

Abstract
Over the last decade, significant advances have been made in the methodology for studying immune responses in vivo. It is now possible to follow almost every aspect of pathogen-specific immunity using in vivo models that incorporate physiological infectious doses and natural routes of infection. This new ability to study immunity in a relevant physiological context will greatly expand our understanding of the dynamic interplay between host and pathogen. Visualizing the resolution of primary infection and the development of long-term immunological memory should also aid the development of new vaccines and therapeutics for infectious diseases. In this review, we will describe the application of in vivo visualization technology to Salmonella infection, describe our current understanding of Salmonella-specific immunity, and discuss some unanswered questions that remain in this model.
2. Introduction
During the 20th century, increased understanding of immunity to infectious disease led to the development of successful vaccines and therapeutics for viral and bacterial infections which plagued previous generations (Amanna and Slifka, 2005; Brines, 1996). Despite this success, there remains a significant need for vaccine and therapeutic development today. The emergence of new pathogens (Woolhouse et al., 2005), the acquired resistance of pathogens to currently effective therapeutics (Martinez-Cajas and Wainberg, 2007; Okeke et al., 2007), a lack of research and development in the diseases of the developing world (Walker, 2005), and the deliberate transmission of infectious agents for ideological purposes (Moran et al., 2008), all generate considerable demand for new vaccines and therapeutics.
If these new vaccines and therapeutics are to be developed in a rational manner, the interplay between microbes and the host immune response must be understood in considerable detail. Fortunately, over the last 10 years or so, the development of new imaging methodologies now allow direct study of the immune response in a physiological context (Germain and Jenkins, 2004; Henrickson and von Andrian, 2007; Huang et al., 2004; Jenkins et al., 2001; Negrin and Contag, 2006). So far, the application of these visualization technologies has been limited to a select number of infectious disease models. However, visualization of immune responses to infection is now becoming more widely used and therefore generates significant potential for discovery in many infectious disease models. In this review, we will focus on the development and use of visualization approaches for studying immunity to Salmonella infection. We will describe how imaging tools have confirmed and extended our knowledge of host immunity to Salmonella, discuss current limitations in our knowledge, and speculate on the potential for future advances in studying this disease.
3. Studying immunity to Salmonella
Salmonella are gram-negative enterobacteria that cause significant human and veterinary disease in the US and elsewhere (Grassl and Finlay, 2008; Parry et al., 2002; Rabsch et al., 2001). The immune response to Salmonella has been studied in infected humans and in animal models of disease.
3.1 Human disease cause by Salmonella
After several nomenclature revisions (Su and Chiu, 2007), the Salmonella genus now contains 3 species, S. enterica, S. bongori, and S. subterranean, but almost all significant human disease is caused by a single sub-species, S. enterica subsp. Enterica. This subspecies contains both the typhi and paratyphi Serovars that cause typhoid and paratyphoid fever in developing countries and over 2000 different serovars that cause Salmonella gastroenteritis in humans and animals (Grassl and Finlay, 2008; Parry et al., 2002; Rabsch et al., 2001). The global incidence of typhoid was recently estimated at 21,650,974 infections and 216,510 deaths per year (Crump et al., 2004; Jones and Falkow, 1996). Numerous closely related Salmonella serovars cause non-typhoidal Salmonellosis, an increasingly important food borne infection in the US (Mead et al., 1999). Both diseases have been recognized as significant bio-threats to the US food and water supply (Jones and Falkow, 1996; Sobel et al., 2002). During a typhoid epidemic in Tajikistan, more than 90% of clinical isolates were multi-drug resistant and 82% of these were resistant to ciprofloxacin (Tarr et al., 1999), the antibiotic of choice for treating typhoid in developed nations. Recent analysis of the bacterial proteome indicates that all major targets of Salmonella metabolism have already been targeted by antibiotic development strategies (Becker et al., 2006), therefore the potential for generating new antibiotics is not encouraging. Understanding the generation of an adaptive immune response to Salmonella is therefore of considerable medical and economic importance.
3.2 Animal models of Salmonella infection
Although S. enterica Serovar typhi causes typhoid fever in humans, it does not cause typhoid in other mammals (Fierer and Guiney, 2001). Therefore, Serovar typhi is usually studied in vitro or using in vivo models that require unusual routes of administration (Bueno et al., 2008; Pasetti et al., 2002; Subramanian and Qadri, 2006). In contrast, oral infection of mice with S. enterica Serovar typhimurium causes a systemic typhoid-like disease involving penetration of intestinal Peyer's patches and rapid dissemination to the liver, spleen and bone marrow (Srinivasan and McSorley, 2006). Therefore, murine infection with S. enterica Serovar typhimurium bears a striking similarity to human typhoid and is the most commonly studied laboratory model of this disease (Mastroeni and Sheppard, 2004; Santos et al., 2001). However, despite the utility of this mouse Salmonella model, it does have some limitations as a model of human disease. First, not all bacterial virulence factors that are important for murine disease are required for the pathogenesis of human typhoid. Therefore, as with most animal models, data from the murine typhoid model should not be over-interpreted and needs to be considered alongside clinical studies of typhoid in humans to develop a complete picture of typhoid immunity (Pasetti et al., 2003). Second, Salmonella infection of mice does not cause diarrhea, yet diarrhea is a prominent feature of Salmonella enterocolitis in humans. Therefore, the mouse model is much more useful for studying typhoid than as a model of Salmonella gastroenteritis. A better model of Salmonella gastroenteritis in humans is the bovine Salmonellosis model where intestinal disease and diarrhea develop rapidly in a similar manner to human disease (Santos et al., 2001).
4. Visualizing Salmonella in vivo
Using the mouse model of typhoid, a number of studies have visualized the colonization and growth of Salmonella in vivo. Our laboratory performed fluorescence microscopy on mid-line sections of whole infected mice to simultaneously visualize Salmonella in all organs (McSorley and Jenkins, unpublished). These studies revealed that the spleen and liver are the major sites of Salmonella replication (Fig. 1). More detailed studies using confocal microscopy demonstrated that bacteria in the liver are initially associated with neutrophil infiltration but subsequently are found exclusively within macrophages (Richter-Dahlfors et al., 1997). Similar studies, some using a Salmonella strain expressing green fluorescent protein (GFP), demonstrated that Salmonella in the spleen are also found within macrophages (Matsui et al., 2000; Salcedo et al., 2001). Thus, visualization studies in mice confirm that Salmonella is an intracellular pathogen that replicates extensively within macrophages found in the spleen and liver. A more recent study of Salmonella in the liver found that the bacteria replicate intracellularly in discrete foci, each of which are likely to arise from the clonal expansion of a single bacterium (Sheppard et al., 2003). Thus, individual foci of Salmonella appear to grow independently until reaching a certain threshold, after which the bacteria redistribute to new foci.
Figure 1. Salmonella in the splenic red pulp of infected mice.
