

Abstract
Aims
Our aim was to determine whether the repeated, binge administration of 3,4-methylenedioxymethamphetamine (ecstasy; MDMA) produces structural and/or functional changes in the myocardium that are associated with oxidative stress.
Methods and results
Echocardiography and pressure–volume conductance catheters were used to assess left ventricular (LV) structure and function in rats subjected to four ecstasy binges (9 mg/kg i.v. for 4 days, separated by a 10 day drug-free period). Hearts from treated and control rats were used for either biochemical and proteomic analysis or the isolation of adult LV myocytes. After the fourth binge, treated hearts showed eccentric LV dilation and diastolic dysfunction. Systolic function was not altered in vivo; however, the magnitude of the contractile responses to electrical stimulation was significantly smaller in myocytes from rats treated in vivo with ecstasy compared with myocytes from control rats. The magnitude of the peak increase in intracellular calcium (measured by Fura-2) was also significantly smaller in myocytes from ecstasy-treated vs. control rats. The relaxation kinetics of the intracellular calcium transients were significantly longer in myocytes from ecstasy-treated rats. Ecstasy significantly increased nitrotyrosine content in the left ventricle. Proteomic analysis revealed increased nitration of contractile proteins (troponin-T, tropomyosin alpha-1 chain, myosin light polypeptide, and myosin regulatory light chain), mitochondrial proteins (Ub-cytochrome-c reductase and ATP synthase), and sarcoplasmic reticulum calcium ATPase.
Conclusion
The repeated binge administration of ecstasy produces eccentric LV dilation and dysfunction that is accompanied by oxidative stress. These functional responses may result from the redox modification of proteins involved in excitation-contraction coupling and/or mitochondrial energy production. Together, these results indicate that ecstasy has the potential to produce serious cardiac toxicity and ventricular dysfunction.
Keywords: Pressure–volume loops, Cardiac myocytes, Proteomics, Ecstasy, Tyrosine nitration, MDMA
1. Introduction
3,4-methylenedioxymethamphetamine (ecstasy or MDMA) is a commonly used recreational drug. Despite the popular misconception that ecstasy is ‘safe’, an increasing number of clinical reports and animal studies indicate that MDMA can produce brain, kidney, and liver toxicity.1–7 In addition, accumulating clinical evidence indicates that MDMA use is associated with hypertension, cardiac arrhythmias, aortic dissection, myocardial infarction, and sudden death.8–14 Histological examination of the hearts from five MDMA-associated fatalities revealed contraction band necrosis, myocyte necrosis, and inflammation characterized by neutrophil and macrophage infiltration.9 We have shown that repeated binge administration of MDMA in rats produces myocarditis with inflammatory infiltrates and areas of necrosis and disrupted cytoarchitecture.15 Whether the observed inflammatory and histological changes lead to structural and functional changes in the myocardium has not been systematically studied. In a retrospective study of autopsies, the hearts from MDMA users were found to be heavier than those from individuals who had not used the drug.16 Whether these differences reflected MDMA-mediated cardiac hypertrophy or were associated with functional deficits was not determined. In contrast, methamphetamine, an amphetamine analogue that is structurally similar to MDMA, produces inflammatory myocarditis and left ventricular (LV) dilation and dysfunction.17–19 Therefore, the first goal of this study was to test the hypothesis that the repeated administration of MDMA would produce structural and functional deficits in the rat myocardium. For these studies, we used a repeated binge pattern of dosing characterized by periods of drug administration followed by periods of abstinence. Binging is a typical pattern of recreational MDMA use.20 The 9 mg/kg dose of MDMA was chosen because it is within the range of doses producing neurotoxicity and cardiac inflammation and cytoarchitectural changes in rats.15
The mechanism(s) underlying MDMA-induced cardiac toxicity are unknown; however, there is evidence that oxidative stress plays an important role in MDMA-mediated brain, kidney, and liver toxicity.1–7 Antioxidants such as alpha lipoic acid protect against MDMA-mediated neurotoxicity.3 In cultured rat cardiac myocytes, exposure to MDMA activated myocardial NF-kappaB, disrupted cytosolic calcium and mitochondrial homeostasis, and altered gene transcription,21 all of which can occur in response to increased oxidative stress. Finally, several studies have shown that metabolites of MDMA are capable of redox cycling.22 Whether MDMA increases oxidative stress in the heart in vivo has not been determined. Therefore, the second goal of this study was to test the hypothesis that MDMA increases oxidative stress in the heart.
2. Methods
2.1. Experimental model
Male Sprague–Dawley rats (200–225 g; Harlan, Indianapolis, IN) were housed in a temperature- and humidity-controlled room with a 12-h light/dark cycle. Standard rat chow and tap water were available ad libitum. All procedures were performed in accordance with National Institutes of Health Guidelines for the Care and Use of Experimental Animals and approved by the Institutional Animal Care and Use Committee at Louisiana State University Health Sciences Center.
Rats were anaesthetized with a mixture of ketamine and xylazine (100/10 mg/kg i.p.) and a venous cannula (Micro-renathane, 0.033-inch o.d.×0.014-inch internal diameter (i.d.); Braintree Scientific, Braintree, MA) placed into the femoral vein.15 Five to seven days later, the rats were randomly selected to receive i.v. MDMA or saline. MDMA (9 mg/kg in 20–40 µL with a 100 µL saline flush) was administered twice daily (morning and afternoon) for 4 days. This ‘binge’ was followed by a 10 day drug-free period. This pattern of binge administration and abstinence was repeated three more times. Control rats received volume-matched i.v. injections of saline (0.9%) according to the same schedule. All injections were made over 10–15 s. The 9 mg/kg dose of MDMA was chosen because it is within the range of doses producing neurotoxicity in rats23,24 and is equivalent to a neurotoxic dose in primates.25 A 10 mg/kg dose of MDMA administered to rats produces a plasma MDMA concentration of 6.3 nmol/mL, 45 min post-injection,2 this value is within the range occurring in humans following MDMA ingestion.26
2.2. Echocardiographic studies
Echocardiograms (Toshiba Aplio at 8.5 MHz) were performed under isoflurane (1.5–2%) anaesthesia one day before the first binge and one day after the second and fourth binges in MDMA (n = 10) and saline-treated rats (n = 9). Two-dimensional and M-mode echocardiographic measurements were made of posterior wall (PW) and left ventricle diameter during systole (LVDS) and diastole (LVDD) in the parasternal long axis view of the left ventricle and in the parasternal short axis view at the level of the papillary muscles. Left ventricle systolic function was assessed by changes in fractional shortening (FS = LVDD−LVDS/LVDD). Doppler measurements of mitral inflow velocity were recorded in the apical four-chamber view with the pulsed-wave Doppler sample volume placed at the tips of the leaflets. Peak early ventricular filling (E) and atrial contraction (A) velocities were determined from these data. A change in the E/A ratio was used to assess left ventricle diastolic function. All measurements were performed on three distinct cardiac cycles and the values averaged at each time point.
