

Abstract
Osteosarcoma is the most frequent type of primary bone cancer in children and adolescents. These malignant osteoid forming tumors are characterized by their uncontrolled hyperproliferation. Here, we investigate the role of Ca2+/calmodulin-dependent protein kinase II (CaMKII) in the growth of human osteosarcoma. We show that α-CaMKII is expressed in human osteosarcoma cell lines and in primary osteosarcoma tissue derived from patients. The pharmacologic inhibition of CaMKII in MG-63 and 143B human osteosarcoma cells by KN-93 resulted in an 80 and 70% decrease in proliferation, respectively, and induced cell cycle arrest in the G0/G1 phase. The in vivo administration of KN-93 to mice xenografted with human osteosarcoma cells significantly decreased intratibial and subcutaneous tumor growth. Mechanistically, KN-93 and α-CaMKII siRNA increased p21(CIP/KIP) gene expression, protein levels, and decreased the phosphorylation of retinoblastoma protein and E2F transactivation. Furthermore, the inhibition of CaMKII decreased membrane-bound Tiam1 and GTP-bound Rac1, which are known to be involved in p21 expression and tumor growth in a variety of solid malignant neoplasms. Our results suggest that CaMKII plays a critical role in the growth of osteosarcoma, and its inhibition could be an attractive therapeutic target to combat conventional high-grade osteosarcoma in children.
Keywords: osteosarcoma, CaMKII, cell cycle, osteoblasts, p21, Rac1
Osteosarcomas are among the most frequent primary bone cancers in humans, excluding hemopoietic malignancies, affecting mainly children and adolescents. They are highly aggressive and lethal if left untreated.1 These malignant tumors commonly occur in the metaphyseal region of long bones and are divided into three broad categories, depending on the predominant matrix produced.2,3 About 50% show large amounts of osteoid or bone produced by ‘osteoblast-like’ cells, and is described as ‘osteoblastic osteosarcoma’. About 35% of all osteosarcomas have a predominantly chondroid matrix mixed with osteoid.4 The remainder, ‘fibroblastic osteosarcoma’, show a predominant spindle cell pattern, where osteoid production is found only in small foci.5
Osteosarcomas provide one of the best examples of uncontrolled proliferation and deregulated cellular growth. Cell cycle progression is regulated by cyclins, cyclin-dependent kinases (CDKs) and CDK inhibitors (CDKIs).6,7 CDKs are serine/threonine kinases, which associate with specific cyclin partners and phosphorylate key substrates to facilitate cell cycle progression. Cyclin/CDK complexes phosphorylate a number of specific targets; the best characterized being the retinoblastoma family proteins, Rb, p107 and p130. Phosphorylated Rb proteins dissociate from E2F family members, allowing active E2F to regulate the expression of genes required for cell cycle progression.6,7 This process is counteracted by two CKI families that prevent the activation of the kinases and uncontrolled replication, the Cip/Kip (p21, p27 and p57) and the INK4 (p15, p16, p18 and p19) families.7 These regulatory proteins are reported to play a role in the uncontrolled growth of osteosarcoma, including p21. The expression of p21 is inversely associated with osteosarcoma tumor growth.8 Furthermore, hyperproliferative osteosarcoma tumors were shown to lack p21 expression while the overexpression of p21 in osteosarcoma cell lines was shown to induce growth inhibition.9
Ca2+ is a ubiquitous intracellular messenger responsible for controlling numerous cellular processes. Calcium/calmodulin-dependent kinase II (CaMKII) is a multifunctional serine/threonine kinase that is critical for decoding Ca2+ signal transduction, and is highly expressed in neuronal cells as well as osteoblasts.10,11 The role of CaMKII in cancer is also been described. CaMKII activates Akt and regulates survival of c-Myc overexpressing mouse mammary carcinoma.12 Furthermore, it has been shown that the inhibition of CaMKII reduces cyclin D1 and enhances the association of p27 with CDK2 that results in a G1 phase arrest in fibroblasts.13 These reports suggest that CaMKII plays a central role in the control of cell cycle machinery, through regulation of cyclins and CDKs.
Rac1 is a member of Rho-GTPases family that includes RhoA, RhoB, RhoC, Rac1, Rac2 and Cdc42. These proteins are involved in the regulation of cytoskeletal reorganization, membrane trafficking, transcriptional regulation and cellular growth and proliferation.14,15 Rho-GTPases family is regulated by the binding of guanine nucleotides. They are inactive when bound to GDP and active when bound to GTP. Guanine nucleotide exchange factors (GEFs) exchange GDP for GTP, thus rendering the G proteins active.16 T-lymphoma and metastasis gene 1 (Tiam1) is the specific GEF that activates Rac1.16 Tiam1 is predominantly localized in the cytoplasm and upon phosphorylation (activation) by kinases such as CaMKII, it translocates to the cell surface membrane.17,18 The phosphorylation of Tiam1 and the activation of Rac1 have been shown to promote the growth of several tumors including mammary carcinoma.18–20
The current study examined the role of α-CaMKII in the growth of a human osteosarcoma in vivo and in vitro. Here, we show that α-CaMKII was expressed in osteosarcoma cell lines and primary human osteosarcoma tissue derived from patients. The pharmacologic inhibition of CaMKII by KN-93 arrested osteosarcoma cells in G0/G1 phase of the cell cycle. Furthermore, the in vivo inhibition of CaMKII by KN-93 significantly reduced the size of osteosarcoma tumors. KN-93 increased p21 expression and decreased the phosphorylation of Rb and E2F transactivation. Finally, the inhibition CaMKII decreased the activation of Tiam1 and its substrate, Rac1. Taken together, our work increases the understanding of the mechanisms by which CaMKII regulates the growth of osteosarcoma, and should have important implications in the development of therapeutic intervention strategies to combat the life-threatening condition of osteosarcoma while dramatically improving patients' survival.
Materials and Methods
Reagents
Antibodies against α-CaMKII, p-α-CaMKII (Thr287), p21, Rb, p-Rb (Thr876/877), cyclin D1, cyclin D2, cyclin D3, CDK 4, Tiam1, integrin β1 and actin; α-CaMKII and p21 siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 2-[N-(2-Hydroxyethyl)]-N-(4-methoxybenzene-sulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (KN-93), a cell-permeable inhibitor of CaMKII that selectively binds to the CaM binding site of CaMKII and prevents the association of CaM with CaMKII and 2-[N-(4-Methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (KN-92), an inactive analogue of KN-93 were obtained from Calbiochem (San Diego, CA, USA) and were dissolved in dimethylsulfoxide (DMSO).
Cells and Cell Culture
Human osteosarcoma cells (MG-63 and 143B) and human mesenchymal stem cells (hMSCs) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and Cambrex Corp (East Rutherford, NJ, USA), respectively. Cells were maintained in DMEM supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA, USA). All cell cultures were maintained at 37°C with 5% CO2.
RNA Extraction and RT-PCR
Total RNA was extracted using the Trizol method as recommended by the manufacturer (Invitrogen, Carlsbad, CA, USA). A 1 μg weight of RNA was reverse-transcribed using Maloney murine leukemia virus reverse transcriptase (M-MLV RT), according to the instruction provided by the manufacturer (Clontech, Mountain View, CA, USA), and the equivalent of 100 ng was used for RT-PCR. The sequences and conditions for the specific primers used in this study were previously described.21 Amplified products were then electrophoretically separated on 1.5% agarose gel. Digital images were obtained and analyzed using the Kodak Digital Science one-dimensional image analysis software (Eastman Kodak Co., Rochester, NY, USA). TaqMan real-time RT-PCR analysis was performed as described previously.22 Expression of the 18S rRNA subunit served as a control. The sequences and conditions for the specific primers used in this study were as previously described.23
MTT Cell Proliferation Assay
MG-63 or 143B cells were seeded in 96-well plates at a density of 1 × 104 cells/well and cultured for 24 h, followed by KN-93 (10 μM) treatment for 24 or 48 h. At the end of incubation, 20 μl of 5 mg/ml MTT was added to each well, followed by incubation for another 4 h. The medium was then aspirated and 150 μl DMSO was added into each well. Optical density was measured at 550 nm using an ELISA plate reader.