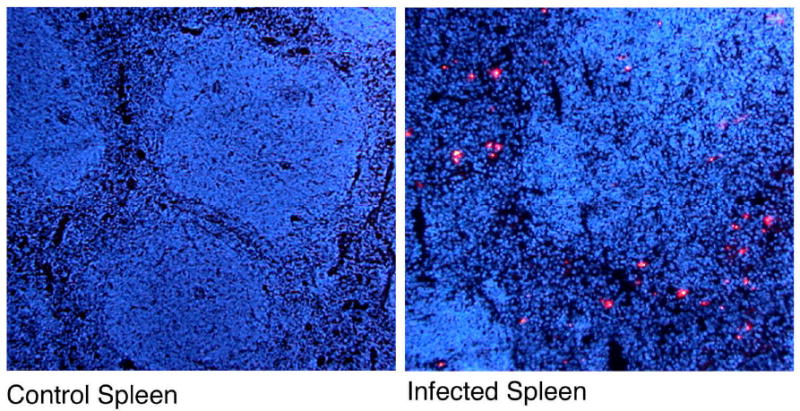
C57BL/6 mice were infected orally with 5×109 attenuated Salmonella, SL3261 and whole body sections were taken three days later. Sections were blocked and stained with an antibody specific to Salmonella LPS and the signal amplified using biotinyl tyramide and Streptavidin-Cy5 (red). Tissue was counterstained will DAPI (blue) to stain nuclei. Images show staining of spleens from control or Salmonella-infected mice.
5. Adaptive immunity in the murine typhoid model
The mouse model of Salmonella infection has long been of interest to scientists trying to understand bacterial pathogenesis and immunity to infection (Blanden et al., 1966; Gowen and Calhoun, 1943; Zinkernagel, 1976). As Salmonella preferentially infects and resides within macrophages, the activation of these cells by IFN-γ produced by Th1 cells plays a prominent role in bacterial killing (Mastroeni, 2002; Mittrucker and Kaufmann, 2000; Wick, 2003). Indeed, nude mice (Sinha et al., 1997), mice lacking TCR β (Hess et al., 1996; Weintraub et al., 1997), CD4 (Hess et al., 1996), CD28 (McSorley and Jenkins, 2000; Mittrucker, 1999), MHC class-II (Hess et al., 1996), IFN-γ (VanCott et al., 1998), IFN-γR (Hess et al., 1996), or the Th1 transcription factor T-bet (Ravindran et al., 2005), all fail to resolve primary infection with a live vaccine strain (LVS) of Salmonella. Furthermore, depletion of IFN-γ (Mastroeni et al., 1992; Nauciel and Espinasse-Maes, 1992), IL-12 (Mastroeni et al., 1996), or TNF-α (Mastroeni et al., 1992; Nauciel and Espinasse-Maes, 1992), abrogates or reduces the protective immunity conferred by LVS-immunization. The relevance of these findings in the murine model are strengthened by clinical reports of increased susceptibility to Salmonella in patients with primary immune deficiencies in IL-12 or IFN-γ receptor signaling (Cleary et al., 2003; Jouanguy et al., 1999). Thus, a considerable amount of data in mouse and human typhoid indicates that CD4 Th1 cells play an important protective role.
In a somewhat surprising finding for an intracellular pathogen, our laboratory and others have demonstrated that Salmonella-specific antibody participates in immunity to typhoid (Mastroeni et al., 2000; McSorley and Jenkins, 2000; Mittrucker et al., 2000). Although B cell-deficient mice survive vaccination with LVS-Salmonella, they do not acquire protective immunity to secondary typhoid, indicating that antibody is absolutely required for effective immunity (Mastroeni et al., 2000; McSorley and Jenkins, 2000; Mittrucker et al., 2000). It is currently unclear how antibody is protective against an intracellular pathogen. One possibility is that serum antibody prevents cell-cell transmission of Salmonella following macrophage apoptosis (Ravindran and McSorley, 2005). Alternatively, an antibody response could alter the processing of Salmonella antigens to enhance antigen presentation to CD4 T cells and subsequent cellular immunity (Bueno et al., 2007; Mastroeni et al., 2001; Ugrinovic et al., 2003). Another possibility is that Salmonella-specific mucosal IgA secreted into the intestine can inhibit initial bacterial penetration of epithelial cells or simply reduce the overall load of bacteria in the gut lumen (Wijburg et al., 2006). Whatever the mechanism, effective immunity against typhoid appears to require the combined activity of Salmonella-specific Th1 cells and antibody.
The role of CD8 T cells in adaptive immunity to Salmonella is less clear. While CD8 CTL responses to Salmonella have been characterized (Lo et al., 1999; Mastroeni et al., 1992; Nauciel, 1990), mice lacking CD8 T cells display only a mild reduction in their ability to resolve infections with attenuated Salmonella (Hess et al., 1996; Lo et al., 1999).
6. Tracking antigen-specific responses in Salmonella infection
The importance of adaptive immunity in mediating host resistance to Salmonella has fueled extensive research into Salmonella-specific T cell responses (Ravindran and McSorley, 2005)(Bueno et al., 2007). As with other models of infectious disease, these studies have benefited greatly from the development of techniques to track antigen-specific T cells in mice (Jenkins et al., 2001). The application of these techniques to in vivo models of Salmonella pathogenesis has revealed valuable information about where, when, and how Salmonella-specific T cells are activated during the course of infection (Ravindran and McSorley, 2005). However, with this knowledge comes a host of new questions, providing the impetus for further development of immunological tools to improve studies of Salmonella in vivo.
6.1 Target antigen and epitope identification
The relationship between Salmonella-specific T and B cells and the nature of the antigens they recognize constitutes a critical parameter determining the outcome of an infection. Thus, one of the most relevant issues concerning the study of adaptive immune responses to Salmonella is the precise identification of antigenic epitopes recognized by host lymphocytes. Explicit knowledge of these epitopes is essential for detailed characterization of T and B cell responses to Salmonella and may also provide potential candidates for a future subunit typhoid vaccine.
The challenge of Salmonella epitope-discovery is a daunting one, as the genome of Serovars typhi and typhimurium contain around 4,300 open reading frames (McClelland et al., 2001; Parkhill et al., 2001), each with the potential to encode multiple peptide epitopes recognized by T cells. Moreover, the timing and localization of bacterial protein expression adds a further layer of complexity with regard to antigenic targets for T cells during different stages of the disease (Becker et al., 2006; Eriksson et al., 2003; Rollenhagen and Bumann, 2006). Nevertheless, a handful of defined CD8 and CD4 T cell epitopes recognized by Salmonella-specific T cells have been successfully identified (Table 1).