2.3. Left ventricle pressure–volume relationships
Left ventricle performance was assessed in vivo using pressure–volume (p–v) loops. Rats were anaesthetized with isoflurane (4%), intubated with an 18 gauge catheter, and ventilated (isoflurane 2% and oxygen 3 L/min). Tidal volume (3–5 mL) and respiratory rate (60–70 breaths/min) were adjusted to maintain arterial blood pH between 7.35–7.45. Body temperature was maintained at 37°C using a heat lamp. Cannulae (micro-renathane, 0.033 in o.d.) were placed into the femoral artery and right jugular vein to record arterial pressure and administer drugs, respectively. A microtipped p–v catheter (SPR-838; Millar Instruments) was introduced into the carotid artery and advanced into the left ventricle. After a 15 min equilibration period, baseline cardiac parameters were acquired using a minimum of three consecutive p–v loops (sampling rate 1000 Hz; MPVS-400, Millar Instruments). Measures of peak LV systolic pressure, LV end-diastolic pressure, maximal slope of the systolic pressure increment (+dP/dt), diastolic decrement (−dP/dt), ejection fraction (EF), stroke volume (SV), cardiac output, and stroke work (SW) were computed using the Millar PVAN analysis system. Transient occlusion of the inferior vena cava was used to alter preload in order to access load independent measures such as the end-systolic pressure volume relationship (ESPVR), end-diastolic pressure volume relationship (EDPVR), and SW index.
The p–v conductance catheter was calibrated using whole blood from saline and MDMA-treated rats to generate standard curves of relative volume units that were used to verify volume measurements. To determine the absolute LV volume 35 µL of hypertonic saline (15%) was injected i.v. to measure the parallel conductance of the system, which was then subtracted from the total volume.
2.4. Adult rat cardiac myocyte isolation
Adult LV myocytes (ALVM) were enzymatically isolated from three rats per group. Hearts were removed and retrogradely perfused via the aorta with perfusion buffer (Alliance for Cell Signaling, Protocol PP00000125) for 2–3 min until spontaneous contraction ceased. Digestion buffer was then perfused (perfusion buffer containing 12.5 µM CaCl2, 0.14 mg/mL trypsin, and 0.25 mg/mL of Liberase Blendzyme 4; Roche). After digestion, the left ventricle was removed and minced in perfusion buffer with 12.5 µM CaCl2 and 5% BSA. Cells were resuspended in perfusion buffer with 5% BSA, in which the concentration of CaCl2 was gradually increased over 20 min to achieve a final concentration of 1 mM. ALVM were plated on dishes or coverslips (coated with laminin) in Minimal Essential Media with Hanks’ salts and 2 mM L-glutamine, supplemented with 5% calf serum, 10 mM 2,3-butanedione monoxime, and 100 U/mL penicillin. After 1 h incubation, ALVM were rinsed and placed in culture medium (serum-free MEM with 0.1% BSA, 100 U/mL penicillin, and 2 mM l-glutamine). Myocytes with obvious sarcolemmal blebs or spontaneous contractions were not used.
2.5. Contractility and [Ca2+]i transients in adult left ventricular myocytes
Mechanical properties of ALVM were assessed using a SoftEdge video-based edge-detection system (IonOptix Corporation, Milton, MA).27,28 Briefly, cells were placed in a Warner chamber mounted on the stage of an inverted microscope (Olympus, IX-70) and incubated with Tyrode's buffer (NaCl 137 mM, KCl 5.4 mM, CaCl2 1.2 mM, MgCl2 0.5 mM, HEPES 10 mM, and glucose 10 mM, pH 7.4, 37°C) containing 0.5 µM Fura 2-AM (Molecular Probes) for 10 min and then washed with Tyrode's buffer to remove excess dye. Myocytes were field stimulated with suprathreshold voltage (20–25 V) at 0.5 Hz (3 ms duration). Sarcomere length (SL) was monitored from a red-light bright-field image (650-nm long-pass filter) and SL was measured using an IonOptix MyoCam camera. For [Ca2+]i imaging, fura-2-loaded myocytes were excited at 360 ± 6.5 and 380 ± 6.5 nm with an ultraviolet xenon lamp. Emission fluorescence was measured at 510 ± 15 nm.
Contractility and changes in [Ca2+]i were analysed using an IonWizard data acquisition system (IonOptix). Absolute twitch amplitude was measured as the difference between the systolic and the diastolic SL; SL shortening % was expressed as the ratio of absolute twitch amplitude to diastolic SL. Twitch duration was measured from the onset of contraction to peak shortening and the time from peak shortening to 90% relaxation.
2.6. Western blot analysis and two-dimensional polyacrylamide gel electrophoresis
Tissues from the mid wall of the LV were flash frozen in liquid nitrogen and stored in −80°C. LV lysates from control and MDMA-treated rats were prepared by homogenization in lysis buffer (0.1% Triton X-100, 0.1% deoxycholate, 25 mM HEPES, pH.7.4, 50 mM NaCl, 1 mM MgCl2, 2 mm EGTA, 10 mM pyrophosphate, 10 µg/mL aprotinin, 10 µg/mL leupeptin, 0.5 mM PMSF, and 500 µM Na3VO4.) and sonicated. Samples were centrifuged at 10 000 rpm for 10 min at 4°C. Protein concentrations in the supernatant were assessed using a bicinchonic acid assay (Pierce). For western blot analysis, equal amounts of protein (50 µg) were separated by SDS–PAGE (4–12%) and transferred to PVDF membranes. Membranes were blocked with 3% (w/v) BSA-TBST and incubated for 1 h at room temperature with anti-3-nTyr antibody (Cayman), mouse monoclonal SERCA-2a (Biomol), or phospholamban antibodies (1:1000) in 3% (w/v) BSA-TBST buffer for 1 h. Membranes were rinsed with TBST and then incubated with anti-mouse Ig-HRP-conjugated secondary antibodies (1:2000) for 1 h at room temperature. The immunoblots were visualized by chemiluminescence using the ECL (GE Healthcare, Piscataway, NJ) according to the manufacturer's protocol. The images were captured using Molecular Imager VersaDoc Imaging Systems (Bio-Rad).