Cell Cycle Analysis by Flow Cytometry
At the end of the study, cells were collected in cold PBS and fixed with −20°C ethanol for 1 h then stained with a propidium iodide (PI) solution (50 μg/ml PI, 0.5 mg/ml RNase in TBST buffer) for 30 min. DNA profiles of PI-stained cells were analyzed with a Becton Dickinson FACScan (San Jose, CA, USA) and the number of cells in each cell cycle phase was determined by analyzing data using ModFit LT 3.1 software.
Colony Formation Assay
MG-63 cells were seeded at 2 × 104 cells/well in 0.3 ml growth factor reduced Matrigel™ Matrix (BD Biosciences, Bedford, MA, USA) in a 24-well plate. The Matrigel™ was covered with DMEM medium supplemented with DMSO, 10 μM KN-92 or 10 μM KN-93. Medium and treatment were replaced every day for 7 days. Images of tumor colonies were obtained at × 200 magnification.
Transient Transfections and Luciferase Reporter Assay
MG-63 cells were plated at a density of 2 × 104 cells/cm2 in six-well plates. Twenty-four hours after plating, cells were transfected with 1.5 μg of luciferase reporter construct driven by the E2F consensus sequences,24 and expression vectors containing wild-type (WT) α-CaMKII, dominant-negative mutant α-CaMKII K42M25 or a CMV-β-galactosidase reporter construct (as a control). Reporter activity was measured using a luciferase (Promega) or β-galactosidase (Clontech) assay system and a luminometer.10
Gene Silencing by siRNA
MG-63 cells were plated at a density of 2 × 104 cells/cm2 in six-well plates. α-CaMKII, p21 or control non-functional siRNA (Santa Cruz Biotechnology) were transfected into the cells using TransIT-TKO transfection reagent as recommended by the manufacturer (Mirus, Madison, WI, USA). The medium was changed 24 h post-transfection, and the cells were cultured with fresh medium. Cells were harvested 72 h later.
Animals and Tumor Cell Inoculation
Six-week-old male Balb/c (nu/nu) mice were used in these studies, with the approval of the UAB Institutional Animal Care and Use Committee. MG-63 cells were prepared from sub-confluent cultures.26 Anesthetized mice were prepared or tumor injection. Cells (2 × 105 cells in 20 μl PBS) were intratibially injected using a TB syringe and a 27 g needle.27 The knee was flexed, and the needle inserted into the tibia, boring the needle through the epiphysis and epiphyseal growth plate for delivery of the cells into the metaphysis. In addition, MG-63 osteosarcoma cells were resuspended in fresh culture media, mixed 1:1 with BD Matrigel™ (1×106 cells/100 μl), and then injected in the flank of the mice using a 23-gauge needle. Mice were divided into two groups that either received a daily intraperitoneal injection of vehicle alone (PBS) or 1 mg/kg of water-soluble KN-93 (Calbiochem, EMD Biosciences, USA). Subcutaneous tumors were evaluated by measuring their volume 6 and 42 days after tumor injections. At the end of the study (after 42 days), complete autopsies were conducted (on all animals) with a dissecting microscope. Subcutaneous tumors were excised, formalin fixed, EDTA decalcified and paraffin embedded. For the intratibial tumors, the hind limbs were excised and microtomography (μ-CT) analysis was performed on selected samples. Limbs were formalin fixed, EDTA decalcified and paraffin embedded. All tissues were sectioned and stained with hematoxylin and eosin (H&E) for histological evaluation of the tumors.28
Immunohistochemistry and Human Tissue Samples
Primary human osteosarcoma tissue samples from four patients were obtained from the Pathology Department of the University of Alabama at Birmingham Hospital, with the approval of the Institutional Review Board. Tissues (human and animal) were formalin fixed, decalcified in EDTA and histologically processed for paraffin embedding and immunostaining, as previously described.29
Rac1 Activity Assay
Rac1 activity was determined by a pull-down assay according to the manufacturer's instructions (Rac activity assay kit; Upstate Biotechnology). Total lysates and precipitates were examined by performing SDS-PAGE and Western blot analysis using mouse monoclonal antibody against Rac1 (Upstate).
Statistical Analysis
Statistical analyses were performed using the Student's t-test. Experiments were repeated at least three times, unless otherwise stated. Values were expressed as mean±s.e. Significance was confirmed at P<0.05.
Results
The Expression and Activation of α-CaMKII is Increased in Human Osteosarcoma
To characterize the expression of α-CaMKII in osteosarcoma, we first examined the gene expression of CaMKII isoforms in well-characterized osteosarcoma cell lines (MG-63 and 143B) and compared it to primary hMSCs that are non-transformed pre-osteoblastic cells. Cells were cultured for 48 h until they reached 80% confluence and then processed for RNA and cytoplasmic protein extraction. In Figure 1a we show, by RT-PCR, that all four isoforms of CaMKII (α, β, γ, δ) were expressed in MG-63 cells, 143B cells as well as in hMSCs. However, the levels of the α-isoform gene expression were significantly increased (460%) in MG-63 cells, but not in 143B osteosarcoma cells, as compared to hMSCs (Figure 1a). As discrepancies between gene expression and protein levels in several types of cancer have previously been reported, we then examined the levels of total and active α-isoform proteins in hMSC and osteosarcoma cell lines. We show by Western blotting techniques that MG-63 and 143B osteosarcoma cells express high levels of the total and the phosphorylated and active form of α-CaMKII as compared to hMSCs (Figure 1b). Furthermore, by immunohistochemistry, the phosphorylated α-CaMKII protein was also increased in osteosarcoma cell lines and displays distinct speckled peri-nuclear localization (Figure 1c). Interestingly, hMSC, which are osteoblast progenitors, also displayed a similar pattern of expression but with less intensity. Negative control (NC) studies were performed using rabbit IgG, instead of primary antibody, confirming the specificity of the p-α-CaMKII antibody.
Figure 1.
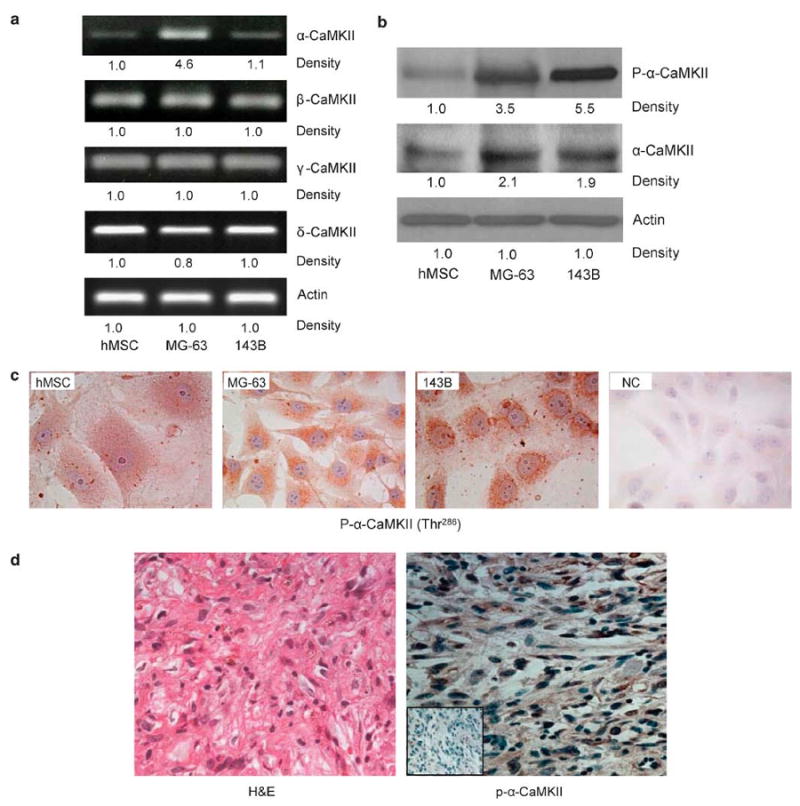
Human osteosarcoma expresses high levels of α-CaMKII. MG-63,143B and hMSC cells were used. Representative images from three experiments of (a) RT-PCR using CaMKII isoforms or actin-specific primers. (b) Western blots using antibodies directed against phosphorylated and total α-CaMKII and actin (c) Immunohistochemistry staining using a specific antibody directed against p-α-CaMKII. (d) Primary human osteosarcoma tissue was surgically removed, formalin-fixed and paraffin-embedded. H&E staining (left) shows osteosarcoma. Immunohistochemical staining (right) was performed using an antibody against p-α-CaMKII (brown), counterstained with hematoxylin (blue). NC, without immunoreactivity, is shown in the low left inset. Photomicrographs were obtained at × 400 magnification, and are representative of the four different patients.
