

Abstract
Large Hydrophobic Residues (LHR) such as phenylalanine, isoleucine, leucine, methionine and valine play an important role in protein structure and activity. We describe the role of LHR in complete set of protein sequences in 15 different species. That is the distribution of LHR in different proteins of different species is reported. It is observed that the proteins prefer to have 27% of large hydrophobic residues in total and all along the sequence. It is also observed that proteins accumulate more LHR in its active sites. A window analysis on these protein sequences shows that the 27% of LHR is more frequent at window length of 45 amino acids. The influenza virus and P. falciparum show a random distribution of LHR in its proteins compared to other model organisms.
Keywords: LHR, large hydrophobic residues, protein analysis, sequence analysis
Background
Proteins are the working force in all living systems. These proteins that are translated from mRNA are evolved to have a defined structure and specified function. There is lot of work gone into proteins to understand the ultimate truth of real hideous information [1–6]. A thorough understanding of protein evolution from sequence information is now possible to study as many whole genome and proteome sequences are made available online. From various statistical and computational approaches it can be studied that which is making a portion of amino acid or mRNA sequences unchanged during evolution.
White and Jacobs [6] reported the statistical distribution of hydrophobic residues along the protein chains and its implications to protein folding and evolution. They have tested for randomness of hydrophobic residues in the proteins with limited sequences. Currently most of the protein sequences of several model organisms are available for thorough understanding, which we have carried out here. The authors argue that the distribution of hydrophobic residues along a sequence cannot be distinguished from that expected for a random distribution and suggests that functional proteins may have originated from random sequences, the folding of proteins into compact structures may be much more permissive with less sequence specificity than previously thought and the clusters of hydrophobic residues along chains which are revealed by hydrophobicity plots are a natural consequence of a random distribution.
Brooks et al. [7] reported the evolution of amino acid frequencies in proteins over time and its order in genetic codes. Recently, we have reported that the protein coding frames of mRNA sequences prefers to have 27% of XTX (X= A, T, G or C) [8] that code for only Large Hydrophobic Residues (LHR) such as phenylalanine, isoleucine, leucine, methionine and valine. This paper analyzes further this in proteins by studying the complete sets of protein sequences of 15 different species.
Methodology
Dataset
The complete sets of protein sequences different species including human, chimpanzee, dog, cow, mouse, chicken, fruitfly, honeybee, mosquito, zebrafish, worm, plant, yeast, Influenza virus and P.falciparum are taken from ftp://ftp.ncbi.nlm.nih.gov/genomes/. The numbers of protein sequences in each set are listed in the table 1 (See Supplementary material).
Definitions and analysis
The distribution of LHR in different proteins of different species is studied using our program written ’C‘. That is the number of LHR residues in a defined window length (number of amino acids between 10 and 100) counted and grouped as shown in Figure 1. The amount of LHR at which there is a maximum number of windows is taken as the probable amount of LHR. All this procedure is repeated for different species and different window length. A plot of probable amount of LHR versus window length for different species is plotted in Figure 2 and discussed.
Figure 1.

Flow diagram showing how the distribution of LHR is computed.
Figure 2.
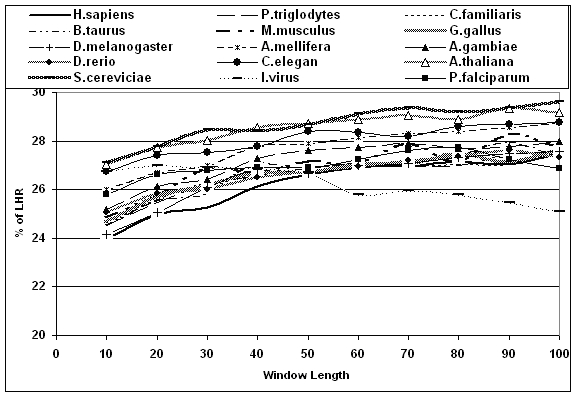
Amount of LHR in different windows and different species.
