

Abstract
Nitric oxide (NO) is a gaseous molecule that plays many key roles in the cardiovascular system. Each of the enzymes that generate NO—neuronal, inducible and endothelial NO synthase—has been genetically disrupted in mice. This review discusses the cardiovascular phenotypes of each of the NO synthase (NOS) gene knockout mice, and the insights gained into the roles of NO in the cardiovascular system. Mice lacking the endothelial isoform are hypertensive, have endothelial dysfunction and show a more severe outcome in response to vascular injury, to stroke and cerebral ischaemia, and to diet-induced atherosclerosis. Mice lacking the neuronal isoform show a less severe outcome in response to stroke and cerebral ischaemia but have increased diet-induced atherosclerosis. Mice lacking the inducible isoform show reduced hypotension to septic shock. Together, NOS gene knockout mice have been useful tools that complement our other approaches to studying the multiple roles of NO in the cardiovascular system.
Keywords: Nitric oxide, Gene knockout, Targeted disruption, Animal models, Pathophysiology, Atherosclerosis, Vascular dysfunction, Cerebral ischaemia, Mouse models
1. Introduction
Nitric oxide (NO) is a key signaling messenger in the cardiovascular system.1 In addition to its role as endothelium-derived relaxing factor (EDRF), NO serves many important biological functions in cardiovascular physiology. NO maintains vascular integrity by inhibiting platelet aggregation,2,3 leukocyte–endothelium adhesion4–6 and vascular smooth muscle proliferation.7 In addition, NO is produced in cardiac smooth muscle, where it regulates cardiac contractility.8 Adequate levels of endothelial NO are important to preserve normal vascular physiology—in the face of diminished NO bioavailability, there is endothelial dysfunction, leading to increased susceptibility to atherosclerotic disease.9–11 Atherosclerosis, hypertension, hypercholesterolemia, diabetes mellitus, congestive heart failure, thrombosis and stroke have all been linked to abnormalities in NO signaling.10,12,13 Genetic manipulation of the enzymes that generate NO in mice has contributed significantly to our understanding of its many roles, both in physiology and in disease pathogenesis.
2. Nitric oxide synthases
NO is produced by nitric oxide synthase (NOS) enzymes, of which there are three main isoforms: neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS).14 Table 1 shows a comparison of these isoforms. The three NOS isoforms are encoded on separate chromosomes by separate genes. They share homology in regions involved in cofactor binding (for example, FAD, FMN, and NADPH ribose and adenine binding sites), and have similar enzymatic mechanisms that involve electron transfer for oxidation of the terminal guanidino nitrogen of l-arginine. However, their expression patterns differ, as do the detailed regulations of their activity. nNOS is predominantly expressed in certain neurons and in skeletal muscle, whereas eNOS is predominantly expressed in endothelial cells. iNOS is expressed by macrophages and cells of macrophage/monocyte lineage. Despite their names, a variety of cell types express these isoforms, with many tissues expressing more than one isoform. Furthermore, the innervation and vasculature in all tissues have the potential to express nNOS and eNOS, while circulating blood elements may express iNOS.
Table 1.
Characteristics of NOS Isoforms
| Isoform | nNOS | iNOS | eNOS |
|---|---|---|---|
| Other names | NOS-1, NOSI, Type I NOS | NOS-2, NOSII, Type II NOS | NOS-3, NOSIII, Type III NOS |
| Human chromosomal location | 12q24.2-12q24.3 | 17cen-q11.2 | 7q35–7q36 |
| Human gene structure and size | 29 exons | 26 exons | 26 exons |
| locus region >200 kbp | 37 kbp | 21–22 kbp | |
| Human monomer size (predominant form) | 161 kDa | 131 kDa | 133 kDa |
| Splice variants | Yes | Yes | No |
| Typical site of expression | Neurons | Macrophages | Endothelial cells |
| Other major sites of expression | Smooth muscle | Smooth muscle | Smooth muscle |
| Skeletal muscle | Liver | Platelets | |
| Gene expression | Constitutive and inducible | Inducible | Constitutive and inducible |
| Ca2+ dependency | Ca2+-dependent | Practically Ca2+-independent | Ca2+-dependent |
| Covalent modifications | Phosphorylation | Myristoylation | |
| Palmitoylation | |||
| Phosphorylation | |||
| Protein–protein interactions | hsp90, caveolin, NOSIP | hsp90, caveolin | |
| Subcellular localization | Neuromuscular junction | Soluble | Caveolae |
| Soluble | |||
| Sarcoplasmic reticulum | |||
| Means of localization | N-terminal PDZ domain (for membrane association) | N/A | N-terminal myristoylation |
| NO production (range) | Moderate (nM to μM) | High (μM) | Low (pm to nM) |
Both nNOS and eNOS are generally constitutively expressed; their activities are primarily regulated by intracellular Ca2+/calmodulin levels. In contrast, iNOS expression is induced in activated macrophages as an immune response. For enzymatic activity, NOS proteins must bind cofactors and dimerize.14 NOS proteins first bind to the cofactors FAD and FMN. The additions of l-arginine, BH4 and heme allow the NOS protein to form dimers. eNOS and nNOS dimers formed this way are inactive, and depend on calmodulin binding stimulated by increases in intracellular calcium. In contrast, the iNOS dimers bind calcium/calmodulin and are active even at low (resting intracellular) concentrations of calcium. Thus, the main switch for activity for nNOS and eNOS is a transient increase in intracellular calcium concentration, whereas the main switch for iNOS is at the level of transcription.
The DNA and protein sequences of the NOS isoforms are conserved between species, with nNOS, eNOS and iNOS sharing up to 96%, 93% and 80% amino acid sequence identity between mice and humans. Within each species, the NOS isoforms share about 51–59% amino acid sequence identity. Each isoform has notable structural features. The nNOS gene encodes a PDZ domain in exon 2 that is required for membrane association.15 Several nNOS splice variants lack exon 2, resulting in expression of cytoplasmic nNOS that lacks subcellular localization sequences.16,17 In endothelial cells, eNOS is localized to caveolae by N-terminal fatty acid modifications—myristoylation and palmitoylation,18–21 as well as interactions with heat shock protein hsp90 and caveolins.12,22 Caveolins (caveolin-1 in endothelial cells and caveolin-3 in cardiac muscle) bind to eNOS and inhibit its activity. eNOS is also regulated by phosphorylation at multiple sites, including serine 1179 and threonine 497.
In addition to nNOS, eNOS, and iNOS, there is a constitutively active NOS isoform present in mitochondria, referred to as mtNOS.23,24 mtNOS is located in the inner mitochondrial membrane, and likely plays key roles in modulating mitochondrial respiration and mitochondrial transmembrane potential. However, whether mtNOS corresponds to one of the three known isoforms is not known.
3. Molecular targets of NO
In many cells and for many of the biological signaling roles of NO, the physiologic target is soluble guanylate cyclase.25,26 NO activates guanylate cyclase by binding to its heme moiety, resulting in increased cGMP levels. This is responsible for events in the brain following NMDA receptor activation. Garthwaite first described that cultures of cerebellar cells produce cGMP in response to the excitatory amino acid neurotransmitter glutamate.27 In the vasculature, cGMP mediates NO-dependent relaxation of vascular smooth muscle, resulting in vasodilation. Similarly, NO produced as a neurotransmitter in the autonomic nervous system innervating the gastrointestinal tract, urinary tract, and the respiratory tract, mediates smooth muscle relaxation in these tissues by increases in cGMP production. These effects are likely mediated by the phosphorylation of downstream proteins by cGMP-dependent protein kinases, including myosin light chain.