In order to examine the in vivo expression of α-CaMKII in primary osteosarcoma tissue, human samples from four patients diagnosed with osteosarcoma were obtained from the Pathology Department at the University of Alabama at Birmingham after appropriate IRB approval. H&E staining (Figure 1d, left photograph) confirmed the diagnosis of osteosarcoma. In the right panel we show that, similar to MG-63 and 143B cells, primary human osteosarcoma expressed high levels of active α-CaMKII (Figure 1d, right photograph).
The CaMKII Antagonist, KN-93, Inhibits Growth of Human Osteosarcoma Cells
Osteosarcoma is characterized by its uncontrolled proliferative capability and CaMKII has been previously shown to be critical for cell proliferation in several biological systems.30 In order to determine the effects of CaMK antagonist (KN-93) on α-CaMKII activation; we examined the phosphorylation of α-CaMKII after KN-93 treatment. We also used an inactive analogue of KN-93 (KN-92) as an additional control. MG-63 osteosarcoma cells were treated with DMSO (0.1% v/v), KN-92 (10 μM) or KN-93 (10 μM) for 24 h followed by protein extraction. Western blotting for phosphorylated α-CaMKII was performed using antibody directed against phosphorylated α-CaMKII (Thr-286). As is clearly shown in Figure 2a, KN-93 effectively inhibited the phosphorylation of α-CaMKII in MG-63 osteosarcoma cells as compared to DMSO- or KN-92-treated cells. Next, we examined the effects of KN-93 on the proliferation of MG-63 and 143B osteosarcoma cells by an MTT assay. Here, we show that treatment with KN-93 (10 μM) for 24 and 48 h resulted in 40 and 80% decrease in the proliferation of MG-63 cells, and 35 and 70% decrease in the proliferation of 143B osteosarcoma cells (Figure 2b). This inhibition of proliferation by KN-93 is due to a cell cycle arrest and results in a 40 and 57% increase in the number of cells that are in the G0/G1 phase of the cell cycle, compared to untreated and KN-92-treated cells after 24 and 48 h, in MG-63 osteosarcoma cells, respectively (Figure 2c). This treatment did not alter apoptosis in these cells, as determined by Annexin V-FITC and PI staining followed by flow cytometric analyses (data not shown). Finally, in order to determine the effects of CaMK inhibition by KN-93 on tumor colony formation, MG-63 cells were cultured in growth factor reduced Matrigel and treated with KN-93 (10 μM) for 7 days. We demonstrate that KN-93 significantly decreased (87%) the number and size of tumor colonies when MG-63 cells are cultured in a three-dimensional (3D) growth factor-reduced Matrigel matrix as compared to untreated or KN-92-treated osteosarcoma cells (Figure 2d).
Figure 2.
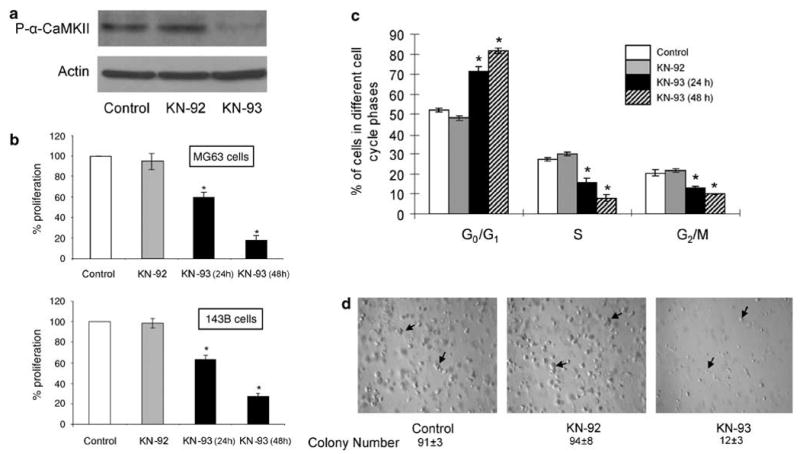
The CaMKII antagonist, KN-93, arrests the growth of human osteosarcoma cells. (a) MG-63 cells were treated with either DMSO (control), KN-92 (10 μM) or KN-93 (10 μM) for 24 h. Cells were then lysed and protein extracts were separated by SDS-PAGE. Western blots were conducted using antibodies directed against p-α-CaMKII or actin. (b) MG-63 or 143B cells were cultured for 24 h and then treated for 24 and 48 h with DMSO (0.1% v/v) (control), KN-92 (10 μM) or KN-93 (10 μM). At the end of the study, an MTT proliferation assay was performed. Values were obtained from three separate experiments and represent the mean±s.e. *P<0.01. (c) MG-63 Cells were stained with PI and flow cytometric analysis of the DNA content was performed. Analysis of the cell cycle was performed using ModFit LT 3.1 software. Values were obtained from three separate experiments and represent the mean±s.e. *P<0.01. (d) MG-63 cells were seeded in growth factor-reduced Matrigel, supplemented with DMSO, 10 μM KN-92 or 10μM KN-93. Medium and treatment were replaced every day for 7 days. Arrows indicate tumor colonies. Photographs are representative of three different experiments. Colonies were counted from three randomly selected microscopic fields from three different experiments.
The CaMKII Antagonist, KN-93, Modulates the Expression and Activation of Cell Cycle Regulatory Proteins in MG-63 Osteosarcoma Cells
To elucidate the mechanism by which CaMKII inhibition causes cell cycle arrest in osteosarcoma cells, we examined the effects of KN-93 on cell cycle regulatory proteins, specifically those related to the transition from G0/G1 to the S phase. Our data demonstrate that the inhibition of CaMKII activation by KN-93 increases p21 protein levels in a time- and dose-dependent manner (Figure 3a, upper and lower panels). The increase in p21 protein levels in response to KN-93 treatment was due to an increase in p21 gene expression, because the general inhibition of transcription by actinomycin D prevents KN-93 from increasing p21 protein levels (Figure 3b, lower panel). Interestingly, the expression of other cell cycle regulatory molecules such as cyclin D1, D2, D3, CDK 4 and p27 did not change in response to KN-93 treatment (data not shown).
Figure 3.
