

Abstract
Summary: The ProteoWizard software project provides a modular and extensible set of open-source, cross-platform tools and libraries. The tools perform proteomics data analyses; the libraries enable rapid tool creation by providing a robust, pluggable development framework that simplifies and unifies data file access, and performs standard proteomics and LCMS dataset computations. The library contains readers and writers of the mzML data format, which has been written using modern C++ techniques and design principles and supports a variety of platforms with native compilers. The software has been specifically released under the Apache v2 license to ensure it can be used in both academic and commercial projects. In addition to the library, we also introduce a rapidly growing set of companion tools whose implementation helps to illustrate the simplicity of developing applications on top of the ProteoWizard library.
Availability: Cross-platform software that compiles using native compilers (i.e. GCC on Linux, MSVC on Windows and XCode on OSX) is available for download free of charge, at http://proteowizard.sourceforge.net. This website also provides code examples, and documentation. It is our hope the ProteoWizard project will become a standard platform for proteomics development; consequently, code use, contribution and further development are strongly encouraged.
Contact: darren@proteowizard.org; parag@ucla.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Historically, three factors have led to a significant overhead in developing proteomics software. The first challenge of writing analysis tools is supporting data generated by different vendors; each vendor typically encodes their data in a vendor-specific, proprietary, closed format, with access restricted to a particular operating system. To simplify cross-vendor software development two open formats were created (Pedrioli et al., 2004), which led to the second challenge: reading and writing of these open formats. No single, natively-compiling cross-platform, open-source software library, has emerged to allow academic and commercial groups to encode or access encoded data. Despite this, the existence of open standards greatly simplified development, and numerous open source software projects, such as the Trans-Proteomic Pipeline (TPP) (Keller et al., 2005), the OpenMS Proteomic Pipeline (TOPP) (Kohlbacher et al., 2007) and the Computational Proteomics Analysis System (CPAS) (Rauch et al., 2006) were built upon the open formats. More recently, the HUPO-PSI and Institute for Systems Biology have taken an important step in simplifying proteomics software development by creating the mzML data format standard to be released in June 2008 (Orchard et al., 2007). The third challenge is the lack of a single, standard cross-platform library that performs common calculations, such as protein digestion, mass computation, peak integration, charge state detection and isotope deconvolution. Consequently, these calculations have been repeatedly implemented in the course of developing more sophisticated tools.
2 DESIGN AND IMPLEMENTATION
ProteoWizard has been designed from the ground-up with testability in mind. Each code module has a unit test that runs automatically during the build process. In addition, the data model has ‘diff’ calculations built in, for validating data after format conversion or preprocessing.
As illustrated in Figure 1, ProteoWizard is built from many independent libraries, grouped together in dependency levels. Each library depends only on libraries in lower levels of the hierarchy. The lowest layer, the utility layer, contains independent classes that perform computations applicable in a wide variety of situations such as binary to text encoding, XML parsing and mathematical calculations that are common in data analysis. Abstract interfaces are used in analysis modules to facilitate comparison between different algorithms. Below we describe the data and analysis layers in greater detail.
Fig. 1.

ProteoWizard architecture.
2.1 The data layer
The ProteoWizard data layer abstracts the source data file, hiding any format-specific details. The underlying data model of the data layer is a one-to-one translation from mzML data elements to C++ data structures. This mapping is shown in the data model details in the Supplementary Material. Metadata fields are encoded in <cvParam> elements, which refer to terms (by their accession number) in a controlled vocabulary (CV) maintained in a central repository by the HUPO-PSI. ProteoWizard parses the CV file at compile time and generates C++ code, which allows convenient, typesafe handling of the CV terms.
The data model uses a virtual interface for accessing spectrum lists, to allow lazy evaluation when accessing the spectra contained in a data file, improving data access time, as files can be read incrementally as needed.
Vendor proprietary formats are handled with a plug-in reader interface. ProteoWizard currently supports reading of mzML, mzXML and Thermo RAW files. As vendor libraries are subject to platform restrictions some features of ProteoWizard may only be available if a vendor library is found at runtime. Collaboration with other labs (Vanderbilt, Nashville, TN, ISB, Seattle, WA) are underway to implement readers for all vendor formats.
Data writing is also handled in a modular, extensible manner, with C++ iostream serialization currently supported for mzML and mzXML. Because ProteoWizard has been released with a permissive license, it can be incorporated in commercial software projects.
2.2 The analysis layer
The analysis layer contains all scientific computation, in reusable modules that are independent of platform and data format. Currently available are modules for convenient access to the underlying data model, spectrum data caching, data and metadata extraction, selected ion chromatogram calculation and pseudo-2D-gel image creation. There are also independent modules for handling chemical formulas, peptide calculations and isotope envelopes. More analysis modules are currently in development, with an emphasis on establishing standard interfaces for common proteomics computations, such as peak picking, isotope de-convolution and precursor estimation. Our goal is to work collaboratively to create an analysis infrastructure in which experts in a particular area will be able to contribute a module that can then be plugged into various software tools. This will allow, for example, an expert in signal processing to create and contribute a peak picker, without having to handle details about file formats, operating system or command line configuration.
2.3 The tools layer
The tools layer code is responsible only for regulating interaction between the user and the analysis modules. Several tools have been implemented using the ProteoWizard Library including:
msConvert: data format conversion from vendor proprietary formats to mzML and mzXML.
msDiff: comparison of two data files, for validation of conversion and preprocessing.
msAccess: command line access to mass spec data files, including spectrum binary data and metadata, selected ion chromatograms. The modular layout of msAccess is shown in Figure 2.
msPicture: pseudo-2D gel image creation tool. msPicture can read from a variety of formats to generate dataset images. In addition, if given a pepXML file, msPicture can denote triggered MS/MS, colored by peptideProphet score. We show an example output in Figure 3.
The hello_analyzer program has been provided in the code example supplement as a demonstration of how easily one can use Proteowizard for creating analysis tools.
Fig. 2.