Another target for NO is sulfhydryl groups on proteins, to form nitrosothiol compounds.28 One such protein is hemoglobin, which may serve as a natural carrier for NO.29 In cardiac muscle, NO S-nitrosylates the ryanodine receptor on the sarcolemmal membrane and is required for its normal activity.30 NO also nitrosylates critical residues in N-ethylmaleimide-sensitive factor, which is important to the regulation of exocytosis.31 NO can react with superoxide anion to form peroxynitrite anion.32–34 Large quantities of NO, made for instance by iNOS, also can directly inhibit mitochondrial complexes I and IV.35–37 Finally, NO can activate the enzyme poly-ADP ribose polymerase (PARP), resulting in depletion of cellular energy stores.38–40 Generally, these latter mechanisms underlie some of the toxicity of NO, while effects on soluble guanylate cyclase and S-nitrosylation of proteins mediate many of the biological signaling roles of NOS. In addition to NO, NOS enzymes are capable of generating reactive oxygen species. When there is insufficient BH4, electron transfer in eNOS becomes ‘uncoupled,’ so the enzyme produces superoxide . This superoxide may react with NO to form peroxynitrite (ONOO−). Under conditions of limited l-arginine bioavailability, eNOS may generate H2O2.41
4. Genetic approaches
NOS enzymes can be inhibited by pharmacologic agents, including arginine analogs substituted at the terminal guanidino nitrogens. These arginine analogs bind to NOS, but cannot serve as substrate, so they compete with l-arginine and inhibit the enzyme. Such pharmacologic NOS inhibitors have yielded a tremendous amount of valuable information. Indeed, blockade of a biological process by l-nitro-arginine (L-NA) or l-N-arginine-methyl-ester (L-NAME), and outcompetition of this effect by an excess of l-arginine, provides very strong evidence for the involvement of NO in that process. One potential limitation of pharmacologic inhibitors, however, is that they may inhibit more than one NOS isoform. There are also structurally distinct inhibitors of NOS that are not arginine analogs (e.g. 7-nitroindazole or aminoguanidine), but some these agents have unrelated effects as well.
A complementary approach is to manipulate the genes that encode the NOS enzymes to generate knockout mice in which a particular NOS gene has been disrupted. This approach complements pharmacologic approaches because its specificity is at the genetic level. It pinpoints the roles of individual NOS genes, since many tissues contain all three of the major NOS isoforms. Further, it allows the study of how chronic absence of the NOS isoform affects physiology in intact animals. Table 2 shows some of the functions of the NOS isoforms, and the phenotypes of NOS knockout mice.
Table 2.
Functions of NOS Isoforms and Phenotypes of NOS Knockout Mice139
| Isoform | Function | Phenotype of Knockout Mice |
|---|---|---|
| nNOS | Signal transduction | Enlarged stomach, pyloric stenosis |
| Neurotransmission | Normal CNS development | |
| Toxicity (at high levels) | Aggressive behavior | |
| Decreased neuronal injury after stroke (acute) | ||
| No protection from rapid cerebral ischaemic preconditioning | ||
| Increased diet-induced atherosclerosis | ||
| Reduced cardiac contractile response to β-adrenergic stimulation | ||
| iNOS | Defense against pathogens | Susceptible to tuberculosis & other infections |
| Inflammation | Reduced hypotension in sepsis | |
| Decreased neuronal injury after stroke (delayed) | ||
| Decreased diet-induced atherosclerosis | ||
| eNOS | Vasodilation | Hypertension |
| Modulation of platelet aggregation | Absence of EDRF | |
| Modulation of leukocyte–endothelial interactions | Increased vascular response to injury | |
| Increased neuronal injury after stroke (acute) | ||
| No protection from rapid cerebral ischaemic preconditioning | ||
| Increased diet-induced atherosclerosis | ||
| Enhanced cardiac contractile response to β-adrenergic stimulation |
Several issues unique to the genetic approach should be kept in mind, as they can potentially confound studies using knockout animals. First, there may be developmental abnormalities due to the gene knockout. If one of the NOS isoforms plays a critical role in embryonic development, its absence may lead to other secondary abnormalities that are difficult to predict. Second, additional phenotypes may emerge from changes to pathways that act upstream or downstream to the gene product of interest. Third, other isoforms or gene products, acting in parallel, may compensate for the absent gene product and mask possible phenotypes. Finally, embryonic stem cells used to generate knockout mice are often derived from particular strains like the SV129 strain that are better sources of pluripotent embryonic stem cells. These knockout mice have mixed genetic background, which may itself lead to phenotypic abnormalities. For this reason, the knockout mice are usually backcrossed for 10 generations, often to the standard C57BL/6 strain, to minimize the effects of genetic background variation.
5. nNOS knockout mice
The first line of nNOS knockout mice was established by disrupting exon 2 of nNOS using homologous recombination.42 This region contains the sequence for the ATG initiation codon and the PDZ domain that is responsible for membrane association.16 Therefore, these mutant mice do not express nNOSα, the predominant splice form of nNOS, as detected by Western blot analysis or NADPH diaphorase staining. They have significantly diminished NO production in the brain, as measured by NOS enzymatic assay, cGMP levels, and measurement of NO by spin trapping.43–46 nNOS splice variants (nNOSβ, nNOSγ, and the testicular isoforms) that lack exon 2 are still expressed and account for less than 5% of all nNOS catalytic activity in the brain. These splice variants are soluble, since they lack the PDZ domain.
The most apparent phenotype of nNOS knockout mice is enlargement of the stomachs, often to several times the normal size, demonstrating a role for nNOS in smooth muscle relaxation of the pyloric sphincter. nNOS knockout mice are also resistant to focal and global cerebral ischaemia, consistent with a role for nNOS-derived NO in cellular injury following ischaemia.46–49 These mice are fertile and viable, although male mutants are more aggressive than their wild-type littermates.42,50 A separate line of nNOS knockout mice was established by ablating exon 6, which encodes the heme-binding domain of nNOS required for catalytic activity.51 These mice have a more severe pyloric stenosis phenotype and also reproductive endocrine abnormalities. [3H]citrulline measurements indicate that nNOS activity in exon 6-deficient mice is 0.3% of that in wild-type, compared to 5% in exon 2-deficient mice.
6. eNOS knockout mice
The first eNOS knockout mice were generated by disrupting the region that encodes for the NADPH ribose and adenine binding sites, which are essential for catalytic activity.52 These mice are viable, fertile and exhibit no gross anatomic abnormalities, despite the absence of detectable eNOS mRNA, protein or enzymatic activity. As outlined below, eNOS knockout mice show abnormalities in vascular relaxation, blood pressure regulation, and cardiac contractility. They are a useful animal model for endothelial dysfunction, as they show increased propensity to form neointima in response to vessel injury,53,54 and accelerated and more severe diet-induced atherosclerosis in the apolipoprotein E (apoE) knockout mouse model.55,56
Several additional strains of eNOS knockout mice have also been reported. In one strain, the eNOS gene was disrupted at the calmodulin binding site, encoded by exons 12 and 13,57 In another strain, the NADPH ribose and adenine binding sites were disrupted,58 similar to the first eNOS knockout mice.52 All three known eNOS knockout mice have similar phenotypes with hypertension and vascular abnormalities. The fact that independently generated mice, particularly those in which different parts of the eNOS gene were targeted, have similar phenotypes, adds confidence that the observed phenotypes are specific.
7. iNOS knockout mice
Three separate groups independently disrupted the iNOS gene. MacMicking et al. deleted the promoter region and the first four exons, including the initiation codon ATG.59 Wei et al. in an attempt to delete the first five exons of the gene, created a genomic rearrangement of the iNOS gene that results in an aberrant transcript, but no detectable iNOS activity.60 Laubach et al. disrupted the calmodulin, FAD, and FMN binding domains of iNOS, and found no detectable iNOS mRNA or protein.61 In all cases, expression of iNOS cannot be induced in the iNOS mutant mice under conditions that induce iNOS in wild-type animals. Peritoneal macrophages from all of the iNOS mutant mice are deficient in NO and nitrite production. None of the mutants demonstrate abnormalities in growth, fertility, or gross histopathology.
Initial characterization of these iNOS mutant animals centered on two proposed functions of iNOS: cell-mediated resistance to pathogens, and hemodynamic responses to septic shock. Inducible NOS mutant mice are more sensitive to the intracellular pathogen Listeria monocytogenes59 and to the intracellular protozoan parasite Leishmania major60 than are wild-type mice. Both are pathogens that elicit cell-mediated immune responses, and the increased susceptibility of iNOS mutant mice demonstrates the importance of iNOS to host defenses against these pathogens.