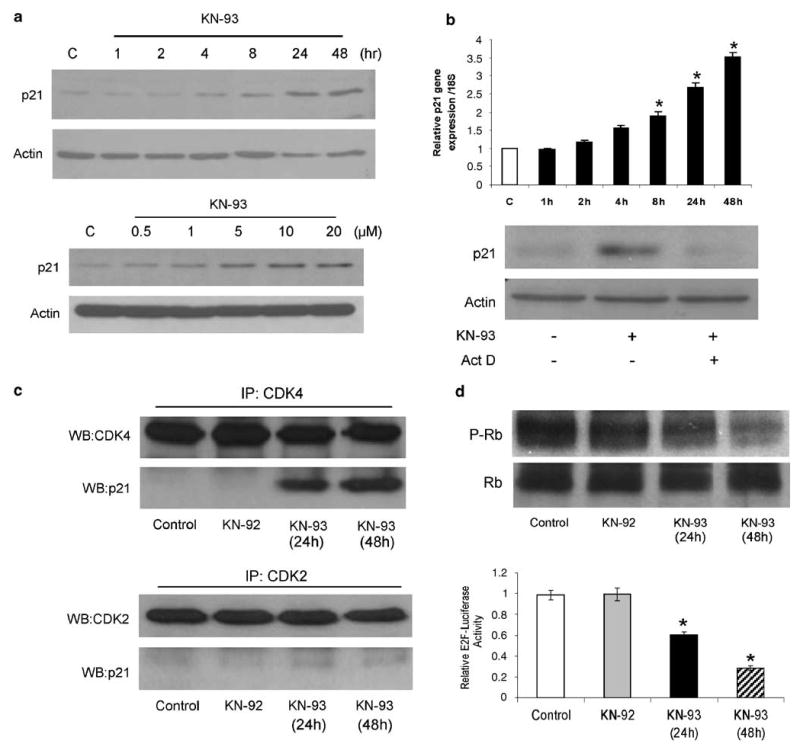
The CaMKII antagonist, KN-93, controls the expression and activation of cell cycle regulatory proteins in osteosarcoma. (a) MG-63 cells were treated with 10 μM KN-93 for the indicated time (upper panel), or treated for 24 h with the indicated concentrations of KN-93 (lower panel). A representative immunoblot of p21 and actin from three separate experiments is shown. (b) Real-time PCR was performed using primers specific for p21 or 18S rRNA (upper panel). Values were obtained from three separate experiments and represent the mean±s.e. of p21 messenger RNA expression relative to 18 S rRNA expression. *P<0.01. (Lower panel) MG-63 cells were treated with DMSO (0.1%), KN-93 (10 μM) or KN-93 (10 μM) + actinomycin D (Act D) (0.8 μM) for 12 h. Cells were then lysed and immunoblotting was performed using p21- and actin-specific antibodies. Representative images from three separate experiments are shown. (c) MG-63 cells were treated with DMSO (control), KN-92 (10 μM) or KN-93 (10 μM) for the indicated time. Protein was extracted and immunoprecipitation was performed using anti-CDK2 or anti-CDK4 antibodies, followed by Western blotting for p21 or CDK4 (upper panel), and p21 or CDK2 (lower panel). Representative images from three separate experiments are shown. (d) MG-63 cells were treated with either DMSO (control), KN-92 (10 μM) or KN-93 (10 μM) for 24 or 48 h. (Upper panel) Western blots using antibodies directed against p-Rb or total Rb were performed. (Lower panel) MG-63 cells were transfected with luciferase reporter plasmids driven by the E2F sequences and a CMV-β-galactosidase reporter constructs using Lipofectamine for 24 h. Cells were then treated with DMSO (control), KN-92 (10 μM) or KN-93 (10 μM) for 24 h. Reporter activity was then measured. Data are expressed relative to an internal control (CMV-β-galactosidase) and are the means±s.e. of three separate experiments, each performed in triplicate; *P≤0.01.
Furthermore, we examined the interaction of p21 and CDK in response to KN-93 treatment. MG-63 cells were treated with KN-93 (10 μM) for 24 and 48 h, protein was extracted and immunoprecipitation was performed using anti-CDK2 or anti-CDK4 antibodies. Immunoprecipitates were then examined by Western blotting using p21 antibody. Our results demonstrate that CDK4 and CDK2 did not interact with p21 in MG-63 osteosarcoma cells, most likely due to the lack of p21 expression in these cells. In contrast, p21 became part of the CDK4 complex 24 and 48 h after treatment with KN-93, without any change in p21/CDK2 binding (Figure 3c).
It has been shown that the Rb proteins repress the transcription of genes required for transition from G1 to S phase by directly binding to the transactivation domain of E2F and by binding to the promoter of these genes as a complex with E2F.6,7 Therefore, the relevance of the increase in p21 expression and its binding to CDK4 in MG-63 osteosarcoma cells was determined by examining the phosphorylation of Rb and the transactivation of E2F in response to KN-93 treatment. MG-63 cells were treated with DMSO (0.1% v/v), KN-92 (10 mM) or KN-93 (10 μM) for 24 and 48 h, followed by protein extraction. Western blotting for phosphorylated and total Rb was performed. Here, we show that KN-93 decreased the phosphorylation of Rb in MG-63 osteosarcoma cells compared to DMSO- or KN-92-treated cells (Figure 3d, upper panel). To examine the effect of KN-93 on E2F transactivation, MG-63 cells were transfected with an E2F luciferase reporter construct (generously provided by Dr Peter Glazer, Yale University, New Haven, CT, USA24). MG-63 cells were then treated with KN-93 for 24 and 48 h, followed by measurement of luciferase activity. E2F transactivation was decreased 40 and 70% after treatment with KN-93 for 24 and 48 h, respectively. Treatment with either DMSO or KN-92 did not change E2F transactivation over the same time period (Figure 3d, lower panel).
Specific Inhibition of the α-Isoform of CaMKII Increases p21 Levels and Decreases Cell Growth
KN-93 is a general CaMK antagonist that inhibits the activation of different CaMK isoforms. Therefore, in order to examine the specific role of α-CaMKII in the growth of osteosarcoma, we transfected MG-63 cells with an siRNA that specifically silences the expression of α-CaMKII. Furthermore, we transfected MG-63 with a ‘kinase-dead’ α-CAMKII (K42 M) that was generously provided by Dr Chunhai Hao, Emory University School of Medicine, Atlanta, GA, USA.25 This kinase has been previously described and extensively used to inhibit the ability of α-CaMKII to phosphorylate its substrates. Overexpression of this mutant construct exerts a dominant-negative effect, as it blocks the function of the endogenous α-CaMKII.25,31 Here, we show that α-CaMKII siRNA was effective in inhibiting the activation of the α-CaMKII enzyme compared to control siRNA-transfected cells (Figure 4a, upper panel). Furthermore, we demonstrate that the overexpression of a WT α-CaMKII and a kinase-dead α-CaMKII (K42M) constructs in MG-63 cells results in expression of equal levels of α-CaMKII protein levels compared to non-transfected cells (NT) (Figure 4a, lower panel). Our data also show that similar to KN-93, the specific inhibition of α-CaMKII expression or activation increased p21 protein levels (Figure 4b).
Figure 4.
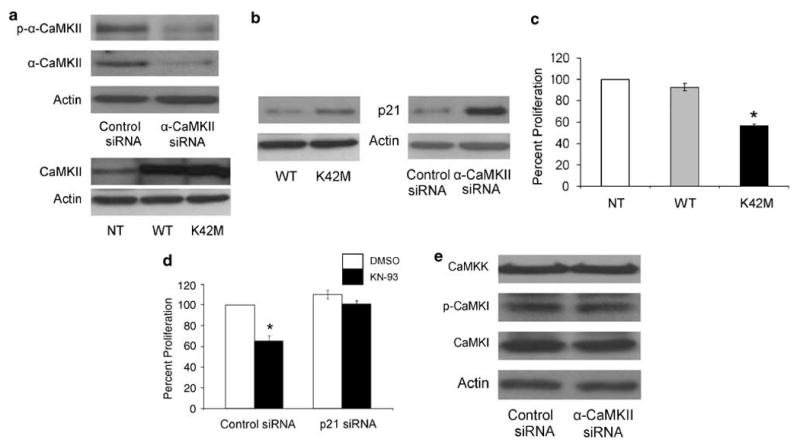
The α-isoform of CaMKII regulates the proliferation of MG-63 osteosarcoma cells and p21 protein levels. MG-63 cells were transfected with control or α-CaMKII siRNA, WT or dominant-negative α-CaMKII (K42M) plasmids 24 h after plating, and were harvested 72 h after transfection. Cells were lysed for whole-cell protein extraction. Immunoblots were developed using antibodies directed against (a) p-α-CaMKII, total α-CaMKII and actin or (b) p21 and actin. The blots are representative of three separate experiments. (c) MG-63 cells were either transfected with WT or dominant-negative α-CaMKII (K42M) plasmids 24 h after plating, or cultured without transfection (NT). MTT proliferation assay was performed 48 h after transfection. Values were obtained from three separate experiments and represent the mean±s.e; *P<0.01. (d) MG-63 cells were transfected for 24 h with control or p21 siRNA. Cells were then treated with DMSO or 10 μM KN-93 for 24 h, followed by an MTT proliferation assay. Values were obtained from three separate experiments and represent the mean±s.e. *P<0.01. (e) MG-63 cells were transfected with control or α-CaMKII siRNA. Immunoblots were developed using antibodies directed against CaMKK, p-CaMKI, CaMKI and actin. The blots are representative of three separate experiments.
