

Abstract
We examined the emission intensity and wavelength of 40 nm diameter silver particles covalently coated with organic fluorophores with different absorption and emission wavelengths. The objective of this study is to use the interactions of fluorophores with the plasmon in the metal particles to create the brightest possible probes. We refer to the complexes as plasmon-coupled fluorescence probes (PCPs). The fluorophores were separated from the metal cores by 10 nm long polymer backbones. The fluorescence was observed to be enhanced for seven fluorophores with emission wavelength from 450 to 700 nm. The enhancement efficiency was shown to approximately increase with long wavelengths for the silver particle-bound fluorophores. When comparing a single fluorophore free in solution and bound to the silver particle, the emission intensity increases 3- to 17-fold. The relationship between the enhancement efficiency and loading number of fluorophore on each silver particle was studied to optimize the conditions for PCP brightness. Compared with the free single fluorophores in the absence of metal, the optimized single labeled silver particles were even more than 1000-fold brighter, showing their potentials in the applications of sensitive clinical and biological assays.
Introduction
Fluorescence is a central enabling technology of the biosciences.1 While the fluorescence detection is highly sensitive, there is a continuous need for improving fluorophores with increased intensity and photostability. The need for such enhanced probes can be seen from single molecule detection (SMD).2–4 In these experiments, the fluorophores are typically immobilized on a surface to prevent their diffusion. Even with the immobilized fluorophores, it is necessary to use diffraction limited optics to detect the single molecule emission above the background due to the samples and optical components. These optical constants are the results of the limited brightness of a single molecule. This limitation can be circumvented by some extent using labeled polymer beads,5 phycobiliproteins,6 or quantum dots.7 In this paper, we describe an alternative approach to obtain bright particles using complexes of fluorophores with metal nanoparticles. For the reasons given below, we refer to these metal-associated fluorophores as plasmon-coupled fluorescence probes (PCPs).
Metal nanoparticles are known to display bright colors due to absorption and scattering of surface plasmons.8–12 Our development of PCPs is based on the interactions of fluorophores with the metal particles. Such metal particles are known to display plasmon resonances, which are a collective localized oscillation of electrons. Plasmon can be created by the illumination of a metal particle with light or by the interaction of fluorophores with the particle.13–16 These interactions can increase the emission intensity of fluorophores by factors of 10 to 100. This effect is typically called metal-enhanced fluorescence (MEF). When the fluorophores are bound on the metal nanoparticles, the metal particles labeled with a single fluorophore become brighter than the single free fluorophores in the absence of metal.17 Beside the increase of brightness as described above, the metal-associated fluorophores have been shown to display shortening of lifetime, increase of photostability, and reducing of photoblinking as compared with the free fluorophores.17,18
In recent publication, we have studied the effect of silver particles on nearby fluorophores. The studies include the effect of particle size, fluorophore-to-metal distance, and to a limited extent the effect of emission wavelength.19,20 In the current paper, we use the interactions of fluorophores with metals to develop the bright particles that can be used as probes for sensing, diagnosis, or cell imaging. Our approach is to covalently attach fluorophores to the surfaces of solid silver spheres. We examined a range of fluorophores, emission wavelengths, and the extent of surface labeling. We selected a single silver particle size of 40 nm, which we previously found to display the maximum fluorescence enhancement.17,21 In these particles, both excitation and emission are affected by the surface plasmon, and we refer to this class of probes as metal-associated fluorophores.
Experimentally, we synthesized the silver particles with an average core size of 40 nm, at a size which we previously show to provide optimal fluorescence enhancement.21b The silver particles were coated with N-(2-mercapto-propionyl)glycine (abbreviated as tiopronin).22,23 These metal particles are known to be water soluble and display good chemical stability. They were fluorescently labeled via three-step surface reactions: succinimidylation via ligand exchange, terminal-amination by diamine polymer via condensation, and labeling with succinimidyl fluorophore via condensation (Scheme 1). There were seven succinimidyl fluorophores with the excitation and emission wavelengths including Alexa Fluor 350 (AL350), fluorescein (FL), pacific blue (PB), Alexa Fluor 514 (AL514), Rhodamine 6G (Rh6G), Alexa Fluor 647 (AL647), and Alexa Fluor 680 (AL680).17 We examined the effect of wavelength on the metal particle brightness. The photophysical properties of these labeled silver particles were determined by ensemble fluorescence spectroscopy and single molecule spectroscopy. The enhancement efficiencies of various fluorophores bound on the metal particles were estimated by the ratios of emission intensities of labeled metal particles over the free fluorophores in the absence of metal at the same concentrations. Additionally, we also studied the influence of the average loading number of fluorophore/metal particles on the emission intensity. The experimental results of fluorophore-dependent MEF in this study are compared with the present understanding of fluorophore–metal interactions.
SCHEME 1.

Succinimidylated Silver Particles Were Covalently Bound by Diamine Polymer Molecules and Fluorescently Labeled by Succinimidyl Fluorophore Derivatives
Experimental Section
All reagents and spectroscopic grade solvents were used as received from Fisher or Aldrich. Succinimidylated fluorophore derivatives were purchase from Molecule Probe. RC dialysis membrane (MWCO 50,000) was obtained from Spectrum Laboratories, Inc. Nanopure water (>18.0 MΩ cm) purified using Millipore Milli-Q gradient system was used in all experiments. (2-mercapto-propionylamino) acetic acid 2,5-dioxo-pyrrolidin-1-ylester was synthesized as previous reported.23
Preparing Succinimidylated Tiopronin-Coated Metal Nanoparticles
Tiopronin-coated silver nanoparticles were prepared by chemical reduction of silver nitrate using ascorbic acid.21 To begin, 10 mg of silver nitrate and 30 mg of trisodium citrate were codissolved in 50 mL of water. A 20 μL sample of 0.1 N NaOH solution was added dropwise under stirring for 2 min. A 20 mg sample of ascorbic acid in 10 mL water was then added dropwise for 5 min, and the solution was stirred for an additional 1 h. Then 50 mg of tiopronin in 5 mL water was added at pH = 7.0, and the solution was stirred for 1 h. The solution was centrifuged at 8000 rpm to remove the suspension. After washing with water, the residue solids were redispersed in 50 mL of water.