Table 1. Identified Salmonella epitopes.
| Peptide | MHC restriction | Reference |
|---|---|---|
| GroEL 192-200 | Qa-1b | (Lo et al., 2000) |
| OmpC 73-80 | Kb | (Diaz-Quinonez et al., 2004) |
| OmpC 132-139 | Kb | (Diaz-Quinonez et al., 2004) |
| FliC 80-94 | I-Ak | (Bergman et al., 2005) |
| FliC 339-350 | I-Ak | (Cookson and Bevan, 1997) |
| FliC 427-441 | I-Ab | (McSorley et al., 2000) |
| FliC 455-469 | I-Ab | (Bergman et al., 2005) |
| SipC 381-394 | I-Ad | (Musson et al., 2002) |
Early studies demonstrated a role for CD8 T cells in immunity to Salmonella and led to a search for MHC class-I restricted peptide epitopes (Lo et al., 1999; Mastroeni et al., 1992; Nauciel, 1990). Lo and colleagues found that a high proportion of CD8 CTL responses in mice infected with attenuated Salmonella recognized epitopes presented by non-classical MHC class Ib molecules, particularly Qa-1b (Lo et al., 1999). MALDI mass spectroscopy of peptides eluted from Qa-1b molecules on Salmonella-infected cells identified the 192-200 peptide of the GroEL protein as an immunodominant epitope for CTL responses (Lo et al., 2000). In a different study, specialized software was used to predict Kb-binding peptides within the sequence of the immunogenic Salmonella outer membrane protein C (OmpC) porin, resulting in the identification of residues 73-80 and 132-139 as two immunodominant peptide epitopes (Diaz-Quinonez et al., 2004). However, our laboratory has been unable to detect CD8 T cell responses to either of these epitopes in Salmonella-infected mice (McSorley et al, unpublished observations), although it should be noted that the methodology used was different from that of the original report. Thus, despite the identification of a few class-I Salmonella epitopes, class-I tetramer reagents for tracking Salmonella-specific CD8 T cells have not yet been developed. This is unfortunate, since the development of ex vivo staining reagents that would allow identification of endogenous Salmonella-specific CD8 T cells should enable detailed analysis of the contribution of CD8 T cells to Salmonella immunity. Hopefully, this is an area that can be addressed in the future in both murine and human typhoid research.
Early work on CD4 T cells demonstrated significant protection after the transfer of Salmonella-specific T cell lines, or after immunization with Salmonella porins or other cell surface proteins (Misfeldt and Johnson, 1979; Paul et al., 1985; Paul et al., 1988; Tabaraie et al., 1994); however, the epitopes recognized by CD4 T cells remained undefined. Cookson and Bevan were the first to identify an epitope of Salmonella flagellin recognized by Salmonella-specific CD4 T cells from C3H/HeJ mice in the context of I-Ak (Cookson and Bevan, 1997). Flagellin is the major structural protein of bacterial flagella, is produced in large quantities by cultured bacteria, and is a known ligand for the innate immune receptor TLR5 (Salazar-Gonzalez and McSorley, 2005). Thus it is perhaps not surprising that it is also a target of the adaptive response. The targeting of flagellin by Salmonella-specific CD4 T cells was subsequently confirmed in C57BL/6 mice, and currently 4 different MHC class-II flagellin epitopes have been reported (Bergman et al., 2005; McSorley et al., 2000). These initial studies also demonstrated the presence of long-lived memory T cell responses to flagellin epitopes, suggesting a role for these antigen-specific responses in protective immunity (Cookson and Bevan, 1997; McSorley et al., 2000). Consistent with this observation, immunization of susceptible mice with flagellin conferred resistance to low dose challenge with virulent Salmonella (McSorley et al., 2000; Strindelius et al., 2002). The only other defined epitope recognized by Salmonella-specific CD4 T cells is the I-Ad restricted peptide 381-394 of Salmonella invasion protein C (SipC) (Musson et al., 2002). Interestingly, expression of both flagellin and SipC are expressed by cultured bacteria, but is tightly regulated during the transition to intra-macrophage growth (Eriksson et al., 2003). Whether this is relevant for the targeting of these antigens by CD4 T cell responses is currently unclear.
In a more comprehensive screen for Salmonella target antigens, Rollenhagen and colleagues created a library of Salmonella promoter elements that were used to drive expression of a fluorescent reporter gene (Rollenhagen et al., 2004). Their logic was that identifying the Salmonella proteins that are highly expressed in vivo would include antigens that were most likely to be dominant T cell targets. Using this methodology, a list of candidate Salmonella proteins was generated that may be an important source of epitopes during an immune response. Within this list of highly expressed proteins, Mig-14 and SseB are two antigens that provided protective immunity when used as a subunit vaccine. Efforts are now underway by our colleagues to fine map peptide epitopes from these proteins by screening T cells from immunized mice with libraries of overlapping peptide sequences.
As should be clear from the limited list of candidate antigens discussed above, epitope discovery still remains a major goal in the study of immunity to Salmonella. However, this situation is little different to numerous other infectious disease models that have been somewhat neglected by immunologists. Fortunately, epitope identification in a number of infectious disease models is now receiving greater attention (Peters and Sette, 2007). The identification of new epitopes opens up the possibility for development of antigen-specific reagents such as T cell clones, TCR transgenic mice, and peptide:MHC tetramers that can facilitate visualization of Salmonella-specific T cells in vivo.
6.2 Visualizing Salmonella-specific responses using surrogate antigens
Due to the scarcity of defined T cell epitopes in the Salmonella model, surrogate antigens have been used to track Salmonella-specific T cell responses. Proof of principle for this approach was established by early studies exploring the use of attenuated strains of Salmonella expressing model antigens, either as full-length proteins or short peptide sequences embedded within a native Salmonella protein (Maskell et al., 1987; Newton et al., 1989). B cell, cytotoxic T cell, and helper T cell responses to a wide variety of model antigens were elicited in this manner, thereby demonstrating potential roles for each of these cell populations in host adaptive immunity to Salmonella (Aggarwal et al., 1990; Khan et al., 1994; McSorley et al., 1997). In most of these experiments, freshly isolated T cells from immunized mice were re-stimulated in vitro with model antigens and a common read-out such as cell proliferation or specific lysis of target cells was recorded. These indirect functional assays can indicate the presence or absence of epitope-specific T cells in an immunized or infected animal but they reveal very little about the biology of epitope-specific T cells in vivo.
The development of TCR transgenic mice that contain a peripheral CD4 population with monoclonal antigen specificity allowed investigators to move beyond this low level of resolution. TCR transgenic mice were initially infected with Salmonella expressing the model antigen recognized by peripheral T cells in the host (Verma et al., 1995), but a TCR transgenic host provides a highly artificial environment in which to model an immune response. The power of TCR transgenic technology was realized using an adoptive transfer approach where antigen specific T cells are seeded at relatively low frequency among polyclonal endogenous CD4 T cells in a wild-type mouse (Kearney et al., 1994; Pape et al., 1997). Chen and Jenkins were the first to use this approach to examine the CD4 T cell response to Salmonella (Chen and Jenkins, 1999). In this study, naive OVA-specific T cells from DO11.10 TCR transgenic mice were adoptively transferred into MHC-compatible BALB/c mice. The transferred cells were distinguished from endogenous T cells using an antibody specific for the DO11.10 TCR (Haskins et al., 1983). DO11.10 T cells proliferated extensively in the draining lymph nodes and spleen of both resistant and susceptible strains of BALB/c host mice, demonstrating that susceptibility was unrelated to the frequency of Salmonella-specific T cells but rather was dependent on the ability of these cells to secrete IFN-γ (Chen and Jenkins, 1999).