The role of iNOS in septic shock is supported by the finding that NO is produced in large quantities during infection.62 Inappropriate vasodilation, abnormal regulation of blood flow to organs, myocardial suppression, and interference with cellular respiration all contribute to hypotension and mortality in septic shock. Multiple studies show that NOS inhibitors can reverse the hypotension of patients in septic shock,63,64 or of animals treated with LPS or TNF.65,66 iNOS mutant mice have a blunted hypotensive response to sepsis and LPS.59–61 These findings suggest that NO generated by iNOS contributes to the inappropriate vasodilation and hypotension seen in sepsis.
8. Endothelium-derived relaxing factor activity
In 1980, Furchgott and Zawadzki found that acetylcholine is able to cause relaxation of blood vessels if, and only if, the endothelium is intact. This indicated that acetylcholine does not act directly on vascular smooth muscle, but rather, that the endothelium plays a key role in vasodilation. This led to the proposal of the existence of endothelium-derived relaxing factor, or EDRF.67 Subsequent pioneering work led to the identification of EDRF as nitric oxide.68–71 Pharmacologic inhibitors of NOS such as L-NA and L-NAME also abrogate vasodilation to acetylcholine, and in their presence, acetylcholine actually causes a slight increase in vascular tone.
One of the first experiments in eNOS knockout mice was to replicate the experiments of Furchgott and Zawadzki. In fact, isolated aortic rings from eNOS knockout do not respond to acetylcholine in organ baths.52 These aortic rings do respond to the exogenous NO donor sodium nitroprusside and to papaverine, indicating that the vascular smooth muscle is capable of relaxation. These observations establish that eNOS is an essential source of NO in the vasculature, and it is required for EDRF activity.
9. Blood pressure
There are several interacting homeostatic regulators of blood pressure, including the renin–angiotensin system, the autonomic nervous system, and local mediators such as EDRF.
L-NA and other NOS inhibitors cause a rise in blood pressure in many species, including rats, guinea pigs, rabbits, dogs and mice.72 This effect is consistent with the predicted role for basal NO production in the regulation of blood pressure. Therefore, it was of particular interest to examine basal blood pressure in the eNOS mutant mice to see if other homeostatic mechanisms would compensate for the absence of endothelial NO production.
The blood pressure of eNOS knockout mice is about 30% higher than that of wild-type animals. This is true regardless of the types of anesthesia used (and in the awake state), and is also true for independently generated eNOS knockout mouse strains. Thus, eNOS plays a key role in regulation of blood pressure. However, it is not clear why other homeostatic systems cannot compensate for absence of eNOS. The set point for systemic blood pressure is regulated through integration of cardiac, neuronal, humoral and vascular mechanisms. One possibility is that the renin–angiotensin system and autonomic nervous system evolved to serve primarily as a defense against hypotension, and diminution in their activity is a poor buffer against hypertension. Alternatively, eNOS (and indeed other NOS isoforms) may be involved in establishing the baroreceptor set point.73,74
eNOS mutant mice show a decrease in blood pressure in response to L-NA. This hypotensive effect of L-NA is prevented by l-arginine and is not observed with d-nitro-arginine. This suggests that non-endothelial NOS isoforms may play a direct or indirect role in the maintenance of blood pressure. nNOS knockout mice, whose blood pressure values are similar to wild-type littermates when awake, tend to be hypotensive under anesthesia.45,75 These results suggest an opposite role for nNOS, in raising or maintaining vascular tone. nNOS is present in central vasomotor centers, perivascular nerves, and skeletal muscle, and its effects in these locations may counter the direct vasodilatory effect of NO in vessels. Multiple roles for endothelial and non-endothelial NOS isoforms in vasodilation and vasoconstriction could also explain the observed variability in maximal pressor effects of various NOS inhibitors.
10. Vascular function and dysfunction
NO is critical to the pathophysiology of vascular disease and the concept of endothelial dysfunction. Endothelial dysfunction is defined as impairment of physiologic endothelium-dependent relaxation. It occurs in atherosclerosis, hypertension, diabetes, hypercholesterolemia, and normal aging.9–11 Impairment of endothelial function occurs before structural changes such as intimal hyperplasia or lipid deposition. This is therefore an early event in the pathophysiology of atherosclerosis. Clinically, endothelial function can be tested by using ultrasound to determine the forearm blood flow response to reflow hyperemia. Experimentally, endothelial function can be tested by using a myograph to determine the vasodilator response of an isolated vessel segment to pharmacologic agents such as acetylcholine, bradykinin, and VEGF. Endothelial dysfunction is characterized by diminished endothelial NO levels. Because eNOS knockout mice completely lack endothelial NO production, they serve as a model of extreme endothelial dysfunction.
There are several potential mechanisms for endothelial dysfunction, as outlined in Figure 1.76 These can be separated into three broad categories: reduced eNOS expression levels, reduced eNOS enzymatic activity, and rapid removal of NO. First, changes in eNOS mRNA or protein expression levels can lead to a reduction in eNOS activity.77 However, most evidence from animal models and humans suggests there is an increase in the amount of eNOS with diabetes and atherosclerosis, rather than a decrease. Second, l-arginine, the substrate for NO production, can be limiting in tissues. An endogenous competitive inhibitor, asymmetric dimethylarginine (ADMA) may reduce endothelial NO production even in the presence of adequate l-arginine levels.78,79 Third, eNOS requires FAD, FMN, NADPH, and BH4 as cofactors. BH4, whose synthesis is rate-limited by GTP cyclohydrolase, is a particularly important cofactor, because in its absence, electron transport through eNOS can become ‘uncoupled,’ resulting in generation of superoxide anion.80 Fourth, eNOS requires dimerization and proper intracellular localization to caveolae, mediated in part by interactions with caveolin and hsp90.12,22 Fifth, eNOS is phosphorylated at S1179 by Akt kinase and other kinases.81,82 Sixth, NO produced by eNOS may be rapidly inactivated by reaction with superoxide to form peroxynitrite anion (OONO−).32 This superoxide can be formed by NADPH oxidase,83 or uncoupled eNOS.80 These conditions may all contribute to endothelial dysfunction.
Figure 1.
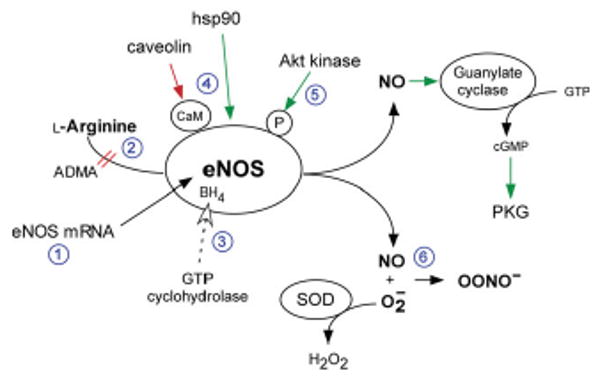
Regulation of eNOS activity and mechanisms for endothelial dysfunction. Several mechanisms can account for endothelial dysfunction, including: (1) changes in eNOS mRNA or protein levels; (2) decreased substrate availability; (3) decreased cofactor availability; (4) improper subcellular localization; (5) abnormal phosphorylation; and (6) scavenging of NO by superoxide to form peroxynitrite anion (ONOO−). ADMA, asymmetric dimethylarginine; SOD, superoxide dismutase; PKG, protein kinase G.
While eNOS plays important roles in vessel function, excessive NO production may contribute to the development of atherosclerosis. iNOS is expressed in activated monocytes and macrophages, and both iNOS and nNOS are expressed in vascular smooth muscle cells in atherosclerotic lesions.84–86 These isoforms may contribute to lesion formation by forming NO which can react with superoxide to form the extremely potent oxidant peroxynitrite. NO and peroxynitrite can both increase oxidative stress and oxidize LDL.87 Human atherosclerotic lesions contain nitrotyrosine, which is evidence for the presence of peroxynitrite. NO can also affect redox-sensitive transcription of genes involved in endothelial cell activation such as VCAM-1.88,89
11. Vascular injury and diet-induced atherosclerosis
Atherosclerosis is driven by biochemical, cellular, and hemodynamic forces in the vessel wall that cause vascular injury, ultimately leading to endothelial dysfunction, cellular proliferation, recruitment of inflammatory cells, and accumulation of oxidized LDL.90 The response of blood vessels to injury is formation of neointima. Vascular smooth muscle cells proliferate in the medial layer and migrate across the internal elastic lamina to form the neointima. NO suppresses smooth muscle proliferation in response to vessel injury,91 suggesting that it normally serves a protective role. In association with other effects such as inhibition of platelet aggregation and adhesion2 and inhibition of leukocyte activation and adhesion,5,92 NO normally suppresses the processes that lead to the development of atherosclerotic plaques. A relative deficiency in vascular NO would reduce these normally protective effects and thereby predispose to atherosclerosis.