These tiopronin-coated silver particles were succinimidylated by ligand exchange (Scheme 1).25,26 (2-mercapto-propionylamino) acetic acid 2,5-dioxo-pyrrolidin-1-ylester and tiopronin-coated silver particles were codissolved in water/methanol mixing solvent at a molar ratio of 1/2, and stirred for 24 h.21 The solution was removed by centrifugation at 5000 rpm at pH= 4. The residual was washed with ethanol and water, respectively, and then dispersed in 50 mL water at pH = 7.
Binding Long Linkers and Fluorophores on Metal Nanoparticles
The 10 nm long linkers were bound on the succinimidylated metal particles by a surface reaction.21b The succinimidylated metal particles were codispersed with an excess amount of poly bis(ethylene glycol) (3-aminopropyl) (MW 1500) in water at a molar ratio of 1/20. The solution was stirred for 2 h, and then ammonium was added dropwise to block the nonreacted terminal succinimidyl esters. The solution was removed by centrifugation at 5000 rpm at pH = 4. The residue was washed with water and then dispersed in water at pH = 7.
The fluorophores were chemically bound on the metal particles by condensation between the terminal amino moieties on the metal particles and the succinimidyl ester moieties on the fluorophore compounds. The fluorophores included Alexa Fluor 350 (AL350), fluorescein (FL), pacific blue (PB), Alexa Fluor 514 (AL514), Rhodamine 6G (Rh6G), Alexa Fluor 647 (AL647), and Alexa Fluor 680 (AL680). The reactions were performed in water for 24 h. The loading numbers were controlled by adjusting the molar ratio of metal particle/fluorophore from 1/20 to 1/800 as listed in Table 1. The suspension was removed by centrifugation at 5000 rpm. The residual particles were washed with water and then dialyzed against water (MWCO 50 000) to remove any unbound impurities in solution for the spectral measurements.
TABLE 1.
Summary of Labeling Conditions, Loading Number of Fluorophores/Per Metal Particle, And Average Distance between Rh6G Molecules on the Surface of the Metal Particlea
| molar ratio of fluorophore/metal particle in the reaction | 20 | 40 | 60 | 100 | 200 | 400 | 800 |
|---|---|---|---|---|---|---|---|
| loading number of fluorophore/per particle | 10 | 18 | 32 | 75 | 98 | 150 | 200 |
| average distance between Rh6G molecules on the metal particle nm) | 25.3 | 18.9 | 14.1 | 9.2 | 8.1 | 6.5 | 5.7 |
The concentration of metal particle was 0.5 nM in solution.
Spectra, Images, and TEM Measurements
Absorption spectra were monitored with a Hewlett-Packard 8453 spectrophotometer. Ensemble fluorescence spectra were recorded in solution with a Cary Eclipse fluorescence spectrophotometer. Lifetimes were measured by PicoQuant Modular fluorescence lifetime spectrometer (Fluo Time 100) using different color LED laser sources with a resolution of 34 ps. A corresponding filter was used to isolate the emission. The collected data were analyzed using PicoQuant Fluofit 3.3 software.
For the fluorescence imaging measurements, the glass coverslips were first soaked in a 10:1 (v:v) mixture of concentrated H2SO4 and 30% H2O2 overnight, extensively rinsed with water, sonicated in absolute ethanol for 2 min, and dried with air stream. Diluted solutions of the labeled metal particles were dispersed on the precleaned coverslips. Single fluorophore or metal particle measurements were performed using a time-resolved confocal microscopy (MicroTime 200, PicoQuant). Briefly, it consists of an inverted microscope coupled to high sensitivity single particle detection. A single-mode pulsed laser diode was used as the excitation source. An oil immersion objective (Olympus, 100×, 1.3 NA) was employed both for focusing laser light onto the samples and for collecting fluorescence emission from the samples. The fluorescence that passed a dichroic mirror was focused onto a 75 μm pinhole for spatial filtering to reject out-of-focus signals. The integration time was 0.6 ms per pixel. The data was stored in the time-tagged-time-resolved (TTTR) mode that allowed recording every photon with its individual timing information with regard to both the location or sample and the time delay between the excitation and the emission
Transmission electron micrographs (TEM) were taken with a side-entry Philips electron microscope at 120 keV. Samples were cast from water solutions onto standard carbon-coated (200–300 Å) Formvar films on copper grids (200 mesh) by placing a droplet of a 1 mg/mL aqueous sample solution on grids. The size distribution of metal core was analyzed with Scion Image Beta Release 2 counting at least 200 particles.
Results and Discussion
The silver particles were first synthesized with the citrate ions on the surfaces. The citrate ions were completely replaced by the tiopronin ligands to form more chemically stable sulfur–metal bonds. These tiopronin-coated silver particles were found to display good solubility in water at neutral and weak basic conditions. Representative TEM images showed individual metal spheres with an average diameter of 40 nm (inset of Figure 1). The size distributions of metal particles were analyzed with Scion Image Beta Release 2 counting at least 200 images, showing heterogeneous distribution of metal core size but with most in ranges of 40 ± 20 nm. Absorbance measurement showed a typical silver plasmon wavelength at 427 nm (Figure 1),27,28 but the band was broadened, consistent with the heterogeneous distribution of metal cores. The composition of the metal particles was estimated to be approximately (Ag)2×106(Tio)2×104 according to the average size of 40 nm for these metal particles.
Figure 1.

(a) Absorbance spectra of initial tiopronin-coated silver particles and aminated metal particles with 40 nm metal cores. Their transmission electron micrographs (TEM) images are given in the insets a and b, respectively. (b) Histogram of size distribution for the tiopronin-coated silver particles achieved from TEM images. At least 200 metal particles were counted from the TEM images.
As described in Experimental Section, the metal particles were fluorescently labeled via three-step surface reactions. They were first succinimidylated by (2-mercapto-propionylamino) acetic acid 2,5-dioxo-pyrrolidin-1-ylester via ligand exchange.21 This reaction usually occurs at a molar ratio of 1:1 on the metal particles.25,26 Since our goal was to obtain bright particles, we labeled the metal cores with multiple fluorophores so that more than one of the succinimidyl ligands was displaced on each metal particle. In the experiment, the succinimidylated thiolate compound and metal particle were codissolved in the reaction solution at the molar ratio of 1000/1. The loaded number of succinimidyl ligand on each metal particle was estimated to be about 200 as discussed later.
