MAP) kinase activation by the ErbB family of receptors. A specific role for SHP-2 in map, but not c-Jun amino-terminal kinase activation. J Biol Chem. 1998;273:16643–16646. doi: 10.1074/jbc.273.27.16643. [DOI] [PubMed] [Google Scholar]
- 25.Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol. 2000;20:8513–8525. doi: 10.1128/mcb.20.22.8513-8525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi ZQ, Yu DH, Park M, Marshall M, Feng GS. Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol Cell Biol. 2000;20:1526–1536. doi: 10.1128/mcb.20.5.1526-1536.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol. 2004;37:449–471. doi: 10.1016/j.yjmcc.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Telemaque S, Miller RE, Marsh JD. High glucose inhibits apoptosis induced by serum deprivation in vascular smooth muscle cells via upregulation of Bcl-2 and Bcl-xl. Diabetes. 2005;54:540–545. doi: 10.2337/diabetes.54.2.540. [DOI] [PubMed] [Google Scholar]
- 29.Sakuma H, Yamamoto M, Okumura M, Kojima T, Maruyama T, Yasuda K. High glucose inhibits apoptosis in human coronary artery smooth muscle cells by increasing bcl-xL and bfl-1/A1. Am J Physiol Cell Physiol. 2002;283:C422–428. doi: 10.1152/ajpcell.00577.2001. [DOI] [PubMed] [Google Scholar]
- 30.Hall JL, Matter CM, Wang X, Gibbons GH. Hyperglycemia inhibits vascular smooth muscle cell apoptosis through a protein kinase C-dependent pathway. Circ Res. 2000;87:574–580. doi: 10.1161/01.res.87.7.574. [DOI] [PubMed] [Google Scholar]
- 31.Yamaguchi H, Igarashi M, Hirata A, Sugae N, Tsuchiya H, Jimbu Y, Tominaga M, Kato T. Altered PDGF-BB-induced p38 MAP kinase activation in diabetic vascular smooth muscle cells: roles of protein kinase C-delta. Arterioscler Thromb Vasc Biol. 2004;24:2095–2101. doi: 10.1161/01.ATV.0000144009.35400.65. [DOI] [PubMed] [Google Scholar]
- 32.Natarajan R, Scott S, Bai W, Yerneni KK, Nadler J. Angiotensin II signaling in vascular smooth muscle cells under high glucose conditions. Hypertension. 1999;33:378–384. doi: 10.1161/01.hyp.33.1.378. [DOI] [PubMed] [Google Scholar]
- 33.Dormond O, Madsen JC, Briscoe DM. The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. J Biol Chem. 2007;282:23679–23686. doi: 10.1074/jbc.M700563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, Ullrich A, Bartram CR, Janssen JW. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene. 1997;14:2619–2631. doi: 10.1038/sj.onc.1201123. [DOI] [PubMed] [Google Scholar]
- 35.Hafizi S, Alindri F, Karlsson R, Dahlback B. Interaction of Axl receptor tyrosine kinase with C1-TEN, a novel C1 domain-containing protein with homology to tensin. Biochem Biophys Res Commun. 2002;299:793–800. doi: 10.1016/s0006-291x(02)02718-3. [DOI] [PubMed] [Google Scholar]
- 36.Fernandes H, Cohen S, Bishayee S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem. 2001;276:5375–5383. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- 37.Whitson KB, Whitson SR, Red-Brewer ML, McCoy AJ, Vitali AA, Walker F, Johns TG, Beth AH, Staros JV. Functional effects of glycosylation at Asn-579 of the epidermal growth factor receptor. Biochemistry. 2005;44:14920–14931. doi: 10.1021/bi050751j. [DOI] [PubMed] [Google Scholar]
- 38.Yu X, Sharma KD, Takahashi T, Iwamoto R, Mekada E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol Biol Cell. 2002;13:2547–2557. doi: 10.1091/mbc.01-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagai K, Matsubara T, Mima A, Sumi E, Kanamori H, Iehara N, Fukatsu A, Yanagita M, Nakano T, Ishimoto Y, Kita T, Doi T, Arai H. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int. 2005;68:552–561. doi: 10.1111/j.1523-1755.2005.00433.x. [DOI] [PubMed] [Google Scholar]
- 40.Faries PL, Rohan DI, Takahara H, Wyers MC, Contreras MA, Quist WC, King GL, Logerfo FW. Human vascular smooth muscle cells of diabetic origin exhibit increased proliferation, adhesion, and migration. J Vasc Surg. 2001;33:601–607. doi: 10.1067/mva.2001.111806. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki LA, Poot M, Gerrity RG, Bornfeldt KE. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: lack of direct growth-promoting effects of high glucose levels. Diabetes. 2001;50:851–860. doi: 10.2337/diabetes.50.4.851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.