For two-dimensional PAGE, nitrated proteins (600 µg left ventricle lysates pooled from five saline and six MDMA-treated rats) were first immunoprecipitated with anti-nTyr antibodies coupled to agarose beads. Immunoprecipitates from the two groups were resuspended in 150 µL of IEF buffer (7 M urea, 2 M thiourea, 2% CHAPS, 1% lauryl-maltoside, 1% Triton X-100, 1% ‘BioLyte’ carrier ampholytes, 30 mM DTT, 1 mM TBP, and 0.05% bromophenol blue. IPG strips (pH 4–7, 7 cm, Invitrogen) were allowed to rehydrate overnight. IEF was performed using an Invitrogen Zoom runner. IEF strips were then equilibrated in buffer containing 375 mM Tris, 50 mM DTT, 3% SDS, pH 8.6. For the two-dimension, strips were placed above standard format mini-gels (5–15% acrylamide gradient, Laemmli buffer system, Invitrogen), and set in place with warm 1% agarose. SDS–PAGE gels were stained with colloidal blue and analysed via PDQuest image analysis software to identify spots of interest. Gel spots were excised, destained, dried in vacuo, and hydrolyzed ‘in-gel’ with trypsin at 37°C overnight. The trypsin digests were then adjusted with alphahydroxy-cinnamic acid. One microlitre of sample was spotted onto a matrix-assisted laser-desorption ionization time-of-flight (MALDI-TOF) sample plate and analysed by MALDI-TOF-mass spectrometry (MALDI-TOF-MS) according to established procedures. The parent polypeptides were identified by comparing the profile of tryptic peptide masses generated by the mass spectrometer with predicted tryptic peptides from all known polypeptides using the MASCOT program.
2.7. Detection of SERCA-2a tyrosine nitration
Six hundred microgram of LV protein were pooled from saline (n = 5) and MDMA-treated (n = 6) rats and immunoprecipitated with anti n-Tyr antibodies coupled to agarose beads. Immunoprecipitated proteins were subjected to western blot analysis with SERCA-2a (Biomol) antibodies.
2.8. Statistical analysis
All data are reported as mean ± SEM. Between group, comparisons of data from echocardiograms and p–v loops from saline and MDMA- treated rats were made using one-way analysis of variance followed by Bonferroni post hoc tests (SigmaStat). Changes in contractility and [Ca2+]i in ALVM and western blot data from saline and MDMA- treated rats were compared using Student's t-test (SigmaStat). P < 0.05 was considered statistically significant.
3. Results
3.1. 3,4-methylenedioxymethamphetamine treatment produce eccentric left ventricular dilation
Baseline echocardiographic parameters between MDMA (n = 10) and saline-treated (n = 9) rats were not significantly different prior to dosing or after the second binge (data not shown). After four binges LVDD and LVDS were significantly greater in MDMA-treated than in saline-treated rats (Table 1). PW during diastole (PWD) was significantly decreased in MDMA-treated rats, whereas PW during systole (PWS) was similar between the two groups (Table 1). The ratio of (2 × PWD)/LVDD, an index of eccentric dilation, was significantly smaller in MDMA- compared with saline-treated rats (Table 1). There were no differences in FS between the two groups; conversely, the E/A ratio was significantly greater in MDMA-treated rats (Table 1). Figure 1 shows representative M-mode echocardiograms of the left ventricle from a saline-treated rat (A) and MDMA-treated rat (B) illustrating the difference in left ventricle dimension and PW thickness after four saline or MDMA binges. We used colour flow Doppler echocardiography to access all four valves, since we did not detect any functional valvular abnormalities, we did not conduct histological studies of the valves.
Table 1.
Echocardiographic parameters obtained from 3,4-methylenedioxymethamphetamine (n = 10) and saline-treated (n = 9) rats after four binges
| Saline group binge 4 | MDMA group binge 4 | |
|---|---|---|
| LVDD (mm) | 7.52 ± 0.15 | 8.35 ± 0.11* |
| LVDS (mm) | 4.99 ± 0.13 | 5.54 ± 0.18* |
| PWD (mm) | 1.38 ± 0.03 | 1.22 ± 0.04* |
| PWS (mm) | 2.28 ± 0.08 | 2.08 ± 0.08 |
| (2*PWD)/LVDD | 0.37 ± 0.012 | 0.29 ± 0.014* |
| E/A ratio | 1.11 ± 0.04 | 1.34 ± 0.09* |
| % FS | 32.19 ± 1.23 | 33.00 ± 1.73 |
LVDD, left ventricle diameter at diastole; LVDS; left ventricle diameter at systole; PWD, posterior wall thickness during diastole; PWS; posterior wall thickness during systole *indicates significant difference (P < 0.05) between groups.
Figure 1.

Representative M-mode echocardiograms from a saline (A) and an 3,4-methylenedioxymethamphetamine (B) treated rat subjected to four binges. LVDD; left ventricle diameter at diastole; LVDS; left ventricle diameter at systole; PWD; posterior wall thickness during diastole; PWS; posterior wall thickness during systole.
3.2. Binge 3,4-methylenedioxymethamphetamine treatment impairs left ventricular relaxation during diastole
Since echocardiographic analysis showed significant differences in LV function and structure between the two groups after the fourth binge, we chose this time point to further evaluate LV function using p–v catheters. Figure 2A compares representative p–v loops from an MDMA and a saline-treated rats. Compared to saline, MDMA significantly increased EDV (Figure 2).
Figure 2.