In order to increase the distance from the bound fluorophore to the surface of metal core, an additional PEG linker was first bound on the metal particle. A diamine PEG derivative was used to react with the succinimidyl moieties on the metal particles. In the reaction, the diamine derivative was in an excess amount to avoid the aggregation of metal particles. The bound PEG backbone is about 10 nm long, which is regarded as an optimal distance for efficient MEF.24 The reaction brought the metal particles terminated with the amino moieties on the surfaces, so they were fluorescently labeled via condensation with succinimidyl fluorophores. Seven kinds of fluorophore were used including Alexa Fluor 350 (AL350), fluorescein (FL), pacific blue (PB), Alexa Fluor 514 (AL514), Rhodamine 6G (Rh6G), Alexa Fluor 647 (AL647), and Alexa Fluor 680 (AL680), which displayed a progressive shift to longer emission wavelengths. The loading numbers of fluorophores were controlled by the molar ratios of fluorophore compound/metal particle in reaction solution in a range of 20–800 (Table 1). By removing the silver cores of particles using NaCN solution as described later, the concentrations of released fluorophores in solution could be measured accurately by the emission intensities. To compare with the concentrations of metal particles in solution, the real loading numbers of fluorophore per particle were estimated as shown in a range of 10–200 (Table 1). A further increase of the fluorophore compound in reaction solution could not increase the loading number, indicating that the metal particles were maximally labeled by roughly 200 fluorophores. On the other hand, we believe that there were about 200 succinimidyl ligands to display on one metal particle in ligand exchange reaction.
The labeling durations of fluorophore onto the metal particles were monitored by the emission spectral changes. A typical example of Rh6G was given in Figure 2a, on which the emission intensity increased with the reaction time until saturation in 1.5 h. With the increase of emission intensity, the emission wavelength was observed to slightly red shift from 560 to 563 nm for the Rh6G-labeled silver particles.29 These surface reactions on the metal particles also led to a slight red-shift of plasmon resonance absorbance from 427 to 432 nm. This small shift value is not due to the aggregation of metal particles.30 TEM images also showed insignificant changes on the shapes and sizes of metal particles after the surface reactions.
Figure 2.

Ensemble emission spectral changes with the reaction time when (a) loading Rh6G on 40 nm silver particles and (b) removing metal cores by NaCN from the metal associated fluorophores.
We compare the wavelength-dependent emission properties of the labeled metal particles in this study. The ensemble emission measurements were performed at the concentration of 0.5 nM of the labeled metal particles (extinct coefficient = 4 × 109 M−1 cm−1 at 427 nm) in solution. Although the maximal loading number of fluorophore could reach to 200 on each metal particle, the loading number here was controlled to be as low as 10 for every fluorophore because a dense loading was expected to bring up a significant self-quenching. It was achieved by adding the succinimidyl fluorophore of 1.0 nM to the metal particle solution (Table 1). The labeled metal particles were excited at their corresponding maximal absorbance wavelengths, and the emission spectra were collected (Figure 3). It was shown that all of these labeled metal particles displayed analogous spectral changes to time-dependent Rh6G: increases of emission intensity. In order to determine the effect of the metal particles on the emission intensities, we removed the metal cores of the labeled metal particle by adding a couple drops of 0.1 N NaCN into solution.31 Consequently, the metal cores were dissolved, which were verified by disappearing of the metal plasmon resonance. So the bound fluorophores were released into solution. The emission intensity of Rh6G-labeled silver particles was observed to decrease with the dissolution of metal core by the NaCN treatments (Figure 2b). We observed the similar effect with the other labeled metal particles that were decreases in intensities upon removing the metal cores with NaCN. In all cases, it appeared that the released fluorophores displayed identical emission spectra to the free fluorophores but with decreased emission intensities. Since the bulk concentration of the fluorophores remained constant when the silver was dissolved, the enhancement efficiency could be determined from the ratio of intensities in the presence and absence of metal silver. We found that the apparent enhancement efficiency displayed an approximate increase with longer emission wavelengths (Figure 4). Although a significant downward derivation from a line is observed, nonetheless, the metal particle leads to a substantial fluorescence enhancement with the longer shift of emission wavelength, indicating that the metal-associated fluorophores can be synthesized in a wide range of colors. More specially, the enhanced fluorescence can occur at wavelengths where the metal particles do not display a strong plasmon resonance.
Figure 3.
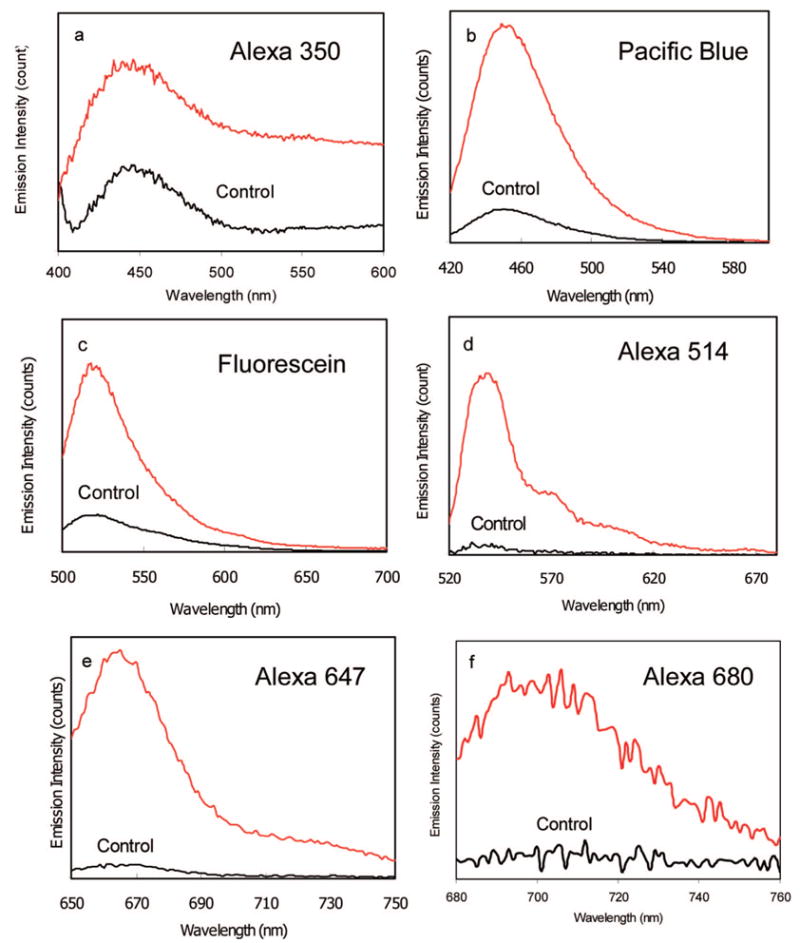
Emission spectra of different fluorophores bound on the 40 nm silver particles with the loading number of near 10 per particle and their controls that were measured after removing the metal cores with NaCN in solution. The fluorophores include (a) Alexa Fluor 350, (b) fluorescein, (c) pacific blue, (d), Alexa Fluor 514, (e) Alexa Fluor 647, and (f) Alexa Fluor 680.