To assess whether eNOS has a role in neointima formation following vascular injury, eNOS knockout mice were subjected to a cuff model of vascular injury.53 eNOS knockout mice show significantly greater neointima formation after cuff injury than wild-type mice. Thus, results from eNOS knockout mice confirm results using pharmacologic agents, and show that a deficiency in the amount of available NO in the vessel wall by itself increases neointimal formation in response to vascular injury.
To mimic human diet-induced atherosclerosis, apoE knockout mice have been a useful mouse model. apoE knockout mice develop spontaneous atherosclerotic lesions in their aortas which are exacerbated by a high fat, ‘Western’ diet.93,94 To study the effects of individual NOS isoforms on diet-induced atherosclerosis, each NOS knockout mouse was bred onto the apoE knockout background, creating double knockout mice.
apoE/eNOS double knockout mice on a Western diet develop atherosclerosis significantly faster than, and have almost twice the atherosclerotic lesion areas as, apoE knockout mice on the same diet.56 apoE/eNOS double knockout mice also show evidence of coronary artery disease, left ventricular dysfunction, aortic aneurysm and aortic dissection. The phenotype of apoE/eNOS double knockout mice more closely resembles the spectrum of cardiovascular complications seen in human atherosclerosis. It is also the first murine model to demonstrate spontaneous distal coronary arteriosclerosis associated with left ventricular dysfunction. These findings support the concept that restoration of eNOS function in patients with atherosclerosis is an important therapeutic goal. apoE/eNOS double knockout mice are hypertensive, but pharmacological control of blood pressure still leads to accelerated atherosclerosis and the development of aortic aneurysms.55 Thus, the effects due to eNOS deficiency are not merely caused by hypertension.
In contrast, apoE/iNOS double knockout mice show significantly smaller lesion areas compared to apoE knockout mice at 16 and 24 weeks.95 The lipoprotein profile, as assessed by FPLC, do not differ between apoE knockout mice and apoE/iNOS double knockout mice. The reduction in atherosclerosis in double knockout animals is associated with decreased plasma levels of lipoperoxides, suggesting that reduction in iNOS-mediated oxidative stress may explain the protection from lesion formation in double knockout animals. Thus, genetic deficiency of iNOS decreases atherosclerosis in Western diet-fed apoE knockout animals.
Like the apoE/eNOS double knockout mice, apoE/nNOS double knockout mice on a Western diet develop greater atherosclerotic lesion areas than apoE knockout mice.96 RT-PCR shows that the predominant nNOSα splice variant is absent, although nNOSγ is present. nNOS deficiency significantly reduces the mean arterial blood pressure in female apoE knockout mice but is unchanged in male mice. apoE/nNOS double knockout mice also have higher mortality rates than apoE knockout mice. Thus, there is evidence that nNOS may serve atheroprotective roles, like eNOS.
12. Cerebral ischaemia
Following cerebral ischaemia, NO levels in the brain rise several orders of magnitude, from baseline nanomolar levels to stimulated micromolar levels.97 nNOS knockout mice reveal that the nNOS isoform mediates this increase, since the knockout mice do not show it.48 When subjected to a middle cerebral artery (MCA) occlusion model of focal ischaemia, nNOS knockout mice develop significantly smaller infarct sizes and have better neurological outcome than wild-type mice. These results confirm that although it normally serves important vascular physiological roles, NO overproduction in the setting of cerebral ischaemia actually contributes to tissue damage.
Measurements of regional cerebral blood flow (rCBF) by laser Doppler flowmetry show that both nNOS knockout mice and wild-type mice have similar reductions in blood flow. Thus, the smaller infarct sizes in the nNOS knockout mice cannot be explained by differences in cerebral blood flow.48 Protection in nNOS knockout mice is also observed in transient and permanent focal ischaemia models and global ischaemia models.47,49 The iNOS isoform is not present in ischaemic brain until days later, as glia and inflammatory cells enter the infarct zone. Like nNOS, iNOS contributes to tissue damage after cerebral ischaemia. Inhibition of iNOS by selective pharmacologic inhibitors, or gene deletion of iNOS, reduces this late damage.98–100 nNOS and iNOS may contribute to neuronal toxicity in several ways, including reaction with superoxide to form peroxynitrite anion or otherwise increasing reactive oxygen species,101 activation of poly-ADP ribose synthase resulting in depletion of cellular energy stores,40 and direct inhibition of mitochondrial complexes I and IV.102
In contrast, eNOS knockout mice subjected to the MCA occlusion model develop larger infarct sizes compared to wild-type mice.103 Laser Doppler flowmetry103 and temporal correlation mapping104 shows that eNOS knockout mice have significantly reduced blood flow than do wild-type mice. This confirms that eNOS normally serves to vasodilate and preserve blood flow in the setting of ischaemia; in its absence, inability to preserve blood flow contributes to the enlarged infarct sizes observed in eNOS knockout mice.
13. Ischaemic preconditioning
Ischaemic preconditioning (IPC) refers to processes by which brief, sublethal episodes of ischaemia stimulate a protective response against subsequent, more severe, ischaemia.105 IPC has been described in many tissues, including the heart, brain, liver, and gastrointestinal tract. Although there are similarities between different tissues, it is not known whether the triggers and mediators of IPC are the same in all tissues. The potential protective mechanisms include alterations in cell death genes,106 heat shock proteins,107 lipid peroxidation,108 inflammation,109 and mitochondrial metabolism.110 IPC has been divided into rapid and delayed forms. Rapid IPC occurs when the preconditioning stimulus precedes the severe ischaemic insult by a short time interval (minutes to several hours), while delayed IPC occurs requires a longer time interval (hours to days) to develop. It has been hypothesized that delayed IPC may involve changes in gene expression and new protein synthesis.
Pharmacologic studies suggest separable roles for each of the NOS isoforms in cerebral IPC. In a newborn rat model of hypoxia-ischaemia, preconditioning by mild hypoxia protects against more severe hypoxia 24 h later.111 IPC protection is prevented by L-NA, but not the iNOS-specific inhibitor 7-nitroindazole or the iNOS specific inhibitor aminoguanidine, suggesting by process of elimination, involvement of the eNOS isoform. In another rat model, both transient cerebral ischaemia and LPS could protect against later ischaemia.112 LPS treatment was associated with increased eNOS expression, and protection was blocked by L-NAME. The anesthetics isoflurane and halothane also protect against cerebral ischaemia 24 h later.113 iNOS induction is required and protection is blocked by the iNOS inhibitor aminoguanidine.
Cerebral IPC is more difficult to study in mice for technical reasons because the vessels are smaller. However, a mouse model of rapid preconditioning has been developed.114 Three episodes of transient middle cerebral artery occlusion (the preconditioning stimulus), each lasting five minutes, protect the brain against damage from permanent vessel occlusion thirty minutes later. Infarct size measured 24 h later is reduced in preconditioned animals. This mouse model allows the study of NOS knockout mice to determine the separate roles of NOS isoforms. While wild-type mice demonstrate a reduction in infarct size following 3 cycles of IPC, neither eNOS knockout mice nor nNOS knockout mice do.115 Baseline absolute blood flow measurements are the same in the three genotypes, so differences in the baseline absolute rCBF do not account for the results. Relative blood flow measurements by laser Doppler flowmetry confirm effective MCA occlusion with each preconditioning episode in each of the three genotypes. These results suggest that both nNOS and eNOS are required for cerebral IPC.