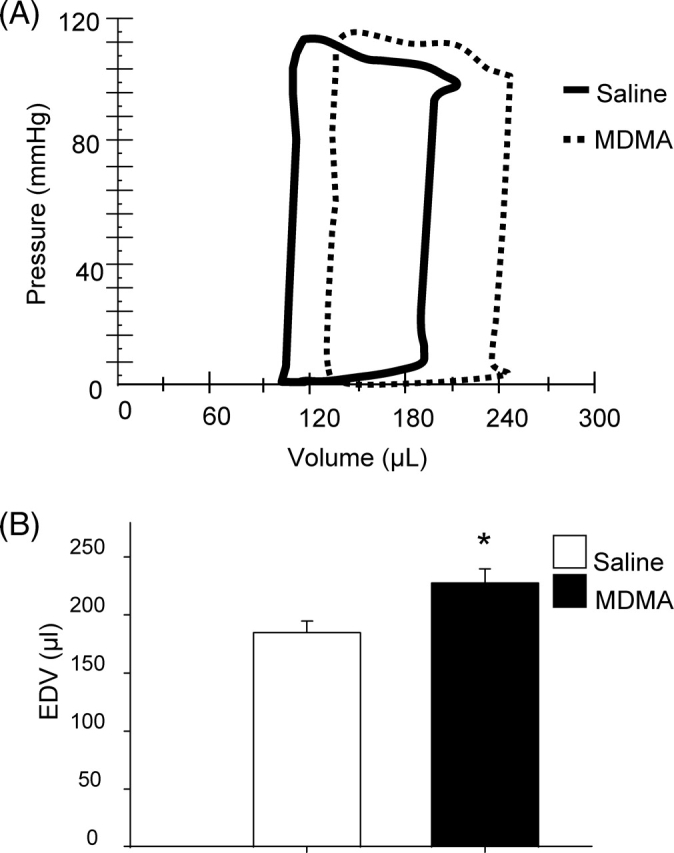
Measurement of left ventricular end-diastolic volumes. (A) Representative p–v loops obtained from a saline and 3,4-methylenedioxymethamphetamine binge rat subjected to four binges. (B) Comparison of EDV between saline (n = 7) and 3,4-methylenedioxymethamphetamine-treated rats (n = 7). EDV; end-diastolic volume. *P < 0.05 between groups.
The effects of MDMA on diastolic and systolic function were assessed by calculating EDPVR and ESPVR while changing preload. Representative p–v loops generated in response to changes in preload in a saline and an MDMA-treated rat are shown in Figure 3A and B, respectively. Compared with saline (n = 7), MDMA (n = 7) treatment significantly increased EDPVR, indicating that the drug decreased LV compliance (Figure 3C). ESPVR, a measure of systolic function, was similar in the two groups (Figure 3D). EF (60 ± 4 vs.52 ± 3%, respectively), SV (116 ± 6 vs. 125 ± 9 µL, respectively), heart rate (274 ± 13 vs. 268 ± 9 bpm, respectively), and Pmax (103 ± 4 vs. 115 ± 5 mmHg, respectively) were not significantly different between the saline and MDMA groups. Although the body weights of the MDMA-treated and control rats were the same before binge 1, the MDMA-treated rats were significantly smaller than control after binge 4 (354 ± 8 vs. 391 ± 7 g, respectively).
Figure 3.

In vivo p–v analysis of systolic and diastolic function. (A and B) Representative p–v loops generated during changes in preload in a saline and 3,4-methylenedioxymethamphetamine-treated rat, respectively. Preload was altered by occluding the inferior vena cava. (C) Comparison of end-diastolic pressure–volume relationships in saline (n = 7) and 3,4-methylenedioxymethamphetamine-treatment groups (n = 7). (D) Comparison end-systolic pressure–volume relationships in the same groups. *P < 0.05 between groups.
3.3. Systolic and diastolic dysfunction of adult left ventricular myocytes from 3,4-methylenedioxymethamphetamine-treated rats
To compare contractile function and changes in [Ca2+]i handling in ALVM isolated from saline and MDMA-treated rats, contractility and [Ca2+]i were simultaneously measured in electrically paced (0.5 Hz) ALVM loaded with Fura-2. Figure 4A compares representative traces of changes in SL in paced ALVM isolated from a saline and an MDMA-treated rat. Contractility (SL shortening %) was significantly smaller in ALVM from MDMA-treated rats (n = 45 cells from 3 rats) compared with saline-treated rats (n = 37 cells from 3 rats) (Figure 5A). Furthermore, MDMA treatment significantly decreased the maximum velocity of relengthening (Figure 5B) and increased the time to 90% relengthening (Figure 5C).
Figure 4.
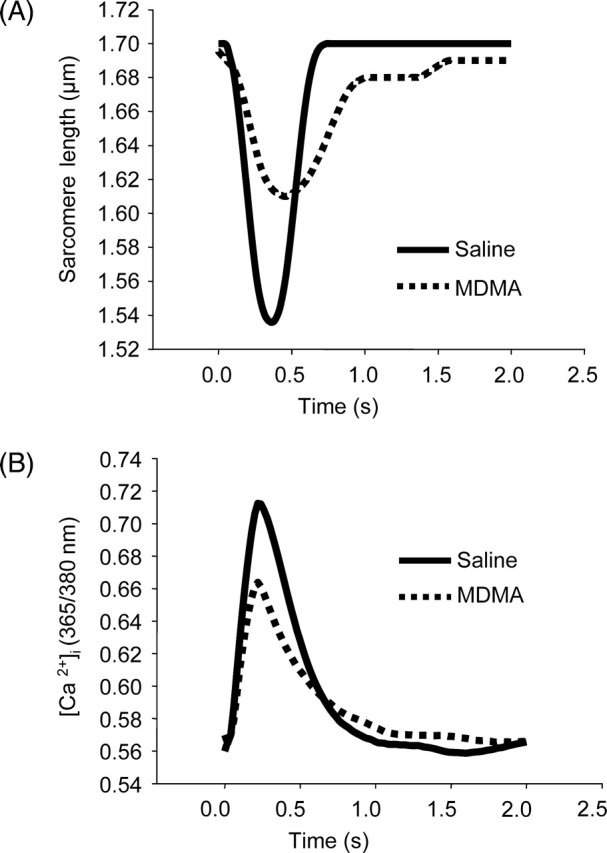
Altered contractility and [Ca2+]i in response to electrical stimulation (0.5 Hz) in adult left ventricular myocytes isolated from rats receiving four 3,4-methylenedioxymethamphetamine or saline binges. (A and B) Representative traces showing simultaneous changes in sarcomere length (contractility) and [Ca2+]i, respectively.