Figure 4.
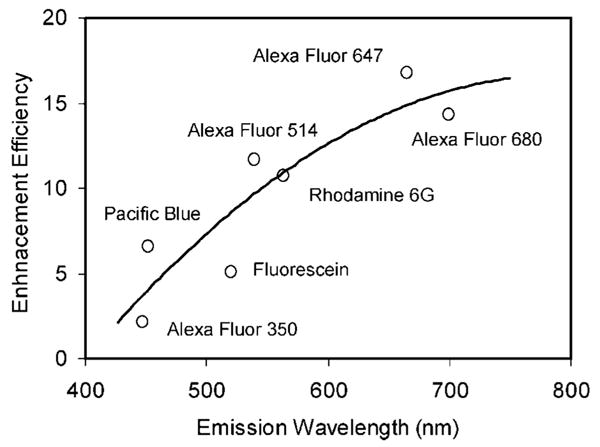
Dependence of the enhancement efficiency on the emission wavelength of fluorophores bound on the metal particles.
At first glance, an increase in enhancement efficiency with increasing the wavelength is surprising because, at this time, fluorescence enhancement is thought to be dependent upon an overlap of the emission spectrum with the metal plasmon resonance.32,33 However, after many experiments on fluorophores near metal particles, we observed enhancement for all fluorophores independent of emission wavelengths. We believe that these observations are the results of two or more effects. First, prepared particles are somehow heterogeneous in size and shape, and the surfaces may be rough rather than smooth. As a result, the plasmon resonances are often broad and extended to long wavelengths. Second, the observation of no plasmon extinction at long wavelength with small particle does not mean that a nearby fluorophore cannot interact with the particle. The absence of a resonance indicates that far-field radiation does not interact with the particle. However, a nearby fluorophore at the same optical frequency (wavelength) can interact with the particle because of the large wavevector components close to the fluorophore. We believe the metal particles can enhance fluorescence, even if there is no plasmon resonance at the emission wavelength of fluorophore, because of these near-field interactions.
As described in Experimental Section, the fluorescence images were collected using scanning confocal microscopy. Figure 5a,b depicts typical fluorescence images of free Rh6G and Rh6G-labeled metal particles with loading number of 10. Figure 5c presents the same image of Figure 5a (free Rh6G) with a lower intensity scale. We found that the emission spots with the labeled metal particles (Figure 5b) were at least 30-fold brighter than those by the free fluorophores in the absence of metal (Figure 5a).
Figure 5.

Fluorescence images of (a) free Rhodamine 6G and (b) 10 Rh6G molecules bound to each silver particle at the scale bar of 600. Image c is for free Rhodamine 6G taken from image a by lowering the brightness scale to 50. Time-traces of free and bound Rh6G on the metal particles is shown in d. The 10 × 10 μm images are 100 × 100 pixels, with in integration time of 0.6 ms per pixel.
It is interesting to conclude the mechanism of emission wavelength-dependent MEF. Generally, a fluorophore can be considered as a dipole to be in the proximity to and interact with a metal particle.34 It appears that MEF is due to at least two effects: an increase in the electric field near the metal particle due to incident light and an increase in the radiative decay rate of the fluorophore due to the metal particle. The first factor provides a stronger excitation rate and the second factor changes the quantum yield and lifetime of fluorophore. We thus can write the apparent fluorescence enhancement factor (Gapp) as the product of two factors: excitation (Gex) and quantum yield (GQY),34a
| (1) |
Gex is a near-field excitation rate of fluorophore at the excitation wavelength and GQY is a factor about the quantum yield of far-field emission. We first tried to interpret the experiment results using the excitation factor on the local electric fields calculated using FDTD simulations.17 However, the calculated average density of local electric field near the metal particles significantly decreased at longer wavelengths, contrary to the experiment results. This suggests that the increase in fluorescence intensity is due in large part to metal-enhanced emission from the excited fluorophore. This mechanism for MEF is closely connected with our suggestion that a excited-state fluorophore with a given optical energy can interact with a metal particle even if a far-field interaction at the same wavelength does not interact with the metal particle.
An increase in the rate of excitation is not expected to change the decay time of a fluorophore. In contrast, an increase in the radiative decay rate of a fluorophore will decrease the lifetime because of an increase in the rate of radiative decay. Hence, the intensity decay provides information on the mechanism of enhancement. For a free-space emission of an excited fluorophore, the quantum yield (Qo) and lifetime (τo) can be expressed,1
| (2) |
| (3) |
where Γ is a radiative decay rate and knr is a nonradiative decay rate of the fluorophore. For a fluorophore localized near a metal particle, both the radiative and the nonradiative decay rates are altered and given by Γ + Γm for the radiative rate and knr + km for the nonradiative rate, in which Γm and km are the additional radiative and nonradiative rate due to the metal substrate. The quantum yield Qm and lifetime τm of the fluorophore near the metal substrate thus become34
| (4) |
| (5) |
These equations show that an increase in the radiative decay rate will increase the quantum yield and decrease the lifetime of a fluorophore. The right-hand sides of eqs 4 and 5 reflect the common assumption that the increase in the radiative decay rate is proportional to the free-space decay rate.