There are several potential mechanisms by which NO mediates IPC.105 First, NO may be required as a trigger to stimulate downstream steps involved in the mechanisms of IPC. Second, NO may be involved as a mediator of protection by affecting neuronal resistance to ischaemic phenomena. NO interacts with at least two signaling pathways important to neuronal survival: the Ras/Raf/MEK/ERK cascade,116,117 and the PI3 kinase/Akt pathway.118,119 These pathways may be unifying mechanisms that underlie protection. Third, as a vasodilator, NO may augment blood flow by vasodilation, reducing leukocyte-endothelial interactions and platelet-endothelial interactions. Together these effects would limit the functional effect of ischaemia.
In the heart, NO is thought to play important protective roles, not only through blood flow effects, but also by directly enhancing cardioprotection. Myocardial ischaemia-reperfusion injury, and cardiac IPC appear to depend on iNOS, rather than nNOS or eNOS.120,121 However, the molecular mechanisms of iNOS protection are not fully understood, and may involve electron transport or the mitochondrial permeability transition pore. iNOS knockout animals may be useful tools to define these processes.
14. Cardiac contractility
Pharmacologic blockade of NOS activity first suggested that NO plays a significant role in regulating cardiac contractility.122–125 NO is produced in at least two major cell types in the heart: endothelial cells and cardiac myocytes. Endothelial cells are rich in eNOS and line the vasculature and endocardium. Cardiac myocytes, which are essential for cardiac excitation-contraction coupling, express both eNOS and nNOS. In cardiac myocytes, eNOS is localized to the sarcolemmal caveolae126 through its interaction with caveolin-3.127 nNOS, however, is localized to the sarcoplasmic reticulum,128 where it is associated with the ryanodine receptor.127 Because eNOS and nNOS reside in different subcellular locations, they can play distinct roles in cardiac function (see Figure 2).
Figure 2.
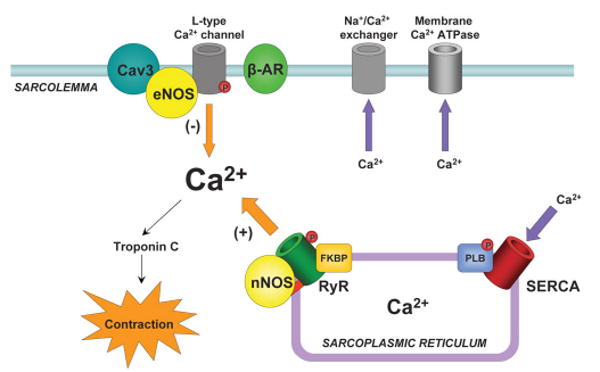
eNOS and nNOS in cardiac excitation–contraction coupling. In cardiac myocytes, eNOS associates with caveolin-3 at the sarcolemma, where it blunts inotropic response to isoproterenol stimulation. nNOS associates with the ryanodine receptor at the sarcoplasmic reticulum and is required for proper Ca2+-mediated Ca2+ release. Arrow thickness reflects magnitude of Ca2+ flux. Cav3, caveolin-3; β-AR, β-adrenergic receptor; RyR, ryanodine receptor; FKBP, FK506-binding protein; PLB, phospholamban; SERCA, sarcoplasmic reticulum Ca2+-ATPase.
Using Langendorff isolated heart preparations and in vivo measurements, Gyurko et al. found that eNOS knockout mice show no difference in basal contractility, but they do show enhanced inotropic and lusitropic responses to isoproterenol stimulation.129 These findings were confirmed by further in vivo studies.127 eNOS gene deletion enhances inotropic response not only to isoproterenol stimulation, but also in basal conditions.127 These results suggest that eNOS blunts the inotropic response to isoproterenol stimulation and its genetic absence enhances contractility.
nNOS knockout mice have been studied for phenotypes related to cardiac contractility, with variable results. In one case, nNOS knockout mice, as well as pharmacologic inhibition of nNOS in wild-type mice, causes enhanced basal LV contraction.130,131 In another study, the same exon 2 nNOS knockout mice did not show significant differences in basal contractility, but showed decreased inotropic response to isoproterenol stimulation.127 nNOS gene deletion has also been associated with more severe left ventricular remodelling after myocardial infarction.132
The subcellular localization of nNOS to the sarcoplasmic reticulum suggests that NO can modulate cardiomyocyte calcium handling. There is direct evidence that nNOS nitrosylates and activates the ryanodine receptor. Thus, nNOS normally maintains function of the ryanodine receptor, and in its absence, contractility will be affected. In patients with congestive heart failure, there is also evidence that nNOS is not properly localized to the sarcoplasmic reticulum.133 When this occurs, nNOS may translocate to the L-type calcium channel at the cell surface, and play an inhibitory role much like eNOS does. The importance of nNOS to cardiac contractility and calcium handling is underscored by the recent finding that long QT syndrome, associated with dangerous ventricular arrhythmias in humans, is closely associated with NOS1-activating protein (NOS1AP), which binds to nNOS.134 This raises the possibility that NOS1AP and nNOS affect not only cardiac contractility, but also propensity to ventricular arrhythmias through effects on the ryanodine receptor and calcium handling.
15. Non-cardiovascular phenotypes
In addition to the cardiovascular phenotypes discussed here, mutant mice have been useful to define the roles of each NOS isoform in other biological processes. nNOS knockout mice, eNOS knockout mice and nNOS/eNOS double knockout mice revealed complex roles for NO as a retrograde messenger in long term potentiation.135,136 In addition to known roles of iNOS in osteoclast function, eNOS knockout mice show that eNOS is essential to bone formation and normal osteoblast activity.137 Given the original enlarged gastrointestinal phenotype of nNOS knockout mice, it is not surprising that they have revealed how NO acts as an neurotransmitter to modulate the inhibitory junctional potential in the stomach and intestines.138
16. Conclusion
Targeted disruption of the nNOS, iNOS and eNOS genes in mice has led to the development of mutant mice that have been useful tools with which to study how NO affects blood pressure regulation, endothelial dysfunction, response to vascular injury, response to stroke and cerebral ischaemia, diet-induced atherosclerosis and cardiac contractility. As we begin to understand better the many diverse roles of NO, we can build upon this foundation to translate these findings into novel clinical approaches to prevent and treat cardiovascular diseases. Because patients with cardiovascular diseases are not totally devoid of either nNOS or eNOS, future work will likely involve more refined approaches to modulate the activity of NOS isoforms short of total gene disruption. Candidate regulatory sites and domains can be mutated, and the effects of these modifications can be studied in vivo. Perspectives from biochemistry, molecular biology, cell biology, physiology, chemistry and pharmacology will all complement genetic approaches to studying the roles of NO in cardiovascular diseases.
Acknowledgments
This work was supported by National Institutes of Health R01 grants NS33335, NS48423, and HL57818 to PLH.
Footnotes
This article was published online by Elsevier on 30 June, 2007.
Conflict of interest: none declared.