Moreover, we examined the effects of inhibiting α-CaMKII activation by K42M on the proliferation of MG-63 osteosarcoma cells. Similar to KN-93, the specific inhibition of α-CaMKII by the K42M kinase-dead construct resulted in 45% inhibition of MG-63 cells' growth compared to non-transfected or WT α-CaMKII-transfected cells (Figure 4c). Finally, we determined if the upregulation of p21 expression in response to KN-93 treatment is indeed responsible for the decrease in the proliferation of osteosarcoma cells. MG-63 osteosarcoma cells were transfected with either control or p21 siRNA for 48 h. Cells were then treated with either vehicle or KN-93 for 24 h. Our results show that KN-93 (10 μM) compared to vehicle, results in a 36% decrease in the proliferation of MG-63 cells that were transfected with control siRNA. While in p21 siRNA-transfected cells, KN-93 treatment did not have any effect on proliferation (Figure 4d). It has been reported that calcium/calmodulin-dependent kinase I (CaMKI) and calcium/calmodulin-dependent kinase kinase (CaMKK) participate in the control of cell cycle progression in cancer cells.32 In order to exclude the unspecific effects of α-CaMKII siRNA on CaMKI and CaMKK, we examined the protein levels of CaMKK and CaMKI, as well as the phosphorylation level of CaMKI in α-CaMKII siRNA-transfected cells. Our results show that α-CaMKII siRNA neither affected the protein levels of CaMKK and CaMKI, nor the phosphorylation level of CaMKI (Figure 4e). Taken together, these data demonstrate that the inhibition of α-CaMKII in osteosarcoma cells caused a cell cycle arrest in a p21-dependent mechanism.
KN-93 Decreases the In Vivo Growth of Human Osteosarcoma
In order to examine the effects of KN-93 on the growth of osteosarcoma cells in vivo, we subcutaneously and intratibially injected MG-63 osteosarcoma cells into 5-week-old male athymic (nude) mice. Animals were either treated with vehicle or KN-93 (1 mg/kg). Treatment started 4 days after tumor cell inoculation and administration was by intraperitoneal injections every other day for 6 weeks. Furthermore, a control group of mice that did not receive tumor cell injection or KN-93 treatment was also maintained. At the end of the study (42 days), mice were euthanized, subcutaneous tumors were excised and tibiae were collected. Tibiae were then examined and scanned by μ-CT and the subcutaneous tumors were processed for immunohistochemical (IHC) staining. Our results demonstrate that KN-93 treatment resulted in a 35.2% reduction in the subcutaneous tumor volume compared to control (data not shown). Furthermore, KN-93 treatment dramatically reduced the size of the intratibial tumor, as indicated by the broken circle in the representative two-dimensional (upper panel) and 3D (lower panel) μ-CT images (Figure 5a). In order to examine the osteosarcoma tumors histologically, H&E and IHC staining of the subcutaneous tumors were performed. H&E staining showed no difference in the cytomorphology between the vehicle- and the KN-93-treated tumors. Furthermore, IHC using antibody directed against p-α-CaMKII demonstrate that treatment with 1 mg/kg is sufficient to inhibit the activation of α-CaMKII in vivo. Similar to the in vitro results, we demonstrate that in vivo treatment with KN-93 increased p21 protein levels and induced the hypo-phosphorylation of Rb protein in osteosarcoma tumors (Figure 5b). These findings strongly validate the role of CaMK in the expression of p21 protein, the phosphorylation of pRb and, most importantly, the control of the growth of osteosarcoma.
Figure 5.
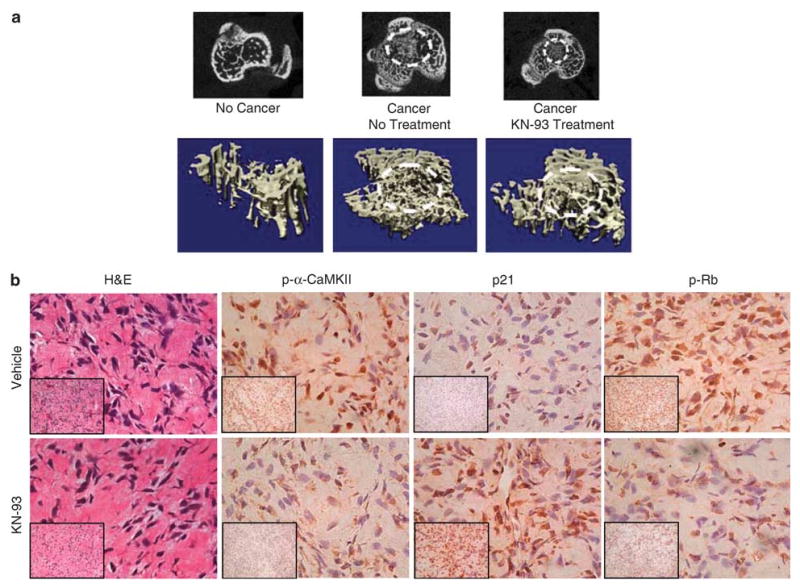
The CaMKII antagonist, KN-93, decreases the in vivo growth of human osteosarcoma. MG-63 cells were subcutaneously or intratibially injected into 5-week-old male athymic nu/nu mice that were divided into untreated and KN-93-treated groups. Mice were treated every other day for 6 weeks. (a) After mice were killed, tibiae were removed, formalin fixed and MicoCT scanning was performed. The upper panel shows a two-dimensional section in the proximal tibia, where osteosarcoma has grown (indicated by broken white circle), whereas the lower panel shows a 3D image of the cross-sectional proximal tibia also showing the tumor. (b) Subcutaneous tumors were removed, formalin fixed and paraffin embedded. H&E and IHC staining were performed for p21, p-Rb and p-α-CaMKII. All images were obtained at × 200 (lower left insets) and × 400. The results showed no change in the cytomorphology comparing the control to the KN-93-treated tumors, but upregulation of p21 and downregulation of p-α-CaMKII and p-pRb, as assessed by IHC.
CaMKII Regulates Rac1 Activation and Tiam1 Translocation in MG-63 Osteosarcoma Cells
To further understand the mechanism by which CaMKII regulates cell cycle progression, and more specifically p21 expression, we examined the activation of Rac1 GTPase and its specific guanine nucleotide exchange factor, Tiam1, in MG-63 osteosarcoma cells, in response to KN-93 treatment. MG-63 cells were cultured for 24 h and then treated with KN-93 for 1, 2, 4, 8 and 24 h. At the end of the study, cells were lysed and cytoplasmic protein was extracted followed by a Rac1 pull-down assay to extract the GTP-bound form of Rac1. In addition, cell membrane proteins were extracted and Western blotting was performed to examine Tiam1 protein levels in cell membranes. Our results demonstrate that the inhibition of CaMKII using KN-93 decreased the GTP-bound form of Rac1 by 38% after 8 h of treatment and 50% after 24 h, without any change in the total Rac1 protein levels (Figure 6a). Likewise, treatment with KN-93 decreased the phosphorylated form (membrane bound) of Tiam1 by 35% after 8 h of treatment and 40% after 24 h, without any change in the levels of another cytoplasmic membrane protein, integrin β1 (Figure 6b). Taken together, our results suggest that the inhibition of CaMKII decreased the phosphorylation of Tiam1 and its membrane localization. This then resulted in a decrease in the activation of Rac1, and ultimately an increase in the expression of p21 protein. This, therefore led to inhibiting cell cycle progression and arrest of osteosarcoma cells in the G1 phase of the cell cycle (Figure 6c).
Figure 6.