The coordinated use of pathogen-expressed surrogate antigens and TCR transgenic T cell adoptive transfer has now become a standard protocol for investigating T cell responses to pathogens (Bertholet et al., 2006; Chen and Jenkins, 1999; Pope et al., 2001). Although this approach certainly provides in vivo antigen-specific information, it also has some limitations. First, the complex regulation of pathogen proteins in response to the host environment is very poorly modeled by the over-expression of a transgene. Indeed, the selection of a promoter for antigen expression in Salmonella can have profound effects on the induction of an adaptive immune response (McSorley et al., 1997). This issue is evident in recent adoptive transfer studies using OVA-specific OT-I TCR transgenic CD8 T cells and Salmonella expressing OVA. A delayed CD8 response to OVA-expressing Salmonella was reported in one study, but was not detected in a second study using a different antigen expression system (Jones-Carson et al., 2007; Luu et al., 2006). Thus, transgenic over-expression studies are heavily dependent on the strength and location of promoter activity in vivo. Second, the high level of model antigen expression in many of these systems can have deleterious effects on the viability of host bacteria (Bumann, 2001b; Wick and Pfeifer, 1996). Forcing a pathogen to devote a large percentage of protein expression machinery to the production of an irrelevant protein is likely to disrupt the natural life cycle of the pathogen and give rise to results that may be difficult to interpret. Third, in many over-expression studies, the visualized CD4 or CD8 response to pathogen-expressed model antigen does not contribute to protective immunity and may therefore provide very little information about the nature of endogenous protective responses to pathogen proteins. However, when dealing with infectious disease models where epitope identification is still in its infancy, the over-expression of model antigens can still be a useful strategy to uncover antigen-specific responses in vivo.
In an attempt to examine both antigen expression and T cell responses simultaneously, an attenuated strain of Salmonella was generated expressing green fluorescent protein (GFP) fused to a minimal peptide sequence containing the immunodominant CD4 T cell epitope of OVA (Bumann, 2001a). In contrast to the earlier study (Chen and Jenkins, 1999), the number of OVA-specific T cells correlated precisely with the presence of antigen in vivo, which led the author to conclude that the transient nature of the T cell response in the former study was due to the enormous initial dose of OVA-expressing Salmonella administered (Bumann, 2003).
6.3 Salmonella-specific TCR transgenic mice
The artificial nature of surrogate antigen expression in visualizing Salmonella-specific immunity provided the motivation for the development of an experimental system to examine T cell responses to endogenous Salmonella epitopes. However, due to the limited number of defined epitopes noted above, almost all these efforts have focused on a single antigen, flagellin.
To provide a source of T cells with specificity to a bona fide Salmonella antigen, a transgenic mouse called SM1 was generated using TCR α and β genes from a CD4 T cell clone specific for residues 427-441 of flagellin in the context of I-Ab (McSorley et al., 2002; McSorley et al., 2000). SM1 mice were backcrossed onto a RAG-2 deficient background to eliminate endogenous TCR rearrangement, which occurs at high frequency in this particular TCR transgenic mouse (McSorley, unpublished observations). SM1 mice have also been crossed to CD90.1 and CD45.1 congenic backgrounds, allowing identification of SM1 cells in adoptively transferred recipients using antibodies specific for these alleles (Srinivasan et al., 2004b; Srinivasan and McSorley, 2007). The advantage of this system over the use of an OVA-specific system (Bumann, 2001a; Chen and Jenkins, 1999) is that SM1 cells allow examination of a CD4 T cell response to an endogenously expressed Salmonella protein. Similar adoptive transfer systems that allow visualization of T cell responses to endogenous bacterial antigens have recently been developed in models of Mycobacterial infection (Reiley et al., 2008; Wolf et al., 2008).
Oral infection of SM1-transferred mice with a virulent strain of Salmonella initiated rapid activation and proliferation of SM1 cells in the intestinal Peyer's patches and mesenteric lymph nodes (McSorley et al., 2002). A similar, highly localized response to antigen produced in the draining lymph nodes was noted in the Mycobacterial model and may therefore be a common feature of mucosal infection (Reiley et al., 2008; Wolf et al., 2008). Interestingly, in the SM1 model the flagellin-specific response remained localized to the Peyer's patch and mesenteric lymph nodes despite systemic spread of the bacteria to the spleen and liver (McSorley et al., 2002). The explanation for this finding is still not clear but several subsequent experiments have addressed some of the potential issues.
A follow-up study indicated that strong competition from endogenous Salmonella-specific T cells contributes to poor persistence of SM1 cells in response to live, but not heat-killed bacteria (Srinivasan et al., 2004a). An intriguingly similar scenario was recently reported in which a CD4 TCR transgenic T cell clone initially proliferated but quickly died off during infection with recombinant Listeria monocytogenes (Williams et al., 2008). In this study, authors argued that the TCR transgenic clones faced competition from endogenous T cell clones specific for the same antigen. However, flagellin only constitutes a fraction of the overall CD4 T cell response to Salmonella (Srinivasan et al., 2004a), and the frequency of naive endogenous T cells specific for the flagellin I-Ab epitope in C57BL/6 mice is very low (Moon et al., 2007). Thus, competition may not be specifically for flagellin:I-Ab ligands but rather access to resources such as essential cytokines or antigen-presenting cells (APC).
Another possibility is that the expression of flagellin itself is simply not maintained following initial infection. Indeed, Cookson and colleagues have reported differential expression of flagellin in the spleen versus intestinal lymphoid tissue (Alaniz et al., 2006; Cummings et al., 2006). However, the transfer of Salmonella-infected splenocytes into naive mice causes activation of SM1 cells, arguing that flagellin is available in the spleen, although this activation was relatively weak suggesting that antigen may indeed be limiting if not absent (Srinivasan et al., 2004b). One possibility is that flagellin is expressed in infected spleens in a sequestered environment away from SM1 cells, but this has not been demonstrated experimentally. Whatever the mechanism for the lack of a systemic SM1 response, flagellin is one of the most abundant proteins expressed by Salmonella, and the evasion of a flagellin-specific T cell response in the spleen may thus represent an important immune evasion tactic by Salmonella.
6.4 Salmonella-specific tetramers
Despite the advantages of using the SM1 system, there are also drawbacks to the use of TCR transgenic adoptive transfer systems. One inherent limitation of this approach is that the epitope-specific T cell population under study is comprised of a single clonotype, raising the possibility that the behavior of a monoclonal population may not accurately reflect the full polyclonal repertoire of endogenous T cells responding to a given epitope. Also, it is now known that modification of the naïve precursor frequency by transfer of TCR transgenic T cells can alter the half-life of T cells, affect the kinetics of T cell activation and have deleterious effects on the development of immune memory (Badovinac et al., 2007; Ford et al., 2007; Foulds and Shen, 2006; Hataye et al., 2006; Marzo et al., 2005). These issues raise the possibility that an endogenous epitope-specific T cell may differ substantially from a relatively high frequency TCR transgenic population.
Direct visualization of endogenous T cell responses in unmanipulated mice would avoid these limitations, but the low frequency of epitope-specific endogenous cells makes this approach technically difficult, and indeed necessitated the development of the TCR transgenic adoptive transfer approach in the first place. However, recent developments in the construction of peptide:MHC tetramers combined with new techniques for detecting low frequency T cell populations now makes endogenous T cell tracking possible (Hataye et al., 2006; Moon et al., 2007). We have recently generated a flagellin427-441: I-Ab tetramer and have used it to examine endogenous flagellin-specific T cell responses in vivo (Moon et al., 2007). Our initial studies have demonstrated that naïve flagellin427-441: I-Ab-specific T cells are found at very low frequency in C57BL/6 mice (around 20 cells/mouse) but expand several hundred fold to a systemic dose of flagellin peptide and LPS. We are currently studying the development of endogenous flagellin-specific T cell responses in Salmonella infected mice using this tracking methodology. With this new technology, the labor-intensive generation of T cell lines and TCR transgenic mice will no longer be needed to study T cell responses to newly identified epitopes in pathogen organisms.