References
- 1.Bredt DS, Snyder SH. Nitric oxide: a physiologic messenger molecule. Ann Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 2.Radomski MW, Palmer RM, Moncada S. Modulation of platelet aggregation by an l-arginine-nitric oxide pathway. Trends Pharmacol Sci. 1991;12:87–88. doi: 10.1016/0165-6147(91)90510-y. [DOI] [PubMed] [Google Scholar]
- 3.Freedman JE, Sauter R, Battinelli EM, Ault K, Knowles C, Huang PL, et al. Deficient platelet-derived nitric oxide and enhanced hemostasis in mice lacking the NOSIII gene. Circ Res. 1999;84:1416–1421. doi: 10.1161/01.res.84.12.1416. [DOI] [PubMed] [Google Scholar]
- 4.Kubes P, Suzuki M, Granger DN. Nitric Oxide: An Endogenous Modulator of Leukocyte Adhesion. Proc Natl Acad Sci USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefer AM, Ma XL. Decreased basal nitric oxide release in hypercholesterolemia increases neutrophil adherence to rabbit coronary artery endothelium. Arterioscler Thromb. 1993;13:771–776. doi: 10.1161/01.atv.13.6.771. [DOI] [PubMed] [Google Scholar]
- 6.Lefer DJ, Jones SP, Girod WG, Baines A, Grisham MB, Cockrell AS, et al. Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am J Physiol. 1999;276:H1943–H1950. doi: 10.1152/ajpheart.1999.276.6.H1943. [DOI] [PubMed] [Google Scholar]
- 7.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:1774–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massion PB, Feron O, Dessy C, Balligand JL. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res. 2003;93:388–398. doi: 10.1161/01.RES.0000088351.58510.21. [DOI] [PubMed] [Google Scholar]
- 9.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 10.Gimbrone MA, Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci. 2000;902:230–239. doi: 10.1111/j.1749-6632.2000.tb06318.x. discussion 9–40. [DOI] [PubMed] [Google Scholar]
- 11.Grange R, Isotani E, Lau K, Kamm K, Huang P, Stull J. Nitric oxide contributes to vascular smooth muscle relaxation in contracting fast-twitch muscles. Physiol Genomics. 2001;5:35–44. doi: 10.1152/physiolgenomics.2001.5.1.35. [DOI] [PubMed] [Google Scholar]
- 12.Gimbrone MA., Jr Endothelial dysfunction and atherosclerosis. J Card Surg. 1989;4:180–183. doi: 10.1111/j.1540-8191.1989.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 13.Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 14.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenman JE, Christopherson KS, Craven SE, McGee AW, Bredt DS. Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. J Neurosci. 1996;16:7407–7415. doi: 10.1523/JNEUROSCI.16-23-07407.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and [alpha]1-syntrophin mediated by PDZ domains. Cell. 1996;84:757. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 17.Silvagno F, Xia H, Bredt DS. Neuronal nitric-oxide synthase-mu, an alternatively spliced isoform expressed in differentiated skeletal muscle. J Biol Chem. 1996;271:11204–11208. doi: 10.1074/jbc.271.19.11204. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamas S, Marsden PA, Li GK, Tempst P, Michel T. Endothelial nitric oxide synthase: molecular cloning and characterization of a distinct constitutive enzyme isoform. Proc Natl Acad Sci USA. 1992;89:6348–6352. doi: 10.1073/pnas.89.14.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Garcia-Cardena G, Sessa WC. Palmitoylation of endothelial nitric oxide synthase is necessary for optimal stimulated release of nitric oxide: implications for caveolae localization. Biochemistry. 1996;35:13277–13281. doi: 10.1021/bi961720e. [DOI] [PubMed] [Google Scholar]
- 21.Robinson LJ, Michel T. Mutagenesis of palmitoylation sites in endothelial nitric oxide synthase identifies a novel motif for dual acylation and subcellular targeting. Proc Natl Acad Sci USA. 1995;92:11776–11780. doi: 10.1073/pnas.92.25.11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaul PW. Regulation of endothelial nitric oxide synthase: Location, location, location. Annu Rev Physiol. 2002;64:749–774. doi: 10.1146/annurev.physiol.64.081501.155952. [DOI] [PubMed] [Google Scholar]
- 23.Ghafourifar P, Richter C. Mitochondrial nitric oxide synthase regulates mitochondrial matrix pH. Biol chem. 1999;380:1025–1028. doi: 10.1515/BC.1999.127. [DOI] [PubMed] [Google Scholar]
- 24.Ghafourifar P, Schenk U, Klein SD, Richter C. Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. J Biol Chem. 1999;274:31185–31188. doi: 10.1074/jbc.274.44.31185. [DOI] [PubMed] [Google Scholar]
- 25.Dawson TM, Dawson VL, Snyder SH. Molecular mechanisms of nitric oxide actions in the brain. Ann NY Acad Sci. 1994;738:76–85. doi: 10.1111/j.1749-6632.1994.tb21792.x. [DOI] [PubMed] [Google Scholar]
- 26.Snyder SH, Bredt DS. Biological roles of nitric oxide. Sci Am. 1992;266:68–71. doi: 10.1038/scientificamerican0592-68. [DOI] [PubMed] [Google Scholar]
- 27.Garthwaite J, Balazs R. Supersensitivity to the cyclic GMP response to glutamate during cerebellar maturation. Nature. 1978;275:328–329. doi: 10.1038/275328a0. [DOI] [PubMed] [Google Scholar]
- 28.Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 29.Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, et al. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- 30.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 31.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 33.Blough N, Zafiriou O. Reaction of superoxide with nitric oxide to form peroxynitrite in alkaline aqueous solution. Inorg Chem. 1985;24:3502–3504. [Google Scholar]
- 34.Beckman JS, Crow JP. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem Soc Trans. 1993;21:330–334. doi: 10.1042/bst0210330. [DOI] [PubMed] [Google Scholar]
- 35.Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- 36.Okada S, Takehara Y, Yabuki M, Yoshioka T, Yasuda T, Inoue M, et al. Nitric oxide, a physiological modulator of mitochondrial function. Physiol Chem Phys Med NMR. 1996;28:69–82. [PubMed] [Google Scholar]
- 37.Takehara Y, Kanno T, Yoshioka T, Inoue M, Utsumi K. Oxygen-dependent regulation of mitochondrial energy metabolism by nitric oxide. Arch Biochem Biophys. 1995;323:27–32. doi: 10.1006/abbi.1995.0005. [DOI] [PubMed] [Google Scholar]
- 38.Dawson TM, Dawson VL, Snyder SH. Nitric oxide as a mediator of neuro-toxicity. NIDA Res Monogr. 1993;136:258–271. [PubMed] [Google Scholar]
- 39.Dawson TM, Zhang J, Dawson VL, Snyder SH. Nitric oxide: cellular regulation and neuronal injury. Prog Brain Res. 1994;103:365–369. doi: 10.1016/s0079-6123(08)61150-4. [DOI] [PubMed] [Google Scholar]
- 40.Endres M, Scott G, Namura S, Salzman AL, Huang PL, Moskowitz MA, et al. Role of peroxynitrite and neuronal nitric oxide synthase in the activation of poly(ADP-ribose) synthetase in a murine model of cerebral ischaemia-reperfusion. Neurosci Lett. 1998;248:41–44. doi: 10.1016/s0304-3940(98)00224-9. [DOI] [PubMed] [Google Scholar]
- 41.Rabelink TJ, Luscher TF. Endothelial Nitric Oxide Synthase: host defense enzyme of the endothelium? Arterioscler Thromb Vasc Biol. 2006;26:267–271. doi: 10.1161/01.ATV.0000196554.85799.77. [DOI] [PubMed] [Google Scholar]
- 42.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 43.Darius S, Wolf G, Huang PL, Fishman MC. Localization of NADPH-diaphorase/nitric oxide synthase in the rat retina: an electron microscopic study. Brain Res. 1995;690:231–235. doi: 10.1016/0006-8993(95)00559-9. [DOI] [PubMed] [Google Scholar]
- 44.Ichinose F, Huang PL, Zapol WM. Effects of targeted neuronal nitric oxide synthase gene disruption and nitroG-L-arginine methylester on the threshold for isoflurane anesthesia. Anesthesiology. 1995;83:101–108. doi: 10.1097/00000542-199507000-00013. [DOI] [PubMed] [Google Scholar]
- 45.Irikura K, Huang PL, Ma J, Lee WS, Dalkara T, Fishman MC, et al. Cerebrovascular alterations in mice lacking neuronal nitric oxide synthase gene expression. Proc Natl Acad Sci USA. 1995;92:6823–6827. doi: 10.1073/pnas.92.15.6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaharchuk G, Hara H, Huang PL, Fishman MC, Moskowitz MA, Jenkins BG, et al. Neuronal nitric oxide synthase mutant mice show smaller infarcts and attenuated apparent diffusion coefficient changes in the peri-infarct zone during focal cerebral ischaemia. Magn Reson Med. 1997;37:170–175. doi: 10.1002/mrm.1910370204. [DOI] [PubMed] [Google Scholar]