Figure 5.

Effects of 3,4-methylenedioxymethamphetamine treatment on contractility and intracellular calcium transient kinetics. Contractility and [Ca2+]i were measured simultaneously in Fura-2-loaded cells from rats treated in vivo with saline (n = 37 cells from three rats) or 3,4-methylenedioxymethamphetamine (n = 45 cells from three rats). (A) Sarcomeric length shortening (sarcomere length shortening %). (B) Relaxation measured as maximum velocity of relengthening (dl/dt). (C) Time to return of sarcomere length to 90% of baseline. (D) Amplitude of the [Ca2+]i transient during contraction. (E) Maximum velocity of return of [Ca2+]i to baseline. (F) time to return of [Ca2+]i to 90% of baseline. *P < 0.05 between groups.
In response to 0.5 Hz stimulation, the amplitude of the peak increase in [Ca2+]i was significantly decreased in ALVM from MDMA-treated rats (Figures 4B and 5D). During relaxation, the maximum velocity of the return of [Ca2+]i to baseline was significantly lower (Figures 4B and 5E) and the return to baseline significantly longer in ALVM from MDMA-treated rats (Figures 4B and 5F).
3.4. Increased oxidative stress in 3,4-methylenedioxymethamphetamine-treated rats
Peroxynitrite is formed by a diffusion-limited reaction of both superoxide and nitric oxide.29 Peroxynitrite leads to nitration of tyrosine residues on a number of different proteins. As a marker of oxidative stress, western blot analysis was used to compare the levels of nitrated tyrosine residues in LV proteins from MDMA (n = 4) and saline-treated (n = 5) rats. Compared with saline, MDMA treatment increased n-Tyr content in the myocardium (Figure 6). To further identify nitrated proteins, n-Tyr immunoprecipitates were separated by two-dimensional PAGE and spots differentially nitrated between the two groups were identified by MALDI-TOF-MS (Figure 7 and Table 2). Proteins differentially nitrated in MDMA-treated rats clustered into two classes: mitochondrial proteins (Ub-cytochrome-c reductase and ATP synthase-D chain) and myofilament proteins (troponin-T, tropomyosin alpha-1 chain, myosin light polypeptide, and myosin regulatory light chain) (Table 2).
Figure 6.

3,4-methylenedioxymethamphetamine treatment increases tyrosine nitration. Left ventricular lysates (75 µg) from saline (n = 5) and 3,4-methylenedioxymethamphetamine-treated rats (n = 4) were subjected to western blot analysis with anti-nTyr antibodies (1:1000). GAPDH was used as a loading control.
Figure 7.

Proteomic identification of tyrosine nitrated proteins in response to in vivo 3,4-methylenedioxymethamphetamine administration. Tyrosine nitrated proteins were immunoprecipitated from pooled lysates (saline n = 5, 3,4-methylenedioxymethamphetamine n = 6) with anti-nTyr antibodies coupled to agarose beads and separated using two-dimensional sodium dodecyl sulphate polyacrylamide gel electrophoresis. Identified spots (F1–F10) were subjected to tryptic digest and identified by matrix-assisted laser-desorption ionization time-of-flight-mass spectroscopy (see Table 2).
Table 2.
Matrix-assisted laser-desorption ionization time-of-flight identification of differentially nitrated proteins in left ventricle of 3,4-methylenedioxymethamphetamine treated rats
| Spot | Protein | Protein score | Protein molecular weight (kDA) |
|---|---|---|---|
| F1 | Ub-cytochrome-c reductase (Complex III) | 229 | 53.5 |
| F2 | Troponin-T, cardiac muscle | 420 | 35.7 |
| F3 | Tropomyosin alpha-1 chain | 499 | 32.7 |
| F7 | ATP synthase-D chain (Complex V) | 153 | 28.8 |
| F9 | Myosin light polypeptide | 552 | 22.3 |
| F10 | Myosin regulatory light chain (ventricular muscle isoform) | 473 | 18.9 |
Western blots of LV homogenates probed for n-Tyr showed an increase in a ∼110 kDa band (Figure 6), similar to the molecular weight of SERCA-2a. To determine if this band represented nitrated SERCA-2a, 600 µg of protein pooled from saline (n = 5) and MDMA-treated (n = 6) rats were immunoprecipitated using n-Tyr antibodies and subjected to western blot analysis with SERCA-2a antibodies. MDMA increased nitrated SERCA-2a vs. saline (Figure 8A). Western blot analysis showed a reduction in SERCA-2a protein expression in the MDMA-treated group; however, this decrease was not statistically significant (P = 0.06). The ratio of SERCA2a/phospholamban was not different (P = 0.1) between the two groups (Figure 8B and C). Phospholamban phosphorylation levels were similar between the two groups (data not shown).
Figure 8.

3,4-methylenedioxymethamphetamine increases tyrosine nitration of SERCA-2a. (A) Nitrotyrosine immunoprecipitates from saline (n = 5 pooled) and 3,4-methylenedioxymethamphetamine (n = 6 pooled) left ventricular lysates were probed by western blot analysis using SERCA-2a antibody. (B) SERCA-2a protein levels normalized to GAPDH (n = 5). (C) SERCA-2a protein levels normalized to phospholamban (n = 5).
4. Discussion
Our in vivo structural and functional data provide the first conclusive evidence that the repeated, binge administration of MDMA produces LV dilation and diastolic dysfunction in rats. Echocardiography revealed a significant decrease in PW thickness during diastole without a significant change in PW thickness during systole (PWS). The ratio of (2*PWD)/LVDD was significantly decreased in MDMA compared with saline-treated animals, indicating that the drug produced eccentric LV dilation. There is a single clinical report linking MDMA use with cardiac hypertrophy.16 In this retrospective study of autopsy data, cardiac hypertrophy was demonstrated by comparing heart weight to body weight ratios in individuals testing positive for MDMA use compared with those not using the drug. Structural measures such as ventricular diameter and/or wall thickness were not reported. To our knowledge, there are no reports of MDMA-mediated ventricular hypertrophy in experimental animals.