7. Development and function of Th1 cells during Salmonella infection
As noted above, numerous studies have demonstrated a requirement for Th1 cells producing IFN-γ for the resolution of Salmonella infection. However, the development of in vivo tracking approaches have allowed more detailed insight into the development of Salmonella-specific Th1 cells and how these cells mediate their effector function in vivo. These studies raise important questions about exactly when Th1 cells contribute to Salmonella immunity and the mechanism by which Th1 cells activate infected macrophages.
7.1 When do Th1 cells activate Salmonella-infected macrophages?
Initial studies tracking SM1 T cells in response to live infection demonstrated that these cells are activated extremely rapidly in intestinal lymphoid tissues of the gut (McSorley et al., 2002). We now know that this early activation is dependent upon the rapid migration of CCR6+ dendritic cells in response to Salmonella penetration of the Peyer's patch epithelium (Salazar-Gonzalez et al., 2006). Thus, Salmonella-specific T cells are activated within a few hours of oral infection. These data are somewhat at odds with earlier studies using gene-deficient mice which suggest that Salmonella-specific T cells only contribute in the late stage of disease resolution. Mice with deficiencies in Th1 cells or the development of Th1 cells all display a profound deficiency in bacterial clearance from the spleen and/or liver (Hess et al., 1996;Ravindran et al., 2005;VanCott et al., 1998; Weintraub et al., 1997), but this was only evident several weeks after infection. Thus, while recent visualization studies indicate activation of T cells within hours, bacterial colonization studies suggest T cells do not affect bacterial growth until several weeks later. The development of Th1 cells and their contribution to Salmonella immunity therefore requires more detailed study. One attractive possibility is that Salmonella actively inhibit the function of Th1 cells in vivo.
The ability of Salmonella to inhibit T cells has been examined in some depth and has generally focused on the ability of bacteria to regulate antigen presentation to avoid T cell activation (Cheminay et al., 2005; Qimron et al., 2004; Svensson et al., 2000; Tobar et al., 2006; Tobar et al., 2004; van der Velden et al., 2003; Yrlid et al., 2000). However, much of this work has been carried out in vitro and the in vivo significance is not readily apparent, especially given the fact that efficient activation of SM1 T cells occurs in response to oral or intravenous infection (McSorley et al., 2002; Salazar-Gonzalez et al., 2006; Srinivasan et al., 2004b). Considerably less attention has been given to the alternative possibility, that Salmonella can inhibit the survival or function of Salmonella-specific T cells after initial expansion and activation has already occurred. Indeed, we have already noted that SM1 T cells expand but fail to survive long-term in mice exposed to live bacteria (Srinivasan et al., 2004a). Our more recent data suggest that these SM1 cells die by apoptosis in a process dependent on bacterial virulence factors (Srinivasan and McSorley, unpublished data). Thus, Salmonella can directly inhibit the function of Salmonella-specific Th1 cells that are activated early in the response. Understanding the nature of this inhibitory effect may lead to the development of more effective therapeutics or more immunogenic vaccine vectors for typhoid.
7.2 How do Th1 cells activate Salmonella-infected macrophages?
Naïve CD4 T cells transit through the blood and secondary lymphoid tissues until they are activated via the TCR by peptides presented by dendritic cells in the context of MHC class-II (Jenkins et al., 2001). After an initial round of expansion, these activated T cells acquire effector functions and the ability to enter non-lymphoid tissues (Lefrancois, 2006; Swain et al., 2006). It is generally assumed that an effector Th1 cell trafficking to the liver will produce IFN-γ following recognition of peptide:MHC presented by a Salmonella-infected macrophage. Therefore, this theoretical model requires two independent recognition events via the TCR; the first in secondary lymphoid tissues, and the second at the infected site. However, recent data raise questions about whether this second recognition event is actually required.
Given the current lack of knowledge about T cell epitopes in Salmonella infection, our laboratory developed a crude means of visualizing endogenous Salmonella-specific T cells in vivo (Srinivasan et al., 2004a). This simply involved the injection of infected mice with a bacterial lysate and examining IFN-γ and TNF-α production directly ex vivo. A sizable population of CD4 and CD8 T cells was found producing these effector cytokines in infected mice but not in naïve mice (Srinivasan et al., 2004a), suggesting that this method allows visualization of polyclonal endogenous CD4 T cells responding to Salmonella. Surprisingly, CD4 and CD8 T cells were also activated to produce IFN-γ when Salmonella-infected mice were injected with ultrapure LPS (Srinivasan et al., 2007), demonstrating that T cell activation in this assay occurred by innate stimulation. Furthermore, previously activated Salmonella-specific T cells could respond and produce IFN-γ even after transfer and stimulation in MHC class-II deficient mice (Srinivasan et al., 2007). This “innate stimulation” of CD4 T cells was only evident during active infection and was partially dependent on IL-18 signaling (Srinivasan et al., 2007). Very similar results have been reported for virus-specific CD8 T cells in infected mice (Beadling and Slifka, 2005; Berg and Forman, 2006; Kambayashi et al., 2003), suggesting that innate stimulation of previously activated T cells is a fairly common phenomenon in infectious disease models. Thus, although TCR ligation is required for initial expansion of Salmonella-specific T cells in secondary lymphoid tissues, it may not be required for elaboration of IFN-γ production in the infected liver. The ability of effector T cells to respond to innate stimuli such as LPS or inflammatory cytokines may be important for amplifying the anti-bacterial effector response in the face of rapid bacterial replication. However, the relative importance of innate activation of CD4 or CD8 T cells versus TCR ligation has not been examined directly in any infectious disease model.
8. Conclusion
Visualization of pathogen-specific immune responses in vivo allows a more detailed understanding of immunity to infectious disease. The development of TCR transgenic adoptive transfer systems and new tetramer approaches will surely expand what we currently know about adaptive immune responses from traditional methodologies. This new knowledge is likely to lead to an in-depth understanding of T cell activation, effector function, and memory development and holds considerable promise for the generation of novel vaccines and therapeutics to treat infectious disease.