- 47.Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab. 1996;16:605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 48.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischaemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 49.Panahian N, Yoshida T, Huang PL, Hedley-Whyte ET, Dalkara T, Fishman MC, et al. Attenuated hippocampal damage after global cerebral ischaemia in mice mutant in neuronal nitric oxide synthase. Neuroscience. 1996;72:343–354. doi: 10.1016/0306-4522(95)00563-3. [DOI] [PubMed] [Google Scholar]
- 50.Nelson RJ, Demas GE, Huang PL, Fishman MC, Dawson VL, Dawson TM, et al. Behavioural abnormalities in male mice lacking neuronal nitric oxide synthase. Nature. 1995;378:383. doi: 10.1038/378383a0. [DOI] [PubMed] [Google Scholar]
- 51.Gyurko R, Leupen S, Huang PL. Deletion of Exon 6 of the neuronal nitric oxide synthase gene in mice results in hypogonadism and infertility. Endocrinology. 2002;143:2767–2774. doi: 10.1210/endo.143.7.8921. [DOI] [PubMed] [Google Scholar]
- 52.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 53.Moroi M, Zhang L, Yasuda T, Virmani R, Gold HK, Fishman MC, et al. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J Clin Invest. 1998;101:1225–1232. doi: 10.1172/JCI1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L, Fishman MC, Huang PL. Estrogen mediates the protective effects of pregnancy and chorionic gonadotropin in a mouse model of vascular injury. Arterioscler Thromb Vasc Biol. 1999;19:2059–2065. doi: 10.1161/01.atv.19.9.2059. [DOI] [PubMed] [Google Scholar]
- 55.Chen J, Kuhlencordt PJ, Astern J, Gyurko R, Huang PL. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein E/endothelial nitric oxide synthase double knockout mice. Circulation. 2001;104:2391–2394. doi: 10.1161/hc4501.099729. [DOI] [PubMed] [Google Scholar]
- 56.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischaemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 57.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Godecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Godecke S, et al. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- 59.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 60.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 61.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccaride-induced death. Proc Natl Acad Sci USA. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stuehr DJ, Marletta MA. Induction of nitrite/nitrate synthesis in murine macrophages by BCG infection, lymphokines, or interferon-gamma. J Immunol. 1987;139:518–525. [PubMed] [Google Scholar]
- 63.Petros A, Lamb G, Leone A, Moncada S, Bennett D, Vallance P. Effects of a nitric oxide synthase inhibitor in humans with septic shock. Cardiovasc Res. 1994;28:34–39. doi: 10.1093/cvr/28.1.34. [DOI] [PubMed] [Google Scholar]
- 64.Schilling J, Cakmakci M, Battig U, Geroulanos S. A new approach in the treatment of hypotension in human septic shock by NG-monomethyl-l-arginine, an inhibitor of the nitric oxide synthetase. Intensive Care Med. 1993;19:227–231. doi: 10.1007/BF01694775. [DOI] [PubMed] [Google Scholar]
- 65.Kilbourn RG, Owen-Schaub LB, Cromeens DM, Gross SS, Flaherty MJ, Santee SM, et al. NG-methyl-l-arginine, an inhibitor of nitric oxide formation, reverses IL-2-mediated hypotension in dogs. J Appl Physiol. 1994;76:1130–1137. doi: 10.1152/jappl.1994.76.3.1130. [DOI] [PubMed] [Google Scholar]
- 66.Thiemermann C. The role of the l-arginine: nitric oxide pathway in circulatory shock. Adv Pharmacol. 1994;28:45–79. doi: 10.1016/s1054-3589(08)60493-7. [DOI] [PubMed] [Google Scholar]
- 67.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 68.Furchgott RF. The 1989 Ulf von Euler lecture. Studies on endothelium-dependent vasodilation and the endothelium-derived relaxing factor. Acta Physiol Scand. 1990;139:257–270. doi: 10.1111/j.1748-1716.1990.tb08923.x. [DOI] [PubMed] [Google Scholar]
- 69.Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- 70.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 72.Sakuma I, Togashi H, Yoshioka M, Saito H, Yanagida M, Tamura M, et al. NG-methyl-l-arginine, an inhibitor of l-arginine-derived nitric oxide synthesis, stimulates renal sympathetic nerve activity in vivo. A role for nitric oxide in the central regulation of sympathetic tone? Circ Res. 1992;70:607–611. doi: 10.1161/01.res.70.3.607. [DOI] [PubMed] [Google Scholar]
- 73.Matsuda T, Bates JN, Lewis SJ, Abboud FM, Chapleau MW. Modulation of baroreceptor activity by nitric oxide and S-nitrosocysteine. Circ res. 1995;76:426–433. doi: 10.1161/01.res.76.3.426. [DOI] [PubMed] [Google Scholar]
- 74.Scrogin KE, Veelken R, Luft FC. Sympathetic baroreceptor responses after chronic NG-nitro-l-arginine methyl ester treatment in conscious rats. Hypertension. 1994;23:982–986. doi: 10.1161/01.hyp.23.6.982. [DOI] [PubMed] [Google Scholar]
- 75.Snyder SH. No endothelial NO. Nature. 1995;377:196–197. doi: 10.1038/377196a0. [DOI] [PubMed] [Google Scholar]
- 76.Huang PL. Unraveling the links between diabetes, obesity, and cardiovascular disease. Circ Res. 2005;96:1129–1131. doi: 10.1161/01.RES.0000170705.56583.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y, Marsden PA. Nitric oxide synthases: gene structure and regulation. Adv Pharmacol. 1995;34:71–90. doi: 10.1016/s1054-3589(08)61081-9. [DOI] [PubMed] [Google Scholar]
- 78.Bode-Boger SM, Boger RH, Kienke S, Junker W, Frolich JC. Elevated l-arginine/dimethylarginine ratio contributes to enhanced systemic NO production by dietary l-arginine in hypercholesterolemic rabbits. Biochem Biophys Res Commun. 1996;219:598–603. doi: 10.1006/bbrc.1996.0279. [DOI] [PubMed] [Google Scholar]
- 79.Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032–2037. doi: 10.1161/01.atv.20.9.2032. [DOI] [PubMed] [Google Scholar]
- 80.Cosentino F, Patton S, d'Uscio LV, Werner ER, Werner-Felmayer G, Moreau P, et al. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J Clin Invest. 1998;101:1530–1537. doi: 10.1172/JCI650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 82.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 84.Aji W, Ravalli S, Szabolcs M, Jiang XC, Sciacca RR, Michler RE, et al. l-arginine prevents xanthoma development and inhibits atherosclerosis in LDL receptor knockout mice. Circulation. 1997;95:430–437. doi: 10.1161/01.cir.95.2.430. [DOI] [PubMed] [Google Scholar]
- 85.Sobey CG, Brooks RM, II, Heistad DD. Evidence that expression of inducible nitric oxide synthase in response to endotoxin is augmented in atherosclerotic rabbits. Circ Res. 1995;77:536–543. doi: 10.1161/01.res.77.3.536. [DOI] [PubMed] [Google Scholar]
- 86.Wilcox JN, Nelken NA, Coughlin SR, Gordon D, Schall TJ. Local expression of inflammatory cytokines in human atherosclerotic plaques. J Atheroscler Thromb. 1994;1 1:S10–S13. doi: 10.5551/jat1994.1.supplemment1_s10. [DOI] [PubMed] [Google Scholar]
- 87.Darley-Usmar VM, Hogg N, O'Leary VJ, Wilson MT, Moncada S. The simultaneous generation of superoxide and nitric oxide can initiate lipid peroxidation in human low density lipoprotein. Free Radic Res Commun. 1992;17:9–20. doi: 10.3109/10715769209061085. [DOI] [PubMed] [Google Scholar]
- 88.Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SVY, Tejani AD, et al. From the Cover: Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci USA. 2004;101:15944–15948. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, et al. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92:1866–1874. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ross R. Atherosclerosis—An Inflammatory Disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 91.Mooradian DL, Hutsell TC, Keefer LK. Nitric oxide (NO) donor molecules: effect of NO release rate on vascular smooth muscle cell proliferation in vitro. J Cardiovasc Pharmacol. 1995;25:674–678. [PubMed] [Google Scholar]
- 92.Bath PM. The effect of nitric oxide-donating vasodilators on monocyte chemotaxis and intracellular cGMP concentrations in vitro. Eur J Clin Pharmacol. 1993;45:53–58. doi: 10.1007/BF00315350. [DOI] [PubMed] [Google Scholar]
- 93.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 94.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 95.Kuhlencordt PJ, Chen J, Han F, Astern J, Huang PL. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation. 2001;103:3099–3104. doi: 10.1161/01.cir.103.25.3099. [DOI] [PubMed] [Google Scholar]