These data also revealed for the first time that binge treatment with MDMA impairs LV relaxation during diastole leading to a less compliant ventricle. Consistent with evidence of diastolic dysfunction in vivo, relaxation was also significantly impaired in ALVM from MDMA-treated rats as evidenced by the decreased return velocity and increased time to return to baseline (Figure 5B and C). The impaired relaxation may be due to decrease in [Ca2+]i uptake since a concomitant decrease in [Ca2+]i transient amplitude, decreased return velocity and an increase in time to baseline was also observed.
In contrast, systolic function in vivo, as indicated by ESPVR, Pmax and EF, was not significantly different between the MDMA and saline-treated animals; however, contractility was significantly reduced in ALVM from rats treated in vivo with MDMA. While the data using ALVM indicate that there are underlying contractile deficits, the reason why these differences are not manifest in vivo after four binges may reflect the sympathomimetic properties of MDMA. MDMA increases heart rate and arterial pressure in several species including humans.30–32 Alternatively, the lack of contractile dysfunction in vivo may reflect early compensatory responses initiated to maintain systolic function in response to the early LV dilation and/or diastolic dysfunction. In view of the contractile deficits observed in vitro, we anticipate that impaired systolic function would have occurred in vivo following continued dosing. We chose to perform the majority of our physiological and biochemical analyses after the fourth MDMA binge to examine the mechanism(s) accompanying the progression of early LV dilation and dysfunction, rather than end-stage disease.
Using radio-telemetry, we previously showed that (3 and 9 mg/kg) binge doses of MDMA produced a biphasic heart rate responses consisting of an initial vagally-mediated bradycardia followed by tachycardia. The bradycardic response increased in magnitude between binges, but exhibited tolerance within binges. MDMA also produced pressor responses (∼40 mmHg) which lasted ∼10 min.15 The magnitude and duration of these responses was remarkably consistent within and between binges. Given the short duration of the pressor responses and the fact that drug treatment did not alter baseline arterial pressure, it is unlikely that increased afterload or other haemodynamic factors were responsible for the observed LV dilation.
While the mechanism(s) responsible for MDMA-mediated LV dysfunction is unknown, our data raise a number of interesting possibilities. Our proteomic analyses showed that MDMA treatment leads to the nitration of tropomyosin alpha-1 chain, troponin-T, myosin light polypeptide and myosin regulatory light chain. Mihm et al.33 have shown using isolated cardiac trabeculae, that peroxynitrite exposure significantly impairs maximum myofilament force generation. These defects were associated with an increased tyrosine nitration of myosin heavy chains and the myofibrillar isoform of creatine kinase. Furthermore, it is well documented that nitration of other myofilament proteins such as desmin, alpha-actinin, and myosin heavy chain is associated with altered cardiac myocytes structure that may contribute to atrial contractile dysfunction in human atrial fibrillation.34 Therefore, it is possible that the observed nitration of different contractile proteins with MDMA treatment may partially account for the observed contractile dysfunction in ALVM.
Calcium pump failure, as well as reduced sarcoplasmic reticulum calcium content, is well documented in heart failure leading to diastolic and/or systolic dysfunction.35 Although there was a trend towards, but not a significant decrease in SERCA-2a protein expression or SERCA-2a/phospholamban ratio (Figure 8B and C), immunoprecipitation assays showed that there was more nitrated SERCA-2a in the MDMA group (Figure 8A). Several laboratories have shown that nitration of SERCA-2a is accompanied by diastolic dysfunction in cardiac myoctyes36 and inhibited SERCA activity in skeletal muscle.37 Therefore, nitration of SERCA-2a may account for the significantly smaller peak increase in [Ca2+]i and decreased relaxation kinetics in ALVM from MDMA-treated rats.
Finally, the observed decreases in contractile function may reflect a decrease in mitochondrial ATP production. Our proteomic analysis revealed increased nitration of mitochondrial cytochrome c reductase (complex III) and ATP synthase (Complex V) in MDMA-treated rats. Nitration of these components of the mitochondrial respiratory chain impairs cellular energy metabolism and may further increase oxidative stress.38–41
A major question that remains is the mechanism(s) by which MDMA produces oxidative stress in the heart. Potential sources of MDMA-mediated oxidative stress include catecholaminergic stimulation, which can ultimately produce myocardial necrosis and infiltration by mechanisms as diverse as ischaemia due to coronary vasoconstriction, calcium overload, and the production of oxygen free radicals by either the auto-oxidation of catecholamines or their degradation by monoamine oxidase.42 Reactive oxygen species may also be produced by mitochondrial dysfunction and leukocyte recruitment and activation,15 and or coronary vasospasm-induced reperfusion ischaemia.15,42 Furthermore, MDMA is metabolized to ortho-quinones, highly redox active molecules, that can undergo redox cycling and generate reactive oxygen and nitrogen species.22 The redox active metabolites of MDMA have been implicated in the toxic effects of MDMA in the heart, kidney, and liver1,7,43,44 in vitro.
In conclusion, these studies demonstrate that the binge administration of MDMA can significantly alter LV structure and function. After repeated dosing, MDMA treatment produced eccentric LV dilatation accompanied by diastolic dysfunction. This LV dysfunction was associated with redox modification of mitochondrial, contractile, and calcium handling proteins. On the cellular level, treatment with MDMA in vivo significantly decreased cardiac myocyte contractile function and impaired the amplitude and relaxation kinetics of the intracellular calcium transient. These data indicate that MDMA can alter cardiac structure and function and these changes are associated with post-translational oxidative damage to mitochondrial, contractile, and calcium handling proteins.
Funding
This work was supported by the American Heart Association Predoctoral Fellowship (615646, S.K.S.), American Heart Association Grant-in-Aid (655796, K.J.V.), NIH HL63318 (P.A.L.) and NCRR P20RR18766 (K.J.V., P.A.L.).
Acknowledgements
We thank Megan Pollman for her technical assistance. MDMA was generously provided by NIDA.
Conflict of interest: none declared.