It is known that the metal-induced decay rate depends on the properties of the metal particle, fluorophore, and surrounding medium including the metal particle size and shape, the fluorophore emission wavelength and orientation as well as the distance from the metal core, and the properties of surrounding medium like refractive index near the fluorophore. Polman, et al.32,35 calculated and evaluated the effects of these factors using the electromagnetic theory and the Gersten–Natzan model. Their calculations on the wavelength-dependent MEF of silver particles are not consistent with our current experimental results. Since the used silver particles display the plasmon resonance at 430 nm, according to Polman’s calculation, the maximum enhancement is expected to occur at about 500 nm. For an ideal metal sphere, the maximal MEF is expected to occur at the wavelength with strong overlap of the plasmon resonance wavelength of metal particle and the emission wavelength of emitter. This prediction has been observed in the experimental results from other laboratories.33,36–38 However, our experiment in this case reveals that the maximal MEF occurs at about 650 nm. But we note that in all of these reports, the experiments were done by shifting the wavelength of plasmon resonance but fixing the emission wavelength instead of shifting the emission wavelength of emitter as we did in this case.
Although the enhancement efficiencies for the pairs of Pacific Blue/Fluorescein, Alexa Fluor 514/Rh6G, and Alexa Fluor 647/Alexa Fluor 680 were contrarily changed, the overall result is a clear tendency of increase with an increase of emission wavelength when binding the fluorophore on the metal particles. The data were confirmed by the ensemble spectra and were reproducible. The differences in the enhancement efficiency for the above-mentioned fluorophore pairs may be the results of their chemical structures that cause their different quantum yields in the space-free states. The metal particles are expected to cause the different enhancement effects of the fluorophores with the different quantum yields. Herein, we believe that the different results from experiment and theory are due to different effects of far- and near-field on the metal particles as well as the chemical structures of the fluorophores. Additionally, we noted that the theory for MEF usually assumes a perfect sphere. It is also likely that the metal particle surface roughness can affect the extents of enhancements.
To further investigate the mechanism of MEF, we measured the lifetime change due to binding of fluorophores to the metal particles.34 The emission intensity decays were collected by a single photon counting spectrometer, and the data were analyzed in terms of single decays for the free fluorophores in the absence of metal and double exponential decays for the fluorophores bound on the metal particles as39
| (6) |
In this expression, τi are the decay times and αi are the amplitudes. The average amplitude-weighted lifetime is represented by
| (7) |
Figure 6 shows representation intensity decay of free Rh6G and that bound to silver particle in solution. The measured lifetimes are listed in Table 2 for free and metal-bound fluorophores. We found that a small fraction of the total emission displayed a long lifetime, close to that of free fluorophores.40 The majority of the intensity is due to the short component with a lifetime shorter than 1 ns. Overall, we found that the metal particles cause a four- to six-fold decrease in the lifetime. We cannot make a perfect correlation between the lifetime and the enhancement efficiency because the lifetime can be shortened by nonradiative effects. Nonetheless, the decrease lifetime and associated increase intensity demonstrate that the metal particles cause an increase in the radiative decay rate of the fluorophore.
Figure 6.
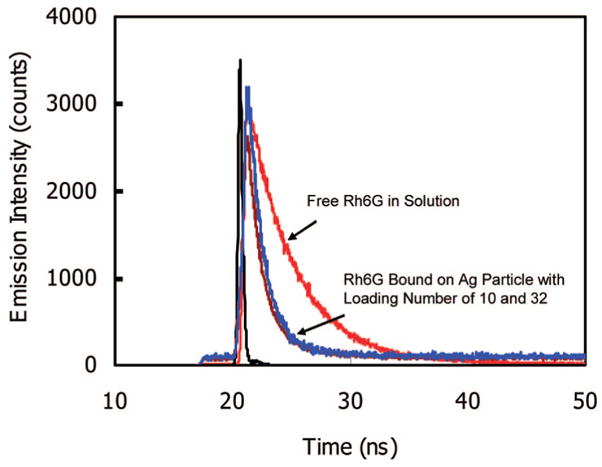
Emission decay curves for the free Rhodamine 6G and bound Rhodamine 6G with loading number of 10 and 32 upon excitation at 470 nm.
TABLE 2.
Lifetime Data for Free Fluorophores (10 nM) and Bound Fluorophores on Metal Particles (0.5 nM) in Water at pH = 7a
| Samples | τi (ns) | αi | 〈τ〉(ns) | χR2 |
|---|---|---|---|---|
| Alexa Fluor Fluor 350 in water | 2.38 | 2.38 | 1.0 | |
| Alexa Fluor Fluor 350 on 40 nm silver particle | 2.88 | 0.13 | 0.69 | 1.3 |
| 0.38 | 0.87 | |||
| Pacific Blue in water | 2.37 | 2.37 | 1.0 | |
| Pacific Blue on 40 nm silver particle | 2.81 | 0.01 | 0.29 | 1.2 |
| 0.25 | 0.99 | |||
| Fluorescein in water | 3.11 | 3.11 | 1.1 | |
| Fluorescein on 40 nm silver particle | 4.09 | 0.09 | 0.73 | 1.2 |
| 0.39 | 0.91 | |||
| Alexa Fluor Fluor 514 in water | 3.89 | 3.89 | 1.3 | |
| Alexa Fluor Fluor 514 on 40 nm silver particle | 3.94 | 0.08 | 0.93 | 1.0 |
| 0.67 | 0.92 | |||
| Rhodamine 6G in water | 4.01 | 4.01 | 1.1 | |
| Rhodamine 6G on 40 nm silver particle | 4.39 | 0.06 | 0.86 | 1.3 |
| 0.64 | 0.94 | |||
| Alexa Fluor Fluor 647 in water | 1.24 | 1.24 | 1.1 | |
| Alexa Fluor Fluor 647 on 40 nm silver particle | 1.93 | 0.05 | 0.42 | 1.3 |
| 0.34 | 0.95 | |||
| Alexa Fluor Fluor 680 in water | 1.23 | 1.23 | 1.1 | |
| Alexa Fluor Fluor 680 on 40 nm silver particle | 2.22 | 0.12 | 0.57 | 0.9 |
| 0.35 | 0.88 |
The loading numbers were close to 20 for all samples.