References
- Aggarwal A, Kumar S, Jaffe R, Hone D, Gross M, Sadoff J. J Exp Med. 1990;172:1083–90. doi: 10.1084/jem.172.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaniz RC, Cummings LA, Bergman MA, Rassoulian-Barrett SL, Cookson BT. J Immunol. 2006;177:3983–93. doi: 10.4049/jimmunol.177.6.3983. [DOI] [PubMed] [Google Scholar]
- Amanna I, Slifka MK. Viral Immunol. 2005;18:307–15. doi: 10.1089/vim.2005.18.307. [DOI] [PubMed] [Google Scholar]
- Badovinac VP, Haring JS, Harty JT. Immunity. 2007;26:827–41. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadling C, Slifka MK. Blood. 2005;105:1179–86. doi: 10.1182/blood-2004-07-2833. [DOI] [PubMed] [Google Scholar]
- Becker D, Selbach M, Rollenhagen C, Ballmaier M, Meyer TF, Mann M, Bumann D. Nature. 2006;440:303–7. doi: 10.1038/nature04616. [DOI] [PubMed] [Google Scholar]
- Berg RE, Forman J. Curr Opin Immunol. 2006;18:338–43. doi: 10.1016/j.coi.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Bergman MA, Cummings LA, Alaniz RC, Mayeda L, Fellnerova I, Cookson BT. Infect Immun. 2005;73:7226–35. doi: 10.1128/IAI.73.11.7226-7235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholet S, Goldszmid R, Morrot A, Debrabant A, Afrin F, Collazo-Custodio C, Houde M, Desjardins M, Sher A, Sacks D. J Immunol. 2006;177:3525–33. doi: 10.4049/jimmunol.177.6.3525. [DOI] [PubMed] [Google Scholar]
- Blanden RV, Mackaness GB, Collins FM. J Exp Med. 1966;124:585–600. doi: 10.1084/jem.124.4.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brines R. Immunol Today. 1996;17:203–4. doi: 10.1016/s0167-5699(96)90203-0. [DOI] [PubMed] [Google Scholar]
- Bueno SM, Gonzalez PA, Carreno LJ, Tobar JA, Mora GC, Pereda CJ, Salazar-Onfray F, Kalergis AM. Immunology. 2008 doi: 10.1111/j.1365-2567.2008.02805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno SM, Gonzalez PA, Schwebach JR, Kalergis AM. Immunol Lett. 2007;111:14–20. doi: 10.1016/j.imlet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Bumann D. Infect Immun. 2001a;69:4618–4626. doi: 10.1128/IAI.69.7.4618-4626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumann D. Infect Immun. 2001b;69:7493–500. doi: 10.1128/IAI.69.12.7493-7500.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumann D. FEMS Immunol Med Microbiol. 2003;37:105–9. doi: 10.1016/S0928-8244(03)00064-6. [DOI] [PubMed] [Google Scholar]
- Cheminay C, Mohlenbrink A, Hensel M. J Immunol. 2005;174:2892–9. doi: 10.4049/jimmunol.174.5.2892. [DOI] [PubMed] [Google Scholar]
- Chen ZM, Jenkins MK. Infect Immun. 1999;67:2025–9. doi: 10.1128/iai.67.4.2025-2029.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary AM, Tu W, Enright A, Giffon T, Dewaal-Malefyt R, Gutierrez K, Lewis DB. J Immunol. 2003;170:597–603. doi: 10.4049/jimmunol.170.1.597. [DOI] [PubMed] [Google Scholar]
- Cookson BT, Bevan MJ. J Immunol. 1997;158:4310–4319. [PubMed] [Google Scholar]
- Crump JA, Luby SP, Mintz ED. Bull World Health Organ. 2004;82:346–53. [PMC free article] [PubMed] [Google Scholar]
- Cummings LA, Wilkerson WD, Bergsbaken T, Cookson BT. Mol Microbiol. 2006;61:795–809. doi: 10.1111/j.1365-2958.2006.05271.x. [DOI] [PubMed] [Google Scholar]
- Diaz-Quinonez A, Martin-Orozco N, Isibasi A, Ortiz-Navarrete V. Infect Immun. 2004;72:3059–62. doi: 10.1128/IAI.72.5.3059-3062.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC. Mol Microbiol. 2003;47:103–18. doi: 10.1046/j.1365-2958.2003.03313.x. [DOI] [PubMed] [Google Scholar]
- Fierer J, Guiney DG. J Clin Invest. 2001;107:775–80. doi: 10.1172/JCI12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ML, Koehn BH, Wagener ME, Jiang W, Gangappa S, Pearson TC, Larsen CP. J Exp Med. 2007;204:299–309. doi: 10.1084/jem.20062319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds KE, Shen H. J Immunol. 2006;176:3037–43. doi: 10.4049/jimmunol.176.5.3037. [DOI] [PubMed] [Google Scholar]
- Germain RN, Jenkins MK. Curr Opin Immunol. 2004;16:120–5. doi: 10.1016/j.coi.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Gowen JW, Calhoun ML. Proc Natl Acad Sci U S A. 1943;29:144–9. doi: 10.1073/pnas.29.5.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassl GA, Finlay BB. Curr Opin Gastroenterol. 2008;24:22–6. doi: 10.1097/MOG.0b013e3282f21388. [DOI] [PubMed] [Google Scholar]
- Haskins K, Kubo R, White J, Pigeon M, Kappler J, Marrack P. J Exp Med. 1983;157:1149–69. doi: 10.1084/jem.157.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Science. 2006;312:114–6. doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- Henrickson SE, von Andrian UH. Curr Opin Immunol. 2007;19:249–58. doi: 10.1016/j.coi.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Hess J, Ladel C, Miko D, Kaufmann SH. J Immunol. 1996;156:3321–3326. [PubMed] [Google Scholar]
- Huang AY, Qi H, Germain RN. Immunity. 2004;21:331–9. doi: 10.1016/j.immuni.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Jenkins MK, Khoruts A, Ingulli E, Mueller DL, McSorley SJ, Reinhardt RL, Itano A, Pape KA. Annu Rev Immunol. 2001;19:23–45. doi: 10.1146/annurev.immunol.19.1.23. [DOI] [PubMed] [Google Scholar]
- Jones BD, Falkow S. Annu Rev Immunol. 1996;14:533–61. doi: 10.1146/annurev.immunol.14.1.533. [DOI] [PubMed] [Google Scholar]
- Jones-Carson J, McCollister BD, Clambey ET, Vazquez-Torres A. Infect Immun. 2007;75:2708–16. doi: 10.1128/IAI.01905-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouanguy E, Doffinger R, Dupuis S, Pallier A, Altare F, Casanova JL. Curr Opin Immunol. 1999;11:346–51. doi: 10.1016/s0952-7915(99)80055-7. [DOI] [PubMed] [Google Scholar]
- Kambayashi T, Assarsson E, Lukacher AE, Ljunggren HG, Jensen PE. J Immunol. 2003;170:2399–408. doi: 10.4049/jimmunol.170.5.2399. [DOI] [PubMed] [Google Scholar]
- Kearney ER, Pape KA, Loh DY, Jenkins MK. Immunity. 1994;1:327–39. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Khan CM, Villarreal-Ramos B, Pierce RJ, Riveau G, Demarco de Hormaeche R, McNeill H, Ali T, Fairweather N, Chatfield S, Capron A, et al. Proc Natl Acad Sci U S A. 1994;91:11261–5. doi: 10.1073/pnas.91.23.11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancois L. Immunol Rev. 2006;211:93–103. doi: 10.1111/j.0105-2896.2006.00393.x. [DOI] [PubMed] [Google Scholar]
- Lo WF, Ong H, Metcalf ES, Soloski MJ. J Immunol. 1999;162:5398–406. [PubMed] [Google Scholar]