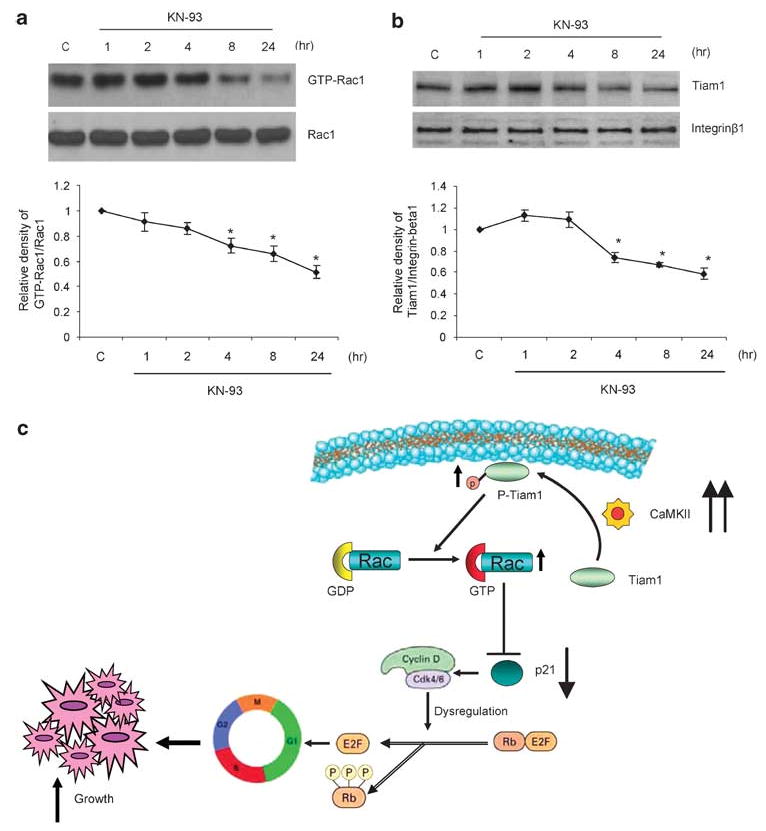
The CaMKII antagonist, KN-93, decreases the activation of Rac1 and Tiam1 in osteosarcoma cells. MG-63 cells were treated with vehicle only (control) or 10 μM KN-93 for 1, 2, 4, 8 and 24 h. (a) Rac1 activation assay was performed by a pulldown assay, whereby the activated (GTP bound) Rac1 protein is pulled down from whole-cell protein lysates with PAK-1 PBD-conjugated agarose beads. (b) Cytoplasmic membrane proteins were extracted and Western blots analyses were performed using antibodies directed against Tiam1 and integrin β1. Integrin β1 was examined as a loading control. Representative images from three experiments are shown. The band intensities of both GTP-Rac1 relative to Rac1 (a, lower panel) and Tiam 1 relative to integrin β1 (b, lower panel) are plotted as a graph. Values were obtained from three separate experiments and represent the mean±s.e; *P<0.05. (c) A proposed mechanism depicting the role of CaMKII in cell cycle progression in osteosarcoma. The increase in the expression/activation of α-CaMKII in osteosarcoma cells results in an increase in the phosphorylation of Tiam1 and its membrane localization. This increases the activation of Rac1 and ultimately inhibits the expression of p21 protein. This leads to a disruption of cell cycle regulation and results in an unregulated growth of these cells, and therefore development of osteosarcoma.
Discussion
Osteosarcoma is the most frequent type of primary bone cancer in children and adolescents. These tumors often demonstrate genetic dysregulation in cell cycle regulatory genes, as characterized by chromosomal losses, gains or mutations, resulting in altered levels and/or activation of proliferation-regulatory proteins such as p53, pRb, cyclin D1, CDK4 and MDM2.33,34
The present study demonstrates that the uncontrolled proliferation of human osteosarcoma is partly due to an increase in the protein levels and activation of α-CaMKII. Our results show that in both osteosarcoma cell lines and in primary human clinical samples, the α-isoform of CaMKII protein is highly expressed. These results are consistent with several reports describing the increased expression of different CaMKII isoforms in several neoplastic cells.12,35 However, we found that the gene expression of α-isoform of CaMKII in 143B osteosarcoma cells is not significantly different from hMSCs, whereas protein levels are higher. This discrepancy between RNA and protein levels has previously been reported in several type cancers and is likely due to differences in post-transcription mRNA half-life regulation.36–38 Our results also demonstrate that not only the expression of α-CaMKII is increased in osteosarcoma cells, but also its phosphorylation. It is known that CaMKII is a Ca2+/CaM-regulated kinase. Therefore, the increase in the expression of α-CaMKII in osteosarcoma could potentially be insignificant if it is not accompanied with a similar increase in activation. Therefore, we also demonstrate that in MG-63 and 143B osteosarcoma cells the activation of α-CaMKII is similarly high. This could be due to a direct increase in the expression levels of the kinase itself, or indirectly in response to an increase in an upstream activator such as intracellular calcium. It has been shown that osteosarcoma cells express high levels of the L-type α-Ca2+ channels.39 Ca2+ is known to be critical for the growth and proliferation of normal cells. Several reports suggest that this is achieved by the activation of the CaMK-dependent signaling pathway in response to Ca2+, which ultimately leads to the activation of cyclin protein expression and acceleration of the rate of cells' entry into the cell cycle.32 In contrast, neoplastic cells are able to proliferate in Ca2+-deficient media, suggesting that this strict requirement for Ca2+ is lost and possibly compensated by an upregulation of Ca2+ downstream targets in these transformed (neoplastic) cells.40 Indeed, several tumors such as neuroblastoma, lung, breast and colon adenocarcinoma have been shown to express high levels of different isoforms of CaMK, specifically CaMKII.35 Many studies that investigate the role of CaMK in cancer suggest that this kinase plays a critical role in the regulation of proliferation in these tumor cells.41 Taken together, these findings may indicate that in osteosarcoma, the combination of an increase in the intracellular calcium with the upregulation of α-CaMKII expression is necessary for the uncontrolled proliferative capability of this tumor.
The general inhibition of CaMK by KN-93 or its specific isoforms by siRNA has been shown to be effective in arresting cell cycle progression in many different cancers such as human breast carcinoma.42 We significantly inhibited the proliferation of MG-63 and 143B osteosarcoma cells by treating them with the CaMK antagonist, KN-93, as well as with an α-CaMKII dominant-negative construct. This decrease in proliferation was not due to an increase in apoptosis (data not shown). Our data show that KN-93 treatment for 48 h induces a growth arrest of osteosarcoma cells in the G1 phase of cell cycle. The precise mechanism by which CaMK regulates the proliferation of transformed cells is not well understood. Many studies have focused on the relationship between CaMK and cyclin D1, due to the importance of cyclin D1 in regulating cell cycle progression from the G1 to S phase. Many studies have also shown that the inhibition of CaMK decreases the accumulation of cyclin D1 in breast cancer cells.42 In contrast, other studies, including ours, have shown that while CaMK inhibition decreases the activation of the cyclin D1/CDK4 complex, it does not affect the total levels of cyclin D1 protein.43 The decrease in cyclin D1/CDK4 activation is generally achieved by an increase in the protein levels of CDKIs such as p21 and p27. In many tumor cells, there are various dysfunctions of p21 that allows neoplastic cells to avoid arrest at the G1/S checkpoint. In this work, although the inhibition of CaMKII by KN-93 and siRNA did not decrease cyclin D1 expression (data not shown), it was successful in increasing the expression of p21 that ultimately decreased the activation of cyclin D1. This observation was supported by the decrease in the phosphorylation of Rb protein and the transactivation of E2F.
Our results demonstrate that the inhibition of CaMKII by KN-93 or by an α-CaMKII dominant-negative construct increases the expression of p21. The cellular levels of p21 are regulated by various mechanisms, including transcriptional regulation, epigenetic silencing, mRNA stability, and ubiquitin-dependent and -independent degradation of the protein.44 In this work, we show that the increase in p21 in response to inhibiting CaMKII in osteosarcoma is due to a transcriptional upregulation of p21 gene expression, as KN-93 was not able to increase p21 protein in the presence of actinomycin D, which binds to DNA and inhibits RNA synthesis. The role of p21 in the progression of the cell cycle has been somewhat controversial. While a consensus exists regarding the inhibition of CDK2-cyclinE/A activation by p21, there are several contrasting reports describing the role of p21 in regulating the activity of CDK4/6-cyclin D.45,46 Our results demonstrate that the inhibition of CaMKII upregulates p21 gene expression and protein levels, as well as its association with CDK4. We also show that the inhibition of p21 by siRNA abrogates the effects of KN-93 on the growth of MG-63 osteosarcoma cells. Our results are consistent with several reports describing the effects of increasing the expression of p21 in different mammalian cells, and demonstrating the ability of p21 to induce a G1 phase arrest, CDK4 activity inhibition and a decrease in Rb phosphorylation.47,48
The antiproliferative effects of CaMKII inhibition were not only limited to our in vitro findings. Our results demonstrate that the in vivo administration of KN-93 significantly decreases the size of osteosarcoma tumors. However, although we demonstrate an increase in p21 expression and decrease in the p-Rb in response to KN-93 treatment in vivo, we cannot conclusively determine that this is the specific mechanism by which KN-93 decreases primary osteosarcoma tumor growth. It has been shown that matrix metalloproteinases (MMP) play an important role in the growth and invasion of osteosarcomas.49 Interestingly, it was previously reported that CaMKII regulates MMP-13 gene expression.50 Therefore, it is possible that the decrease in osteosarcoma tumor size by KN-93 treatment in vivo is in response to a decrease in MMP-13 expression in combination with the upregulation of p21 gene expression. Studies to examine this hypothesis are currently ongoing in our laboratory.