Reductions in the fluorophore lifetimes can show favorable effects on the spectral properties of fluorophores. The photostability is expected to increase because a shorter lifetime allows less time for photobleaching in the excited state.17 Additionally, a shorter lifetime allows a higher emission rate at optical saturation. The presence of multiple fluorophores on one metal particle can also increases the time available for observation. Time-dependent intensity traces were used to compare the photostability (Figure 5d), showing a typical one-step photo-bleaching for a free fluorophore but more gradual decrease in intensity for multiple fluorophores bound to a metal particle. The emission trace from a labeled metal particle is shown to continue for more than 60 s, but that from a free fluorophore is bleached in several seconds indicating that the labeled metal particles become much more photostable.
In order to achieve the brightest PCPs in this system, we study the emission intensity by varying the number of fluorophore/metal particles. A metal particle with a small number of fluorophores is observed to be less bright than the one with a large number of fluorophores. But a too dense loading with the fluorophores can cause a self-quenching of the fluorophores. Here, we take an example of Rh6G bound on the silver particle to optimize the brightness. As described above, the loading number was approximately controlled by the molar ratio of metal particles over succinimidyl fluorophore in the reaction solution and was accurately detected after removing the metal using NaCN treatments as shown in Table 1. The ensemble spectral measurements presented a significant increase in the intensity at 563 nm with an increase of the loading number (Figure 7). However, the increase in the intensity was sublinear with the loading density, as shown by an obvious downward curvature (inset of Figure 7). The enhancement efficiency estimated from the intensity at 563 nm showed a significant decrease with the loading number. A similar phenomenon was also observed in our previous study when incorporating Rh6G into the silica beads and then coating them with the silver thin shells to construct the silica core/silver shell structures.21 The average distance between the Rh6G molecules is calculated on the surfaces of metal particles (Table 1), showing that the maximal loading of fluorophores (number = 200) corresponds to a distance of 5.7 nm, which is short enough for significant self-quenching. However, the minimum loading (number = 10) corresponds to a distance of 25.3 nm, which is too long for the occurrence of self-quenching. We note that the lifetime is not significantly altered with the change of loading number (examples of loading number = 10 and 32 in Figure 6), indicating that the interactions between the fluorophores and the metal cores still play more important roles than the intensities between the fluorophores. Although the enhancement efficiency for the bound fluorophores on the metal particles was reduced with an increase of the loading number, the brightness of the labeled metal particles was observed to continuously increase (inset of Figure 7), indicating that the labeled metal particles can become brighter by the additional effect of multiple fluorophore on the same metal particle.
Figure 7.

Emission spectral changes with the loading number of Rhodamine 6G on the 40 nm silver particles upon excitation at 500 nm. The inset represents the dependence of emission intensity at 563 nm on loading number of fluorophore per metal particle and the enhancement efficiency calculated from the emission intensity at 563 nm.
Summary
In this paper, we discussed the synthesis and spectral properties of silver particles which contained multiple fluorophores bound to the surfaces. It is of interest to consider the advantages of these fluorophore–metal complexes for use as fluorescence probes.
The absorbance and emission wavelengths can be controlled by the choices of fluorophore, not particle size, as is the case with quantum dots.
A single size particle can be used to create different wavelength probes. This is possible because one particle size was found to enhance the fluorescence of fluorophore with the emission wavelengths ranging from 440 to 680 nm.
The brightness of the labeled metal particle can be controlled by the number of surface-bound fluorophores.
The surface chemistry of silver is well-known. It is really possible to alternate protein, DNA, or surface-coating for the clinical application, and different type of molecules can be bound to the same particle.
Silver has low toxicity to cells and organisms.
The presence of multiple fluorophores eliminates the problem due to blinking and will minimize the problem due to photostability. Even if a single fluorophore on the silver particle displays blinking, this effect is not seen with multiple fluorophores per particle. Also, the particle will remain visible even after some fraction of the fluorophores is photobleached.
The advantages for PCPs listed above are described in this report. However, we regard the present PCPs as only the first stage of structures. For example, we can imagine the use of enlarged metal particles that are expected provides larger fluorescence enhancements. Additionally, a rod-like particle can display two plasmon resonances because of the longitudinal and transverse model. It may be possible to couple on the model to the excitation wavelength and on the other model to the emission wavelength, thereby enhancing the processes of both excitation and emission. There is another chance PCPs will be based on the encapsulation of fluorophores with metal shells. We have already observed enhanced emission and more narrow emission spectra for fluorophores in the metal shells.21 The uses of the metal shells will keep the fluorophores from direct contact with diffusion into the samples. It is also possible that these shells protect the fluorophores from photobleaching by restricting the access to oxygen.
In summary, we believe the plasmon-coupled fluorescence probes provide a new choice of a bright and photostable probe for use in analytical and cell biology applications.
Acknowledgments
This research was supported by a grant from NIH (HG-002655, EB-000682, and EB0 065211) and NCRR (RR-08119).