Architecture of msAccess tool (utility and data layers).
Fig. 3.
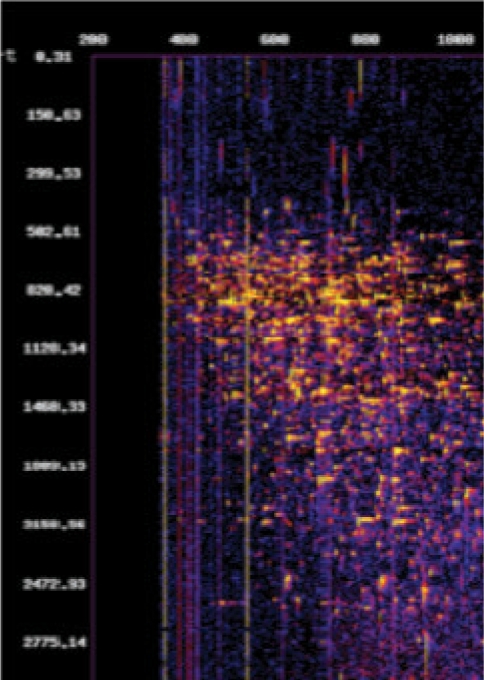
msPicture output.
3 SUMMARY AND FUTURE DIRECTIONS
As the proteomics field continues to grow, an increasing number of research groups are writing custom software for data analysis. Our goal is for ProteoWizard to become a repository where proteomics software developers can share their work, and benefit from the work of others. We welcome contributions of existing code, or new projects that will benefit the proteomics community. Already, the TPP tools will be using ProteoWizard to support mzML in the next major release, in advance of the formal release of mzML at ASMS 2008. SWIG bindings are currently being developed to allow access to the ProteoWizard Library from JAVA, Python, PERL and R.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Roland Luethy and Jonathan Katz for contributing towards the development and application of ProteoWizard, Natalie Tasman for code contributions, Brian Pratt for RAMP implementation and Eric Deutsch, the ISB NHLBI Proteome Center and the HUPO-PSI for help with ProteoWizard's mzML interfaces.
Funding: This work has been supported by the NCI CCNE-TR, the Wunderkinder Foundation and by the Bennioff Foundation. M.C. is supported by NIH Grants: R01 CA126218 and U24 CA126479.
Conflict of Interest: none declared.
REFERENCES
- Keller A, et al. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 2005;1:1–8. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlbacher O, et al. TOPP–the OpenMS proteomics pipeline. Bioinformatics. 2007;23:e191–e197. doi: 10.1093/bioinformatics/btl299. [DOI] [PubMed] [Google Scholar]
- Orchard S, et al. Five years of progress in the standardization of proteomics data 4th annual spring workshop of the HUPO-proteomics standards initiative April 23-25, 2007 Ecole Nationale Superieure (ENS), Lyon, France. Proteomics. 2007;7:3436–3440. doi: 10.1002/pmic.200700658. [DOI] [PubMed] [Google Scholar]
- Pedrioli PG, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- Rauch A, et al. Computational Proteomics Analysis System (CPAS): an extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. J. Proteome Res. 2006;5:112–121. doi: 10.1021/pr0503533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.