Discussion
The probable amount of LHR is observed to be 27%. Though this amount varies from 5 to 55%, the occurrence in proteins is very less. It is observed that given any window length, the amount of LHR is preferably 27%. That is proteins prefer to have 27% of LHR. Generally, in animals the amount of LHR is less compared to fungi and plants. The shortage of LHR in animals is balanced by adding more number of small hydrophobic residues such as Glycine, Alanine, Proline and Cysteine. In another calculation it was found that each LHR is compensated with three small hydrophobic residues. This is the reason why the length of the animal proteins increases. The probable amounts of LHR for different window lengths in all 15 species are plotted in figure 2. The window lengths are studied from 10 to 100 amino acids. Below the window length of 45, the animal's sequences are having less than 27% of LHR and is not so in yeast and plant. The 27% of LHR is observed very much near window length of 45 amino acids. It is also observed that the amount of LHR in active site is higher and proteins prefer to have 27% of LHR. So there is an alteration in amount of LHR in and around active site. This leads to slight changes in the amount of LHR in different window length. In another term an optimum level of LHR is observed when window length is between 40 and 50 i.e., at 45 amino acids. After the window length of 45 the amount of LHR is either not changed or slightly more.
Thymine is responsible for coding LHR in protein. This thymine has one extra methyl group compared to its RNA counterpart uracil. As the animals are hetero sexual, the alteration in thymine content in mRNAs is altered. This leads to reduction in LHR in animal sequences. This is not so in plants and fungus. As argued earlier [6] the distribution of LHR are not randomly distributed but with specific definition. Functional proteins have not originated from random sequences. The clusters of hydrophobic residues along the protein chains are a natural consequence of protein function and not random event. A further atom level understanding in these protein sequences might give clear idea on why it happens at window length of 45 amino acids will be of worth doing which is underway in our lab. Globular proteins are expected to follow this distribution profile. The sequences taken here are complete set of the given species. It may include membrane proteins. However the numbers of such sequences are less. Their impacts in the overall results are minimal and ignored. The influenza virus and P. falciparum show a different trend unlike other species.
Conclusion
The nature of proteins is that except the active site the other portions of the proteins prefer to have a definite amount (27%) of LHR that gives local stability all along the sequence and overall structure. This is in agreement with our earlier work on distribution of XTX in mRNA sequences [8]. Heterosexual animals show a less number of LHR in its proteins compared to fungi and plants. The diseased sequences lack these LHR in total or in some portion along the sequences that leads to malfunctioning of the protein. Globular proteins are expected to follow this behavior. An optimum level of LHR is observed when window length is 45 amino acids. The alteration in amount of LHR leads to malfunctioning of the proteins.
Supplementary material
Footnotes
Citation:Jayaraj et al, Bioinformation 3(9): 409-412 (2009)
References
- 1.Gromiha MM, Ponnusamy PK. Int J Pept Protein Res. 1996;48:452. doi: 10.1111/j.1399-3011.1996.tb00863.x. [DOI] [PubMed] [Google Scholar]
- 2.Venkatachalam CM. Biopolymers. 1968;6:1425. doi: 10.1002/bip.1968.360061006. [DOI] [PubMed] [Google Scholar]
- 3.Huang LT, et al. Biophys Chem. 2006;125:462. doi: 10.1016/j.bpc.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Mirny L, Shakhnovich E. J Mol Biol. 2001;308:123. doi: 10.1006/jmbi.2001.4602. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz R, et al. Protein Sci. 2001;10:1023. doi: 10.1110/ps.33201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White SH, Jacobs RE. Biophys J. 1990;57:911. doi: 10.1016/S0006-3495(90)82611-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooks DJ, et al. Mol Biol Evol. 2002;19:1645. doi: 10.1093/oxfordjournals.molbev.a003988. [DOI] [PubMed] [Google Scholar]
- 8.Anandagopu P, et al. Bioinformation. 2008;2:304. doi: 10.6026/97320630002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.