Several signaling pathways are involved in tumor growth and control of cell cycle, including Rac1. It has been shown that Rac1 promotes cell cycle progression from G1 to S phase, leading to uncontrolled cellular proliferation by increasing cyclin D1 activation, pRb phosphorylation and E2F transactivation.51 Furthermore, it has been reported that Rac1 inhibition induces p21 mRNA expression in different cancers.52 Likewise, our data show that the inhibition of CaMKII decreases the activation of Rac1. This ultimately leads to an increase in p21 expression that contributes to G1/G0 cell cycle arrest.
In order to investigate how CaMKII regulates Rac1 activation in osteosarcoma, we examined the upstream activators of Rac1; Tiam1.53 The phosphorylation of Tiam1 has been shown to play a pivotal role in the activation of Rac and the promotion of tumor cell growth, invasion and metastases in several organs such as the breast.18,19 It has been shown that the phosphorylation of Tiam1 by CaMKII enhances its nucleotide exchange rate toward Rac1.17 Our data demonstrate that the pharmacologic inhibition of CaMKII in MG-63 osteosarcoma cells decreases, in a time-dependent manner, the activation of Tiam1, which leads, in turn, to a decrease in the activation of Rac1. However, this does not exclude the possibility that CaMKII could also be responsible for the activation of other unidentified GEFs beside Tiam1.
Taken together, our results suggest that an increase in the expression/activation of α-CaMKII in osteosarcoma cells results in an increase in the phosphorylation of Tiam1 and its membrane localization. This increases the activation of Rac1, inhibits the expression of p21 and leads to a disruption in cell cycle regulation that results in the unregulated growth of osteosarcoma (Figure 6c). Findings from our studies provide the first documentation of a novel molecular mechanism that describes the role of CaMKII in the growth of osteosarcoma. In addition, although we focus on the role of CaMKII specifically in osteosarcoma, it is possible that this mechanism is also important in promoting the growth of other types of solid tumors. If found to be true, this would provide basis for the development of a new target for drug design and therapeutic intervention to combat the life-threatening condition of osteosarcoma and to dramatically improve patient's survival.
Acknowledgments
We thank Patty Lott at the University of Alabama at Birmingham, Center for Metabolic Bone Disease, Histomorphometry and Molecular Analysis Core Laboratory, NIH Grant P30-AR46031, for help with the histology and for critically reading the manuscript. We also thank the Clinical Nutrition Research Unit, Small Animal Phenotyping Core, NIH Grant P30-DK56336, for help with the μ-CT. Finally, we would like to acknowledge Dr Hyeran Choo for technical assistance with the in vivo studies. This work was supported by the National Institute of Health Grant CA093796, CA098543, the Haley's Hope Memorial Support Fund for Osteosarcoma Research at UAB (GPS), P01-CA098912 (MZ), and an Intramural Pilot and Feasibility Clinical/Translational Grant from the UAB Center for Metabolic Bone Disease/Osteoporosis Prevention and Treatment Clinic (MZ).
References
- 1.Papachristou DJ, Batistatou A, Sykiotis GP, et al. Activation of the JNK-AP-1 signal transduction pathway is associated with pathogenesis and progression of human osteosarcomas. Bone. 2003;32:364–371. doi: 10.1016/s8756-3282(03)00026-7. [DOI] [PubMed] [Google Scholar]
- 2.Dahlin DC, Unni KK. Osteosarcoma of bone and its important recognizable varieties. Am J Surg Pathol. 1977;1:61–72. doi: 10.1097/00000478-197701010-00007. [DOI] [PubMed] [Google Scholar]
- 3.Ballance WA, Jr, Mendelsohn G, Carter JR, et al. Osteogenic sarcoma. Malignant fibrous histiocytoma subtype. Cancer. 1988;62:763–771. doi: 10.1002/1097-0142(19880815)62:4<763::aid-cncr2820620421>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 4.Unni KK. Osteosarcoma of bone. J Orthop Sci. 1998;3:287–294. doi: 10.1007/s007760050055. [DOI] [PubMed] [Google Scholar]
- 5.Klein MJ, Siegal GP. Osteosarcoma: anatomic and histologic variants. Am J Clin Pathol. 2006;125:555–581. doi: 10.1309/UC6K-QHLD-9LV2-KENN. [DOI] [PubMed] [Google Scholar]
- 6.Pestell RG, Albanese C, Reutens AT, et al. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr Rev. 1999;20:501–534. doi: 10.1210/edrv.20.4.0373. [DOI] [PubMed] [Google Scholar]
- 7.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 8.Zhang CL, Liao WM, Li FB, et al. Prognostic significance of p21(WAF1) expression in osteosarcoma. Zhonghua Bing Li Xue Za Zhi. 2005;34:524–527. [PubMed] [Google Scholar]
- 9.Agiostratidou G, Derventzi A, Gonos ES. Over-expression of CDKIs p15INK4b, p16INK4a and p21CIP1/WAF1 genes mediate growth arrest in human osteosarcoma cell lines. In Vivo. 2001;15:443–446. [PubMed] [Google Scholar]
- 10.Zayzafoon M, Fulzele K, McDonald JM. Calmodulin and calmodulin-dependent kinase II\{alpha\} Regulate osteoblast differentiation by controlling c-fos expression. J Biol Chem. 2005;280:7049–7059. doi: 10.1074/jbc.M412680200. [DOI] [PubMed] [Google Scholar]
- 11.Zayzafoon M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem. 2006;97:56–70. doi: 10.1002/jcb.20675. [DOI] [PubMed] [Google Scholar]
- 12.Deb TB, Coticchia CM, Dickson RB. Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J Biol Chem. 2004;279:38903–38911. doi: 10.1074/jbc.M405314200. [DOI] [PubMed] [Google Scholar]
- 13.Morris TA, DeLorenzo RJ, Tombes RM. CaMK-II inhibition reduces cyclin D1 levels and enhances the association of p27kip1 with Cdk2 to cause G1 arrest in NIH 3T3 cells. Exp Cell Res. 1998;240:218–227. doi: 10.1006/excr.1997.3925. [DOI] [PubMed] [Google Scholar]
- 14.Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 15.Gosens R, Schaafsma D, Nelemans SA, et al. Rho-kinase as a drug target for the treatment of airway hyperresponsiveness in asthma. Mini Rev Med Chem. 2006;6:339–348. doi: 10.2174/138955706776073402. [DOI] [PubMed] [Google Scholar]
- 16.Buchanan FG, Elliot CM, Gibbs M, et al. Translocation of the Rac1 guanine nucleotide exchange factor Tiam1 induced by platelet-derived growth factor and lysophosphatidic acid. J Biol Chem. 2000;275:9742–9748. doi: 10.1074/jbc.275.13.9742. [DOI] [PubMed] [Google Scholar]
- 17.Fleming IN, Elliott CM, Buchanan FG, et al. Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem. 1999;274:12753–12758. doi: 10.1074/jbc.274.18.12753. [DOI] [PubMed] [Google Scholar]
- 18.Michiels F, Stam JC, Hordijk PL, et al. Regulated membrane localization of Tiam1, mediated by the NH2-terminal pleckstrin homology domain, is required for Rac-dependent membrane ruffling and C-Jun NH2-terminal kinase activation. J Cell Biol. 1997;137:387–398. doi: 10.1083/jcb.137.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bourguignon LY, Zhu H, Shao L, et al. Ankyrin-Tiam1 interaction promotes Rac1 signaling and metastatic breast tumor cell invasion and migration. J Cell Biol. 2000;150:177–191. doi: 10.1083/jcb.150.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bourguignon LY, Zhu H, Shao L, et al. CD44 interaction with tiam1 promotes Rac1 signaling and hyaluronic acid-mediated breast tumor cell migration. J Biol Chem. 2000;275:1829–1838. doi: 10.1074/jbc.275.3.1829. [DOI] [PubMed] [Google Scholar]