References and Notes
- 1.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer; New York: 2006. [Google Scholar]
- 2.Ambrose WP, Goodwin PM, Jett JH, Van Orden A, Werner JH, Keller RA. Chem Rev. 1999;99:2929. doi: 10.1021/cr980132z. [DOI] [PubMed] [Google Scholar]
- 3.Willets KA, Nishimura SY, Schuck PJ, Twieg RJ, Moerner WE. Acc Chem Res. 2005;38:549. doi: 10.1021/ar0401294. [DOI] [PubMed] [Google Scholar]
- 4.Michalet X, Weiss S, Jager M. Chem Rev. 2006;106:1785. doi: 10.1021/cr0404343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Smith JE, Medley CD, Tang Z, Shangguan D, Lofton C, Tan W. Anal Chem. 2007;79:3075. doi: 10.1021/ac062151b. [DOI] [PubMed] [Google Scholar]; (b) He GS, Tan LS, Zheng Q, Prasad PN. Chem Rev. 2008 doi: 10.1021/cr050054x. ASAP Article. [DOI] [PubMed] [Google Scholar]
- 6.(a) Zhao KH, Ran Y, Li M, Sun YN, Zhou M, Storf M, Kupka M, Bohm S, Bubenzer C, Scheer H. Biochemistry. 2004;43:11576. doi: 10.1021/bi0491548. [DOI] [PubMed] [Google Scholar]; (b) He JA, Hu YZ, Jiang LJ. J Am Chem Soc. 1996;118:8957. [Google Scholar]
- 7.(a) Empedocles S, Bawendi M. Acc Chem Res. 1999;32:389. [Google Scholar]; (b) Penner RM. Acc Chem Res. 2000;33:78. doi: 10.1021/ar9702839. [DOI] [PubMed] [Google Scholar]
- 8.(a) Gryczynski I, Malicka J, Shen YB, Gryczynski Z, Lakowicz JR. J Phys Chem B. 2002;106:2191. doi: 10.1021/jp013013n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aslan K, Huang J, Wilson GM, Geddes CD. J Am Chem Soc. 2006;128:4206. doi: 10.1021/ja0601179. [DOI] [PubMed] [Google Scholar]; (c) Ray K, Badugu R, Lakowicz JR. J Phys Chem C. 2007;111:7091. doi: 10.1021/jp067635q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Sokolov K, Chumanov G, Cotton TM. Anal Chem. 1998;70:3898. doi: 10.1021/ac9712310. [DOI] [PubMed] [Google Scholar]; (b) Shen Y, Swiatkiewicz J, Lin TC, Markowicz P, Prasad PN. J Phys Chem B. 2002;106:4040. [Google Scholar]; (c) Lee IYS, Suzuki H, Ito K, Yasuda Y. J Phys Chem B. 2004;108:19368. [Google Scholar]; (d) Yonzon CR, Jeoung E, Zou S, Schatz GC, Mrksich M, Van Duyne RP. J Am Chem Soc. 2004;126:12669. doi: 10.1021/ja047118q. [DOI] [PubMed] [Google Scholar]
- 10.(a) Song JH, Atay T, Shi S, Urabe H, Nurmikko AV. Nano Lett. 2005;5:1557. doi: 10.1021/nl050813r. [DOI] [PubMed] [Google Scholar]; (b) Brolo AG, Kwok SC, Moffitt MG, Gordon R, Riordon J, Kavanagh KL. J Am Chem Soc. 2005;127:14936. doi: 10.1021/ja0548687. [DOI] [PubMed] [Google Scholar]
- 11.(a) Kawasaki M, Mine S. J Phys Chem B. 2005;109:17254. doi: 10.1021/jp053167t. [DOI] [PubMed] [Google Scholar]; (b) Yu F, Persson B, Lofas S, Knoll W. J Am Chem Soc. 2004;126:8902. doi: 10.1021/ja048583q. [DOI] [PubMed] [Google Scholar]; (c) Ekgasit S, Thammacharoen C, Yu F, Knoll W. Anal Chem. 2004;76:2210. doi: 10.1021/ac035326f. [DOI] [PubMed] [Google Scholar]; (d) Baluschev S, Yu F, Miteva T, Ahl S, Yasuda A, Nelles G, Knoll W, Wegner G. Nano Lett. 2005;5:2482. doi: 10.1021/nl0517969. [DOI] [PubMed] [Google Scholar]
- 12.(a) Futamata M, Maruyama Y, Ishikawa M. J Phys Chem B. 2003;107:7607. [Google Scholar]; (b) Sherry LJ, Chang SH, Schatz GC, Van Duyne RP, Wiley BJ, Xia Y. Nano Lett. 2005;5:2034. doi: 10.1021/nl0515753. [DOI] [PubMed] [Google Scholar]; (c) Oubre C, Nordlander P. J Phys Chem B. 2005;109:10042. doi: 10.1021/jp044382x. [DOI] [PubMed] [Google Scholar]; (d) Wang H, Goodrich GP, Tam F, Oubre C, Nordlander P, Halas NJ. J Phys Chem B. 2005;109:11083. doi: 10.1021/jp051466c. [DOI] [PubMed] [Google Scholar]
- 13.(a) Mertens H, Biteen JS, Atwater HA, Polman A. Nano Lett. 2006;6:2622. doi: 10.1021/nl061494m. [DOI] [PubMed] [Google Scholar]; (b) Hernández FE, Yu S, García M, Campiglia AD. J Phys Chem B. 2005;109:9499. doi: 10.1021/jp050803e. [DOI] [PubMed] [Google Scholar]
- 14.(a) He L, Smith EA, Natan MJ, Keating CD. J Phys Chem B. 2004;108:10973. [Google Scholar]; (b) Stoermer RL, Keating CD. J Am Chem Soc. 2006;128:13243. doi: 10.1021/ja0637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Duan C, Cui H, Zhang Z, Liu B, Guo J, Wang W. J Phys Chem C. 2007;111:4561. [Google Scholar]; (b) Tam F, Goodrich GP, Johnson BR, Halas NJ. Nano Lett. 2007;7:496. doi: 10.1021/nl062901x. [DOI] [PubMed] [Google Scholar]; (c) Ianoul A, Bergeron A. Langmuir. 2006;22:10217. doi: 10.1021/la061894p. [DOI] [PubMed] [Google Scholar]