- Lo WF, Woods AS, DeCloux A, Cotter RJ, Metcalf ES, Soloski MJ. Nat Med. 2000;6:215–8. doi: 10.1038/72329. [DOI] [PubMed] [Google Scholar]
- Luu RA, Gurnani K, Dudani R, Kammara R, van Faassen H, Sirard JC, Krishnan L, Sad S. J Immunol. 2006;177:1516–25. doi: 10.4049/jimmunol.177.3.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cajas JL, Wainberg MA. Antiviral Res. 2007;76:203–21. doi: 10.1016/j.antiviral.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Marzo AL, Klonowski KD, Le Bon A, Borrow P, Tough DF, Lefrancois L. Nat Immunol. 2005;6:793–9. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskell DJ, Sweeney KJ, O'Callaghan D, Hormaeche CE, Liew FY, Dougan G. Microb Pathog. 1987;2:211–21. doi: 10.1016/0882-4010(87)90022-2. [DOI] [PubMed] [Google Scholar]
- Mastroeni P. Curr Mol Med. 2002;2:393–406. doi: 10.2174/1566524023362492. [DOI] [PubMed] [Google Scholar]
- Mastroeni P, Chabalgoity JA, Dunstan SJ, Maskell DJ, Dougan G. Vet J. 2001;161:132–64. doi: 10.1053/tvjl.2000.0502. [DOI] [PubMed] [Google Scholar]
- Mastroeni P, Harrison JA, Chabalgoity JA, Hormaeche CE. Infect Immun. 1996;64:189–96. doi: 10.1128/iai.64.1.189-196.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni P, Sheppard M. Microbes Infect. 2004;6:398–405. doi: 10.1016/j.micinf.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Mastroeni P, Simmons C, Fowler R, Hormaeche CE, Dougan G. Infect Immun. 2000;68:46–53. doi: 10.1128/iai.68.1.46-53.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni P, Villarreal-Ramos B, Hormaeche CE. Microb Pathog. 1992;13:477–91. doi: 10.1016/0882-4010(92)90014-f. [DOI] [PubMed] [Google Scholar]
- Matsui H, Eguchi M, Kikuchi Y. Microb Pathog. 2000;29:53–9. doi: 10.1006/mpat.2000.0370. [DOI] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. Nature. 2001;413:852–6. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- McSorley SJ, Asch S, Costalonga M, Rieinhardt RL, Jenkins MK. Immunity. 2002;16:365–377. doi: 10.1016/s1074-7613(02)00289-3. [DOI] [PubMed] [Google Scholar]
- McSorley SJ, Cookson BT, Jenkins MK. J Immunol. 2000;164:986–93. doi: 10.4049/jimmunol.164.2.986. [DOI] [PubMed] [Google Scholar]
- McSorley SJ, Jenkins MK. Infect Immun. 2000;68:3344–8. doi: 10.1128/iai.68.6.3344-3348.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSorley SJ, Xu D, Liew FY. Infect Immun. 1997;65:171–8. doi: 10.1128/iai.65.1.171-178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM, Tauxe RV. Emerg Infect Dis. 1999;5:607–25. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misfeldt ML, Johnson W. Infect Immun. 1979;24:808–16. doi: 10.1128/iai.24.3.808-816.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittrucker H. J Immunol. 1999;163:6769–6776. [PubMed] [Google Scholar]
- Mittrucker HW, Kaufmann SH. J Leukoc Biol. 2000;67:457–63. doi: 10.1002/jlb.67.4.457. [DOI] [PubMed] [Google Scholar]
- Mittrucker HW, Raupach B, Kohler A, Kaufmann SH. J Immunol. 2000;164:1648–52. doi: 10.4049/jimmunol.164.4.1648. [DOI] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. Immunity. 2007;27:203–13. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran GJ, Talan DA, Abrahamian FM. Infect Dis Clin North Am. 2008;22:145–87. vii. doi: 10.1016/j.idc.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musson JA, Hayward RD, Delvig AA, Hormaeche CE, Koronakis V, Robinson JH. Eur J Immunol. 2002;32:2664–71. doi: 10.1002/1521-4141(200209)32:9<2664::AID-IMMU2664>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Nauciel C. J Immunol. 1990;145:1265–9. [PubMed] [Google Scholar]
- Nauciel C, Espinasse-Maes F. Infect Immun. 1992;60:450–4. doi: 10.1128/iai.60.2.450-454.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrin RS, Contag CH. Nat Rev Immunol. 2006;6:484–90. doi: 10.1038/nri1879. [DOI] [PubMed] [Google Scholar]
- Newton SM, Jacob CO, Stocker BA. Science. 1989;244:70–2. doi: 10.1126/science.2468182. [DOI] [PubMed] [Google Scholar]
- Okeke IN, Aboderin OA, Byarugaba DK, Ojo KK, Opintan JA. Emerg Infect Dis. 2007;13:1640–6. doi: 10.3201/eid1311.070674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape KA, Kearney ER, Khoruts A, Mondino A, Merica R, Chen ZM, Ingulli E, White J, Johnson JG, Jenkins MK. Immunol Rev. 1997;156:67–78. doi: 10.1111/j.1600-065x.1997.tb00959.x. [DOI] [PubMed] [Google Scholar]
- Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, Sebaihia M, Baker S, Basham D, Brooks K, Chillingworth T, Connerton P, Cronin A, Davis P, Davies RM, Dowd L, White N, Farrar J, Feltwell T, Hamlin N, Haque A, Hien TT, Holroyd S, Jagels K, Krogh A, Larsen TS, Leather S, Moule S, O'Gaora P, Parry C, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. Nature. 2001;413:848–52. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- Parry CM, Hien TT, Dougan G, White NJ, Farrar JJ. N Engl J Med. 2002;347:1770–82. doi: 10.1056/NEJMra020201. [DOI] [PubMed] [Google Scholar]
- Pasetti MF, Levine MM, Sztein MB. Vaccine. 2003;21:401–18. doi: 10.1016/s0264-410x(02)00472-3. [DOI] [PubMed] [Google Scholar]
- Pasetti MF, Salerno-Goncalves R, Sztein MB. Infect Immun. 2002;70:4009–18. doi: 10.1128/IAI.70.8.4009-4018.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul C, Shalala K, Warren R, Smith R. Infect Immun. 1985;48:40–3. doi: 10.1128/iai.48.1.40-43.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul CC, Norris K, Warren R, Smith RA. Infect Immun. 1988;56:2189–92. doi: 10.1128/iai.56.8.2189-2192.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Sette A. Nat Rev Immunol. 2007;7:485–90. doi: 10.1038/nri2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C, Kim SK, Marzo A, Williams K, Jiang J, Shen H, Lefrancois L. J Immunol. 2001;166:3402–3409. doi: 10.4049/jimmunol.166.5.3402. [DOI] [PubMed] [Google Scholar]
- Qimron U, Madar N, Mittrucker HW, Zilka A, Yosef I, Bloushtain N, Kaufmann SH, Rosenshine I, Apte RN, Porgador A. Cell Microbiol. 2004;6:1057–70. doi: 10.1111/j.1462-5822.2004.00418.x. [DOI] [PubMed] [Google Scholar]
- Rabsch W, Tschape H, Baumler AJ. Microbes Infect. 2001;3:237–47. doi: 10.1016/s1286-4579(01)01375-2. [DOI] [PubMed] [Google Scholar]
- Ravindran R, Foley J, Stoklasek T, Glimcher LH, McSorley SJ. J Immunol. 2005;175:4603–10. doi: 10.4049/jimmunol.175.7.4603. [DOI] [PubMed] [Google Scholar]