- 96.Kuhlencordt PJ, Hotten S, Schodel J, Rutzel S, Hu K, Widder J, et al. Atheroprotective Effects of Neuronal Nitric Oxide Synthase in Apolipo-protein E Knockout Mice. Arterioscler Thromb Vasc Biol. 2006;26:1539–1544. doi: 10.1161/01.ATV.0000223143.88128.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 98.Iadecola C, Zhang F, Xu X. Inhibition of inducible nitric oxide synthase ameliorates cerebral ischaemic damage. Am J Physiol. 1995;268:R286–R292. doi: 10.1152/ajpregu.1995.268.1.R286. [DOI] [PubMed] [Google Scholar]
- 99.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischaemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang F, Casey RM, Ross ME, Iadecola C. Aminoguanidine ameliorates and l-arginine worsens brain damage from intraluminal middle cerebral artery occlusion. Stroke. 1996;27:317–323. doi: 10.1161/01.str.27.2.317. [DOI] [PubMed] [Google Scholar]
- 101.Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 102.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 104.Lo E, Hara H, Rogowska J, Trocha M, Pierce AR, Huang PL, et al. Temporal correlation mapping analysis of the hemodynamic penumbra in mutant mice deficient in endothelial nitric oxide synthase gene expression. Stroke. 1996;27:1381–1385. doi: 10.1161/01.str.27.8.1381. [DOI] [PubMed] [Google Scholar]
- 105.Nandagopal K, Dawson TM, Dawson VL. Critical role for nitric oxide signaling in cardiac and neuronal ischaemic preconditioning and tolerance. J Pharmacol Exp Ther. 2001;297:474–478. [PubMed] [Google Scholar]
- 106.Shimazaki K, Ishida A, Kawai N. Increase in bcl-2 oncoprotein and the tolerance to ischaemia-induced neuronal death in the gerbil hippocampus. Neurosci Res. 1994;20:95–99. doi: 10.1016/0168-0102(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 107.Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–99. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- 108.Chimon GN, Wong PT. Ischemic tolerance and lipid peroxidation in the brain. NeuroReport. 1998;9:2269–2272. doi: 10.1097/00001756-199807130-00023. [DOI] [PubMed] [Google Scholar]
- 109.Perez-Pinzon MA, Vitro TM, Dietrich WD, Sick TJ. The effect of rapid preconditioning on the microglial, astrocytic and neuronal consequences of global cerebral ischaemia. Acta Neuropathol (Berl) 1999;97:495–501. doi: 10.1007/s004010051019. [DOI] [PubMed] [Google Scholar]
- 110.Dave KR, Saul I, Busto R, Ginsberg MD, Sick TJ, Perez-Pinzon MA. Ischemic preconditioning preserves mitochondrial function after global cerebral ischaemia in rat hippocampus. J Cereb Blood Flow Metab. 2001;21:1401–1410. doi: 10.1097/00004647-200112000-00004. [DOI] [PubMed] [Google Scholar]
- 111.Gidday JM, Shah AR, Maceren RG, Wang Q, Pelligrino DA, Holtzman DM, et al. Nitric oxide mediates cerebral ischaemic tolerance in a neonatal rat model of hypoxic preconditioning. J Cereb Blood Flow Metab. 1999;19:331–340. doi: 10.1097/00004647-199903000-00011. [DOI] [PubMed] [Google Scholar]
- 112.Puisieux F, Deplanque D, Pu Q, Souil E, Bastide M, Bordet R. Differential role of nitric oxide pathway and heat shock protein in preconditioning and lipopolysaccharide-induced brain ischaemic tolerance. Eur J Pharmacol. 2000;389:71–78. doi: 10.1016/s0014-2999(99)00893-6. [DOI] [PubMed] [Google Scholar]
- 113.Kapinya K, Penzel R, Sommer C, Kiessling M. Temporary changes of the AP-1 transcription factor binding activity in the gerbil hippocampus after transient global ischaemia, and ischaemic tolerance induction. Brain Res. 2000;872:282–293. doi: 10.1016/s0006-8993(00)02503-8. [DOI] [PubMed] [Google Scholar]
- 114.Stagliano NE, Perez-Pinzon MA, Moskowitz MA, Huang PL. Focal ischaemic preconditioning induces rapid tolerance to middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 1999;19:757–761. doi: 10.1097/00004647-199907000-00005. [DOI] [PubMed] [Google Scholar]
- 115.Atochin DN, Clark J, Demchenko IT, Moskowitz MA, Huang PL. Rapid cerebral ischaemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke. 2003;34:1299–1303. doi: 10.1161/01.STR.0000066870.70976.57. [DOI] [PubMed] [Google Scholar]
- 116.Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, et al. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischaemic preconditioning. Proc Natl Acad Sci USA. 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochemical J. 2000;351:289–305. [PMC free article] [PubMed] [Google Scholar]
- 118.Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- 119.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 120.Guo Y, Stein AB, Wu WJ, Zhu X, Tan W, Li Q, et al. Late preconditioning induced by NO donors, adenosine A1 receptor agonists, and fdeltag1-opioid receptor agonists is mediated by iNOS. Am J Physiol Heart Circ Physiol. 2005;289:H2251–H2257. doi: 10.1152/ajpheart.00341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 122.Balligand JL, Kelly RA, Marsden PA, Smith TW, Michel T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl Acad Sci USA. 1993;90:347–351. doi: 10.1073/pnas.90.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Balligand JL, Kobzik L, Han X, Kaye DM, Belhassen L, O'Hara DS, et al. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. J Biol Chem. 1995;270:14582–14586. doi: 10.1074/jbc.270.24.14582. [DOI] [PubMed] [Google Scholar]
- 124.Hare JM, Keaney JF, Jr, Balligand JL, Loscalzo J, Smith TW, Colucci WS. Role of nitric oxide in parasympathetic modulation of beta-adrenergic myocardial contractility in normal dogs. J Clin Invest. 1995;95:360–366. doi: 10.1172/JCI117664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kelly RA, Balligand JL, Smith TW. Nitric oxide and cardiac function. Circ Res. 1996;79:363–380. doi: 10.1161/01.res.79.3.363. [DOI] [PubMed] [Google Scholar]
- 126.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells J Biol Chem. 1996;271:22810–22814. doi: 10.1074/jbc.271.37.22810. [DOI] [PubMed] [Google Scholar]
- 127.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 128.Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gyurko R, Kuhlencordt P, Fishman MC, Huang PL. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am J Physiol Heart Circ Physiol. 2000;278:H971–H981. doi: 10.1152/ajpheart.2000.278.3.H971. [DOI] [PubMed] [Google Scholar]
- 130.Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation. 2002;105:3011–3016. doi: 10.1161/01.cir.0000019516.31040.2d. [DOI] [PubMed] [Google Scholar]
- 131.Martin SR, Emanuel K, Sears CE, Zhang YH, Casadei B. Are myocardial eNOS and nNOS involved in the beta-adrenergic and muscarinic regulation of inotropy? A systematic investigation. Cardiovasc Res. 2006;70:97–106. doi: 10.1016/j.cardiores.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 132.Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation. 2005;112:3729–3737. doi: 10.1161/CIRCULATIONAHA.105.539437. [DOI] [PubMed] [Google Scholar]
- 133.Damy T, Ratajczak P, Shah P, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel J, Heymes C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363:1365–1367. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 134.Arking DE, Pfeufer A, Post W, Kao WHL, Newton-Cheh C, Ikeda M, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–651. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- 135.Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, et al. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 136.O'Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, et al. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- 137.Aguirre J, Buttery L, O'Shaughnessy M, Afzal F, Fernandez de Marticorena I, Hukkanen M, et al. Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am J Pathol. 2001;158:247–257. doi: 10.1016/S0002-9440(10)63963-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mashimo H, He XD, Huang PL, Fishman MC, Goyal RK. Neuronal constitutive nitric oxide synthase is involved in murine enteric inhibitory neurotransmission. J Clin Invest. 1996;98:8–13. doi: 10.1172/JCI118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang PL, Fishman MC. Genetic analysis of nitric oxide synthase isoforms: targeted mutation in mice. J Mol Med. 1996;74:415–421. doi: 10.1007/BF00217517. [DOI] [PubMed] [Google Scholar]