References
- 1.Carvalho M, Hawksworth G, Milhazes N, Borges F, Monks TJ, Fernandes E, et al. Role of metabolites in MDMA (ecstasy)-induced nephrotoxicity: an in vitro study using rat and human renal proximal tubular cells. Arch Toxicol. 2002;76:581–588. doi: 10.1007/s00204-002-0381-3. [DOI] [PubMed] [Google Scholar]
- 2.Colado MI, Green AR. The spin trap reagent alpha-phenyl-N-tert-butyl nitrone prevents ‘ecstasy’-induced neurodegeneration of 5-hydroxytryptamine neurones. Eur J Pharmacol. 1995;280:343–346. doi: 10.1016/0014-2999(95)00298-y. [DOI] [PubMed] [Google Scholar]
- 3.Aguirre N, Barrionuevo M, Ramirez MJ, Del Rio J, Lasheras B. Alpha-lipoic acid prevents 3,4-methylenedioxy-methamphetamine (MDMA)-induced neurotoxicity. Neuroreport. 1999;10:3675–3680. doi: 10.1097/00001756-199911260-00039. [DOI] [PubMed] [Google Scholar]
- 4.Gudelsky GA. Effect of ascorbate and cysteine on the 3,4-methylenedioxymethamphetamine-induced depletion of brain serotonin. J Neural Transm. 1996;103:1397–1404. doi: 10.1007/BF01271253. [DOI] [PubMed] [Google Scholar]
- 5.Cadet JL, Ladenheim B, Hirata H, Rothman RB, Ali S, Carlson E, et al. Superoxide radicals mediate the biochemical effects of methylenedioxymethamphetamine (MDMA): evidence from using CuZn-superoxide dismutase transgenic mice. Synapse. 1995;21:169–176. doi: 10.1002/syn.890210210. [DOI] [PubMed] [Google Scholar]
- 6.Montiel-Duarte C, Ansorena E, Lopez-Zabalza MJ, Cenarruzabeitia E, Iraburu MJ. Role of reactive oxygen species, glutathione and NF-kappaB in apoptosis induced by 3,4-methylenedioxymethamphetamine (‘ecstasy’) on hepatic stellate cells. Biochem Pharmacol. 2004;67:1025–1033. doi: 10.1016/j.bcp.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 7.Ninkovic M, Malicevic Z, Selakovic V, Simic I, Vasiljevic I. N-methyl–3,4-methylenedioxyamphetamine-induced hepatotoxicity in rats: oxidative stress after acute and chronic administration. Vojnosanit Pregl. 2004;61:125–131. doi: 10.2298/vsp0402125n. [DOI] [PubMed] [Google Scholar]
- 8.Schifano F, Oyefeso A, Webb L, Pollard M, Corkery J, Ghodse AH. Review of deaths related to taking ecstasy, England and Wales, 1997-2000. BMJ. 2003;326:80–81. doi: 10.1136/bmj.326.7380.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milroy CM, Clark JC, Forrest AR. Pathology of deaths associated with ‘ecstasy’ and ‘eve’ misuse. J Clin Pathol. 1996;49:149–153. doi: 10.1136/jcp.49.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duflou J, Mark A. Aortic dissection after ingestion of ‘ecstasy’ (MDMA) Am J Forensic Med Pathol. 2000;21:261–263. doi: 10.1097/00000433-200009000-00016. [DOI] [PubMed] [Google Scholar]
- 11.Lai TI, Hwang JJ, Fang CC, Chen WJ. Methylene 3, 4 dioxymethamphetamine-induced acute myocardial infarction. Ann Emerg Med. 2003;42:759–762. doi: 10.1016/s0196-0644(03)00511-0. [DOI] [PubMed] [Google Scholar]
- 12.Madhok A, Boxer R, Chowdhury D. Atrial fibrillation in an adolescent–the agony of ecstasy. Pediatr Emerg Care. 2003;19:348–349. doi: 10.1097/01.pec.0000092582.40174.38. [DOI] [PubMed] [Google Scholar]
- 13.Henry JA, Jeffreys KJ, Dawling S. Toxicity and deaths from 3,4-methylenedioxymethamphetamine (‘ecstasy’) Lancet. 1992;340:384–387. doi: 10.1016/0140-6736(92)91469-o. [DOI] [PubMed] [Google Scholar]
- 14.Qasim A, Townend J, Davies MK. Ecstasy induced acute myocardial infarction. Heart. 2001;85:E10. doi: 10.1136/heart.85.6.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Badon LA, Hicks A, Lord K, Ogden BA, Meleg-Smith S, Varner KJ. Changes in cardiovascular responsiveness and cardiotoxicity elicited during binge administration of Ecstasy. J Pharmacol Exp Ther. 2002;302:898–907. doi: 10.1124/jpet.302.3.898. [DOI] [PubMed] [Google Scholar]
- 16.Patel MM, Belson MG, Wright D, Lu H, Heninger M, Miller MA. Methylenedioxymethamphetamine (ecstasy)-related myocardial hypertrophy: an autopsy study. Resuscitation. 2005;66:197–202. doi: 10.1016/j.resuscitation.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 17.Nishida N, Ikeda N, Kudo K, Esaki R. Sudden unexpected death of a methamphetamine abuser with cardiopulmonary abnormalities: a case report. Med Sci Law. 2003;43:267–271. doi: 10.1258/rsmmsl.43.3.267. [DOI] [PubMed] [Google Scholar]
- 18.Varner KJ, Ogden BA, Delcarpio J, Meleg-Smith S. Cardiovascular responses elicited by the ‘binge’ administration of methamphetamine. J Pharmacol Exp Ther. 2002;301:152–159. doi: 10.1124/jpet.301.1.152. [DOI] [PubMed] [Google Scholar]
- 19.Wijetunga M, Seto T, Lindsay J, Schatz I. Crystal methamphetamine-associated cardiomyopathy: tip of the iceberg? J Toxicol Clin Toxicol. 2003;41:981–986. doi: 10.1081/clt-120026521. [DOI] [PubMed] [Google Scholar]
- 20.McCann UD, Ridenour A, Shaham Y, Ricaurte GA. Serotonin neurotoxicity after (+/-)3,4-methylenedioxymethamphetamine (MDMA; ‘ecstasy’): a controlled study in humans. Neuropsychopharmacology. 1994;10:129–138. doi: 10.1038/npp.1994.15. [DOI] [PubMed] [Google Scholar]
- 21.Tiangco DA, Lattanzio FA, Jr, Osgood CJ, Beebe SJ, Kerry JA, Hargrave BY. 3,4-Methylenedioxymethamphetamine activates nuclear factor-kappaB, increases intracellular calcium, and modulates gene transcription in rat heart cells. Cardiovasc Toxicol. 2005;5:301–310. doi: 10.1385/ct:5:3:301. [DOI] [PubMed] [Google Scholar]