- 21.Cavazzin C, Bonvicini C, Nocera A, et al. Expression and phosphorylation of delta-CaM kinase II in cultured Alzheimer fibroblasts. Neurobiol Aging. 2004;25:1187–1196. doi: 10.1016/j.neurobiolaging.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Zayzafoon M, Gathings WE, McDonald JM. Modeled microgravity inhibits osteogenic differentiation of human mesenchymal stem cells and increases adipogenesis. Endocrinology. 2004;145:2421–2432. doi: 10.1210/en.2003-1156. [DOI] [PubMed] [Google Scholar]
- 23.Ohta M, Tateishi K, Kanai F, et al. p53-Independent negative regulation of p21/cyclin-dependent kinase-interacting protein 1 by the sonic hedgehog-glioma-associated oncogene 1 pathway in gastric carcinoma cells. Cancer Res. 2005;65:10822–10829. doi: 10.1158/0008-5472.CAN-05-0777. [DOI] [PubMed] [Google Scholar]
- 24.Bindra RS, Gibson SL, Meng A, et al. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005;65:11597–11604. doi: 10.1158/0008-5472.CAN-05-2119. [DOI] [PubMed] [Google Scholar]
- 25.Xiao C, Yang BF, Song JH, et al. Inhibition of CaMKII-mediated c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Exp Cell Res. 2005;304:244–255. doi: 10.1016/j.yexcr.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Sasaki A, Boyce BF, Story B, et al. Bisphosphonate risedronate reduces metastatic human breast cancer burden in bone in nude mice. Cancer Res. 1995;55:3551–3557. [PubMed] [Google Scholar]
- 27.Yu Y, Harris RI, Yang JL, et al. Differential expression of osteogenic factors associated with osteoinductivity of human osteosarcoma cell lines. J Biomed Mater Res. 2004;70A:122–128. doi: 10.1002/jbm.a.30072. [DOI] [PubMed] [Google Scholar]
- 28.Huang WC, Wu D, Xie Z, et al. {beta} 2-Microglobulin is a signaling and growth-promoting factor for human prostate cancer bone metastasis. Cancer Res. 2006;66:9108–9116. doi: 10.1158/0008-5472.CAN-06-1996. [DOI] [PubMed] [Google Scholar]
- 29.Zayzafoon M, Abdulkadir SA, McDonald JM. Notch signaling and ERK activation are important for the osteomimetic properties of prostate cancer bone metastatic cell lines. J Biol Chem. 2004;279:3662–3670. doi: 10.1074/jbc.M308158200. [DOI] [PubMed] [Google Scholar]
- 30.Baitinger C, Alderton J, Poenie M, et al. Multifunctional Ca2+/calmodulin-dependent protein kinase is necessary for nuclear envelope breakdown. J Cell Biol. 1990;111:1763–1773. doi: 10.1083/jcb.111.5.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juang YT, Wang Y, Solomou EE, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest. 2005;115:996–1005. doi: 10.1172/JCI200522854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madgwick S, Levasseur M, Jones KT. Calmodulin-dependent protein kinase II, and not protein kinase C, is sufficient for triggering cell-cycle resumption in mammalian eggs. J Cell Sci. 2005;118:3849–3859. doi: 10.1242/jcs.02506. [DOI] [PubMed] [Google Scholar]
- 33.Pompetti F, Rizzo P, Simon RM, et al. Oncogene alterations in primary, recurrent, and metastatic human bone tumors. J Cell Biochem. 1996;63:37–50. doi: 10.1002/(SICI)1097-4644(199610)63:1%3C37::AID-JCB3%3E3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen GP, Burns KL, Rosenberg AE, et al. CDKN2A gene deletions and loss of p16 expression occur in osteosarcomas that lack RB alterations. Am J Pathol. 1998;153:159–163. doi: 10.1016/S0002-9440(10)65556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tombes RM, Krystal GW. Identification of novel human tumor cell-specific CaMK-II variants. Biochim Biophys Acta. 1997;1355:281–292. doi: 10.1016/s0167-4889(96)00141-3. [DOI] [PubMed] [Google Scholar]
- 36.Hunt DA, Lane HM, Zygmont ME, et al. MRNA stability and overexpression of fatty acid synthase in human breast cancer cell lines. Anticancer Res. 2007;27:27–34. [PubMed] [Google Scholar]
- 37.Agoston AT, Argani P, Yegnasubramanian S, et al. Increased protein stability causes DNA methyltransferase 1 dysregulation in breast cancer. J Biol Chem. 2005;280:18302–18310. doi: 10.1074/jbc.M501675200. [DOI] [PubMed] [Google Scholar]
- 38.Sun HL, Liu YN, Huang YT, et al. YC-1 inhibits HIF-1 expression in prostate cancer cells: contribution of Akt/NF-kappaB signaling to HIF-1alpha accumulation during hypoxia. Oncogene. 2007;26:3941–3951. doi: 10.1038/sj.onc.1210169. [DOI] [PubMed] [Google Scholar]
- 39.Barry EL, Gesek FA, Froehner SC, et al. Multiple calcium channel transcripts in rat osteosarcoma cells: selective activation of alpha 1D isoform by parathyroid hormone. Proc Natl Acad Sci USA. 1995;92:10914–10918. doi: 10.1073/pnas.92.24.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitfield JF. Calcium signals and cancer. Crit Rev Oncog. 1992;3:55–90. [PubMed] [Google Scholar]
- 41.Kahl CR, Means AR. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr Rev. 2003;24:719–736. doi: 10.1210/er.2003-0008. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez-Mora OG, LaHair MM, McCubrey JA, et al. Calcium/calmodulin-dependent kinase I and calcium/calmodulin-dependent kinase kinase participate in the control of cell cycle progression in MCF-7 human breast cancer cells. Cancer Res. 2005;65:5408–5416. doi: 10.1158/0008-5472.CAN-05-0271. [DOI] [PubMed] [Google Scholar]
- 43.Kahl CR, Means AR. Regulation of cyclin D1/Cdk4 complexes by calcium/calmodulin-dependent protein kinase I. J Biol Chem. 2004;279:15411–15419. doi: 10.1074/jbc.M312543200. [DOI] [PubMed] [Google Scholar]
- 44.Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–3985. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- 45.Harper JW, Adami GR, Wei N, et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H, Hannon GJ, Casso D, et al. p21 is a component of active cell cycle kinases. Cold Spring Harb Symp Quant Biol. 1994;59:21–29. doi: 10.1101/sqb.1994.059.01.005. [DOI] [PubMed] [Google Scholar]
- 47.He G, Siddik ZH, Huang Z, et al. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene. 2005;24:2929–2943. doi: 10.1038/sj.onc.1208474. [DOI] [PubMed] [Google Scholar]
- 48.Munoz-Alonso MJ, Acosta JC, Richard C, et al. p21Cip1 and p27Kip1 induce distinct cell cycle effects and differentiation programs in myeloid leukemia cells. J Biol Chem. 2005;280:18120–18129. doi: 10.1074/jbc.M500758200. [DOI] [PubMed] [Google Scholar]
- 49.Uchibori M, Nishida Y, Nagasaka T, et al. Increased expression of membrane-type matrix metalloproteinase-1 is correlated with poor prognosis in patients with osteosarcoma. Int J Oncol. 2006;28:33–42. [PubMed] [Google Scholar]
- 50.Quinn CO, Rajakumar RA, Agapova OA. Parathyroid hormone induces rat interstitial collagenase mRNA through Ets-1 facilitated by cyclic AMP response element-binding protein and Ca(2+)/calmodulin-dependent protein kinase II in osteoblastic cells. J Mol Endocrinol. 2000;25:73–84. doi: 10.1677/jme.0.0250073. [DOI] [PubMed] [Google Scholar]
- 51.Gjoerup O, Lukas J, Bartek J, et al. Rac and Cdc42 are potent stimulators of E2F-dependent transcription capable of promoting retinoblastoma susceptibility gene product hyperphosphorylation. J Biol Chem. 1998;273:18812–18818. doi: 10.1074/jbc.273.30.18812. [DOI] [PubMed] [Google Scholar]
- 52.Knight-Krajewski S, Welsh CF, Liu Y, et al. Deregulation of the Rho GTPase, Rac1, suppresses cyclin-dependent kinase inhibitor p21(CIP1) levels in androgen-independent human prostate cancer cells. Oncogene. 2004;23:5513–5522. doi: 10.1038/sj.onc.1207708. [DOI] [PubMed] [Google Scholar]
- 53.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]