- Ravindran R, McSorley SJ. Immunology. 2005;114:450–8. doi: 10.1111/j.1365-2567.2005.02140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley WW, Calayag MD, Wittmer ST, Huntington JL, Pearl JE, Fountain JJ, Martino CA, Roberts AD, Cooper AM, Winslow GM, Woodland DL. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0801496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Dahlfors A, Buchan AM, Finlay BB. J Exp Med. 1997;186:569–80. doi: 10.1084/jem.186.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen C, Bumann D. Infect Immun. 2006;74:1649–60. doi: 10.1128/IAI.74.3.1649-1660.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen C, Sorensen M, Rizos K, Hurvitz R, Bumann D. Proc Natl Acad Sci U S A. 2004;101:8739–44. doi: 10.1073/pnas.0401283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Gonzalez RM, McSorley SJ. Immunol Lett. 2005;101:117–22. doi: 10.1016/j.imlet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Salazar-Gonzalez RM, Niess JH, Zammit DJ, Ravindran R, Srinivasan A, Maxwell JR, Stoklasek T, Yadav R, Williams IR, Gu X, McCormick BA, Pazos MA, Vella AT, Lefrancois L, Reinecker HC, McSorley SJ. Immunity. 2006;24:623–632. doi: 10.1016/j.immuni.2006.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo SP, Noursadeghi M, Cohen J, Holden DW. Cell Microbiol. 2001;3:587–97. doi: 10.1046/j.1462-5822.2001.00137.x. [DOI] [PubMed] [Google Scholar]
- Santos RL, Zhang S, Tsolis RM, Kingsley RA, Adams LG, Baumler AJ. Micrtobes Infect. 2001;3:1335–1344. doi: 10.1016/s1286-4579(01)01495-2. [DOI] [PubMed] [Google Scholar]
- Sheppard M, Webb C, Heath F, Mallows V, Emilianus R, Maskell D, Mastroeni P. Cell Microbiol. 2003;5:593–600. doi: 10.1046/j.1462-5822.2003.00296.x. [DOI] [PubMed] [Google Scholar]
- Sinha K, Mastroeni P, Harrison J, de Hormaeche RD, Hormaeche CE. Infect Immun. 1997;65:1566–69. doi: 10.1128/iai.65.4.1566-1569.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel J, Khan AS, Swerdlow DL. Lancet. 2002;359:874–80. doi: 10.1016/S0140-6736(02)07947-3. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Foley J, McSorley SJ. J Immunol. 2004a;172:6884–93. doi: 10.4049/jimmunol.172.11.6884. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Foley J, Ravindran R, McSorley SJ. J Immunol. 2004b;173:4091–9. doi: 10.4049/jimmunol.173.6.4091. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, McSorley SJ. Arch Immunol Ther Exp (Warsz) 2006;54:25–31. doi: 10.1007/s00005-006-0003-5. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, McSorley SJ. J Leukoc Biol. 2007;81:403–11. doi: 10.1189/jlb.0306194. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Salazar-Gonzalez RM, Jarcho M, Sandau MM, Lefrancois L, McSorley SJ. J Immunol. 2007;178:6342–9. doi: 10.4049/jimmunol.178.10.6342. [DOI] [PubMed] [Google Scholar]
- Strindelius L, Degling Wikingsson L, Sjoholm I. Infect Immun. 2002;70:1434–42. doi: 10.1128/IAI.70.3.1434-1442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LH, Chiu CH. Chang Gung Med J. 2007;30:210–9. [PubMed] [Google Scholar]
- Subramanian N, Qadri A. Nat Immunol. 2006;7:583–9. doi: 10.1038/ni1336. [DOI] [PubMed] [Google Scholar]
- Svensson M, Johansson C, Wick MJ. Infect Immun. 2000;68:6311–20. doi: 10.1128/iai.68.11.6311-6320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain SL, Agrewala JN, Brown DM, Jelley-Gibbs DM, Golech S, Huston G, Jones SC, Kamperschroer C, Lee WH, McKinstry KK, Roman E, Strutt T, Weng NP. Immunol Rev. 2006;211:8–22. doi: 10.1111/j.0105-2896.2006.00388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabaraie B, Sharma BK, Sharma PR, Sehgal R, Ganguly NK. Microbiol Immunol. 1994;38:553–9. doi: 10.1111/j.1348-0421.1994.tb01822.x. [DOI] [PubMed] [Google Scholar]
- Tarr PE, Kuppens L, Jones TC, Ivanoff B, Aparin PG, Heymann DL. Am J Trop Med Hyg. 1999;61:163–70. doi: 10.4269/ajtmh.1999.61.163. [DOI] [PubMed] [Google Scholar]
- Tobar JA, Carreno LJ, Bueno SM, Gonzalez PA, Mora JE, Quezada SA, Kalergis AM. Infect Immun. 2006;74:6438–48. doi: 10.1128/IAI.00063-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobar JA, Gonzalez PA, Kalergis AM. J Immunol. 2004;173:4058–65. doi: 10.4049/jimmunol.173.6.4058. [DOI] [PubMed] [Google Scholar]
- Ugrinovic S, Menager N, Goh N, Mastroeni P. Infect Immun. 2003;71:6808–19. doi: 10.1128/IAI.71.12.6808-6819.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velden AW, Velasquez M, Starnbach MN. J Immunol. 2003;171:6742–9. doi: 10.4049/jimmunol.171.12.6742. [DOI] [PubMed] [Google Scholar]
- VanCott JL, Chatfield SN, Roberts M, Hone DM, Hohmann EL, Pascual DW, Yamamoto M, Kiyono H, McGhee JR. Nat Med. 1998;4:1247–52. doi: 10.1038/3227. [DOI] [PubMed] [Google Scholar]
- Verma NK, Ziegler HK, Stocker BA, Schoolnik GK. Vaccine. 1995;13:235–44. doi: 10.1016/0264-410x(95)93308-v. [DOI] [PubMed] [Google Scholar]
- Walker RI. Expert Rev Vaccines. 2005;4:807–12. doi: 10.1586/14760584.4.6.807. [DOI] [PubMed] [Google Scholar]
- Weintraub BC, Eckmann L, Okamoto S, Hense M, Hedrick SM, Fierer J. Infect Immun. 1997;65:2306–12. doi: 10.1128/iai.65.6.2306-2312.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick MJ. Immunol Lett. 2003;85:99–102. doi: 10.1016/s0165-2478(02)00230-4. [DOI] [PubMed] [Google Scholar]
- Wick MJ, Pfeifer JD. Eur J Immunol. 1996;26:2790–9. doi: 10.1002/eji.1830261135. [DOI] [PubMed] [Google Scholar]
- Wijburg OL, Uren TK, Simpfendorfer K, Johansen FE, Brandtzaeg P, Strugnell RA. J Exp Med. 2006;203:21–6. doi: 10.1084/jem.20052093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, Ravkov EV, Bevan MJ. Immunity. 2008;28:533–45. doi: 10.1016/j.immuni.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. J Exp Med. 2008;205:105–15. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse ME, Haydon DT, Antia R. Trends Ecol Evol. 2005;20:238–44. doi: 10.1016/j.tree.2005.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrlid U, Svensson M, Johansson C, Wick MJ. FEMS Immunol Med Microbiol. 2000;27:313–20. doi: 10.1111/j.1574-695X.2000.tb01445.x. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM. Infect Immun. 1976;13:1069–73. doi: 10.1128/iai.13.4.1069-1073.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]