- 22.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 23.Battaglia G, Brooks BP, Kulsakdinun C, De Souza EB. Pharmacologic profile of MDMA (3,4-methylenedioxymethamphetamine) at various brain recognition sites. Eur J Pharmacol. 1988;149:159–163. doi: 10.1016/0014-2999(88)90056-8. [DOI] [PubMed] [Google Scholar]
- 24.Commins DL, Vosmer G, Virus RM, Woolverton WL, Schuster CR, Seiden LS. Biochemical and histological evidence that methylenedioxymethylamphetamine (MDMA) is toxic to neurons in the rat brain. J Pharmacol Exp Ther. 1987;241:338–345. [PubMed] [Google Scholar]
- 25.Ricaurte GA, McCann UD. Neurotoxic amphetamine analogues: effects in monkeys and implications for humans. Ann N Y Acad Sci. 1992;648:371–382. doi: 10.1111/j.1749-6632.1992.tb24586.x. [DOI] [PubMed] [Google Scholar]
- 26.Dowling GP, McDonough ET, 3rd, Bost RO. ‘Eve’ and ‘Ecstasy’. A report of five deaths associated with the use of MDEA and MDMA. Jama. 1987;257:1615–1617. doi: 10.1001/jama.257.12.1615. [DOI] [PubMed] [Google Scholar]
- 27.Rothstein EC, Byron KL, Reed RE, Fliegel L, Lucchesi PA. H2O2-induced Ca overload in NRVM involves ERK1/2 MAP kinases: role for an NHE-1-dependent pathway. Am J Physiol Heart Circ Physiol. 2002;283:H598–H605. doi: 10.1152/ajpheart.00198.2002. [DOI] [PubMed] [Google Scholar]
- 28.Wei S, Rothstein EC, Fliegel L, Dell'Italia LJ, Lucchesi PA. Differential MAP kinase activation and Na/H+ exchanger phosphorylation by H2O2 in rat cardiac myocytes. Am J Physiol Cell Physiol. 2001;281:C1542–C1550. doi: 10.1152/ajpcell.2001.281.5.C1542. [DOI] [PubMed] [Google Scholar]
- 29.Huie RE, Padmaja S. The reaction of no with superoxide. Free Radic Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 30.Liechti ME, Saur MR, Gamma A, Hell D, Vollenweider FX. Psychological and physiological effects of MDMA (‘ecstasy’) after pretreatment with the 5-HT(2) antagonist ketanserin in healthy humans. Neuropsychopharmacology. 2000;23:396–404. doi: 10.1016/S0893-133X(00)00126-3. [DOI] [PubMed] [Google Scholar]
- 31.Mas M, Farre M, de la Torre R, Roset PN, Ortuno J, Segura J, et al. Cardiovascular and neuroendocrine effects and pharmacokinetics of 3, 4-methylenedioxymethamphetamine in humans. J Pharmacol Exp Ther. 1999;290:136–145. [PubMed] [Google Scholar]
- 32.Vollenweider FX, Gamma A, Liechti M, Huber T. Psychological and cardiovascular effects and short-term sequelae of MDMA (‘ecstasy’) in MDMA-naive healthy volunteers. Neuropsychopharmacology. 1998;19:241–251. doi: 10.1016/S0893-133X(98)00013-X. [DOI] [PubMed] [Google Scholar]
- 33.Mihm MJ, Yu F, Reiser PJ, Bauer JA. Effects of peroxynitrite on isolated cardiac trabeculae: selective impact on myofibrillar energetic controllers. Biochimie. 2003;85:587–596. doi: 10.1016/s0300-9084(03)00090-7. [DOI] [PubMed] [Google Scholar]
- 34.Mihm MJ, Yu F, Carnes CA, Reiser PJ, McCarthy PM, Van Wagoner DR, et al. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation. 2001;104:174–180. doi: 10.1161/01.cir.104.2.174. [DOI] [PubMed] [Google Scholar]
- 35.Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca and heart failure: roles of diastolic leak and Ca2+ transport. Circ Res. 2003;93:487–490. doi: 10.1161/01.RES.0000091871.54907.6B. [DOI] [PubMed] [Google Scholar]
- 36.Lokuta AJ, Maertz NA, Meethal SV, Potter KT, Kamp TJ, Valdivia HH, et al. Increased nitration of sarcoplasmic reticulum Ca-ATPase in human heart failure. Circulation. 2005;111:988–995. doi: 10.1161/01.CIR.0000156461.81529.D7. [DOI] [PubMed] [Google Scholar]
- 37.Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schoneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca-ATPase in skeletal muscle. Biochem J. 1999;340:657–669. [PMC free article] [PubMed] [Google Scholar]
- 38.Pearce LL, Kanai AJ, Epperly MW, Peterson J. Nitrosative stress results in irreversible inhibition of purified mitochondrial complexes I and III without modification of cofactors. Nitric Oxide. 2005;13:254–263. doi: 10.1016/j.niox.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Guidarelli A, Fiorani M, Cantoni O. Enhancing effects of intracellular ascorbic acid on peroxynitrite-induced U937 cell death are mediated by mitochondrial events resulting in enhanced sensitivity to peroxynitrite-dependent inhibition of complex III and formation of hydrogen peroxide. Biochem J. 2004;378:959–966. doi: 10.1042/BJ20031167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- 41.Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med. 2002;33:1451–1464. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- 42.Jiang JP, Downing SE. Catecholamine cardiomyopathy: review and analysis of pathogenetic mechanisms. Yale J Biol Med. 1990;63:581–591. [PMC free article] [PubMed] [Google Scholar]
- 43.Carvalho M, Remiao F, Milhazes N, Borges F, Fernandes E, Monteiro Mdo C, et al. Metabolism is required for the expression of ecstasy-induced cardiotoxicity in vitro. Chem Res Toxicol. 2004;17:623–632. doi: 10.1021/tx049960f. [DOI] [PubMed] [Google Scholar]
- 44.Carvalho M, Remiao F, Milhazes N, Borges F, Fernandes E, Carvalho F, et al. The toxicity of N-methyl-alpha-methyldopamine to freshly isolated rat hepatocytes is prevented by ascorbic acid and N-acetylcysteine. Toxicology. 2004;200:193–203. doi: 10.1016/j.tox.2004.03.016. [DOI] [PubMed] [Google Scholar]