- 16.(a) Kummerlen J, Leitner A, Brunner H, Aussenegg FR, Wokaun A. Mol Phys. 1993;80:1031. [Google Scholar]; (b) Antunes PA, Constantino CJL, Aroca RF, Duff J. Langmuir. 2001;17:2958. [Google Scholar]; (c) Kamat PV. J Phys Chem B. 2002;106:7729. [Google Scholar]; (d) Pan S, Wang Z, Rothberg LJ. J Phys Chem B. 2006:110–17383. doi: 10.1021/jp063191m. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Fu Y, Chowdhury MH, Lakowicz JR. J Phys Chem B. 2008;112:18. doi: 10.1021/jp074938r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Fu Y, Zhang J, Lakowicz JR. Langmuir. 2008;24:3429. doi: 10.1021/la702673p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Parfenov A, Gryczynski I, Malicka J, Geddes CD, Lakowicz JR. J Phys Chem B. 2003;107:8829. doi: 10.1021/jp022660r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Kumbhar AS, Kinnan MK, Chumanov G. J Am Chem Soc. 2005;127:12444. doi: 10.1021/ja053242d. [DOI] [PubMed] [Google Scholar]; (b) Bruzzone S, Malvaldi M, Arrighini GP, Guidotti C. J Phys Chem B. 2005;109:3807. doi: 10.1021/jp045451a. [DOI] [PubMed] [Google Scholar]; (c) Hubert C, Rumyantseva A, Lerondel G, Grand J, Kostcheev S, Billot L, Vial A, Bachelot R, Royer P, Chang S-h, Gray SK, Wiederrecht GP, Schatz GC. Nano Lett. 2005;5:615. doi: 10.1021/nl047956i. [DOI] [PubMed] [Google Scholar]; (d) Millstone JE, Park S, Shuford KL, Qin L, Schatz GC, Mirkin CA. J Am Chem Soc. 2005;127:5312. doi: 10.1021/ja043245a. [DOI] [PubMed] [Google Scholar]
- 20.(a) Chen Y, Munechika K, Ginger DS. Nano Lett. 2007;7:690. doi: 10.1021/nl062795z. [DOI] [PubMed] [Google Scholar]; (b) Kelly KL, Coronado E, Zhao LL, Schatz GC. J Phys Chem B. 2003;107:668. [Google Scholar]; (c) Hao E, Li S, Bailey RC, Zou S, Schatz GC, Hupp JT. J Phys Chem B. 2004;108:1224. [Google Scholar]; (d) Dragnea B, Szarko JM, Kowarik S, Weimann T, Feldmann J, Leone SR. Nano Lett. 2003;3:3. [Google Scholar]
- 21.(a) Zhang J, Fu Y, Lakowicz JR. J Phys Chem C. 2007;111:50. doi: 10.1021/jp062665e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang J, Fu Y, Chowdhury MH, Lakowicz JR. J Phys Chem C. 2007;111:11784. doi: 10.1021/jp067887r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Huang T, Murray RW. Langmuir. 2002;18:7077. [Google Scholar]; (b) Huang T, Murray RW. J Phys Chem B. 2001;105:12498. [Google Scholar]
- 23.(a) Zhang J, Malicka J, Gryczynski I, Lakowicz JR. J Phys Chem B. 2005;109:7643. doi: 10.1021/jp0490103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang J, Roll D, Geddes CD, Lakowicz JR. J Phys Chem B. 2004;108:12210. doi: 10.1021/jp037772c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Matveeva E, Gryczynski I, Leonenko Z, Lakowicz JR. J Phys Chem B. 2005;109:7669. doi: 10.1021/jp0456842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Templeton AC, Wuelfing WP, Murray RW. Acc Chem Res. 2000;33:27. doi: 10.1021/ar9602664. [DOI] [PubMed] [Google Scholar]
- 26.Ingram RS, Hostetler MJ, Murray RW. J Am Chem Soc. 1997;119:9175. [Google Scholar]
- 27.Hayat MA. Colloidal Gold: Principles, Methods, and Applications. Academic Press; San Diego: 1991. [Google Scholar]
- 28.Feldheim DL, Foss CA. Synthesis, Characterization and Applications. Marcel Dekker Inc; New York: 2002. Metal Nanoparticles. [Google Scholar]
- 29.Hu J, Zhang J, Liu F, Kittredge K, Whitesell JK, Fox MA. J Am Chem Soc. 2001;123:1464. [Google Scholar]
- 30.(a) Esumi K, Matsushima Y, Torigoe K. Langmuir. 1995;11:3285. [Google Scholar]; (b) Link S, Mohamed MB, El-Sayed MA. J Phys Chem B. 1999;103:3073. [Google Scholar]
- 31.Rosi NL, Mirkin CA. Chem Rev. 2005;105:1547. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 32.Penninkhof JJ, Moroz A, van Blaaderen A, Polman A. J Phys Chem C. 2008;112:4146. [Google Scholar]
- 33.Tam F, Goodrich GP, Johnson BR, Halas NJ. Nano Lett. 2007;7:496. doi: 10.1021/nl062901x. [DOI] [PubMed] [Google Scholar]
- 34.(a) Lakowicz JR. Anal Biochem. 2001;298:1. doi: 10.1006/abio.2001.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lakowicz JR. Anal Biochem. 2005;337:171. doi: 10.1016/j.ab.2004.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Koenderink AF, Hernandez JV, Robicheaux F, Noordam LD, Polman Nano Lett. 2007;7:745. doi: 10.1021/nl0630034. [DOI] [PubMed] [Google Scholar]; (b) Mertens H. PhD Thesis. Utrecht University; Apr, 2007. Controlling plasmon-enhanced luminescence. [Google Scholar]; (c) Mertens H, Koenderink AF, Polman A. Phys Rev B. 2007;76:115123. [Google Scholar]
- 36.Chen Y, Munechika K, Ginger DS. Nano Lett. 2007;7:690. doi: 10.1021/nl062795z. [DOI] [PubMed] [Google Scholar]
- 37.(a) Nakamura T, Hayashi S. Jpn J Appl Phys. 2005;44:6833. [Google Scholar]; (b) Seelig J, Leslie K, Renn A, Kuhn S, Jacobsen V, van de Corput M, Wyman C, Sandoghdar V. Nano Lett. 2007;7:685. doi: 10.1021/nl0627590. [DOI] [PubMed] [Google Scholar]
- 38.(a) Jensen TR, Duval ML, Kelly KL, Lazarides AA, Schatz GC, Van Duyne RP. J Phys Chem B. 1999;103:9846. [Google Scholar]; (b) Jensen TR, Malinsky MD, Haynes CL, Van Duyne RP. J Phys Chem B. 2000;104:10549. [Google Scholar]
- 39.Malicka J, Gryczynski I, Gryczynski Z, Lakowicz JR. Anal Biochem. 2003;315:57. doi: 10.1016/S0003-2697(02)00702-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritman-Meer T, Cade NI, Richards D. Appl Phys Lett. 2007;91:123122. [Google Scholar]