

Abstract
The adipose-derived hormone leptin communicates information about metabolic status to the hypothalamic GnRH neuronal system. It is unclear whether leptin can act directly on GnRH neurons. To examine this, we used three approaches. First, the presence of leptin-induced signal transducer and activator of transcription-3 activation was examined in GnRH neurons in male and female rats. Intracerebroventricular treatment with 4 μg leptin-induced robust signal transducer and activator of transcription-3 expression within the anteroventral periventricular nucleus but not in GnRH neurons. Second, fertility was assessed in male and female CRE-loxP transgenic mice with conditional leptin receptor (Lepr) deletion from either all forebrain neurons or GnRH neurons only. Forebrain neuron LEPR deletion prevented the onset of puberty resulting in infertility in males and females and blocked estradiol-induced LH surge. However, mice with GnRH neuron-selective Lepr deletion exhibited normal fertility apart from a slight puberty delay in males. Lastly, the highly sensitive technique of single-cell nested PCR was used to test for Lepr transcript presence in individual GnRH neurons, identified in situ using GnRH-green fluorescent protein transgenics. Whereas 75% of positive control (proopiomelanocortin) neurons contained Lepr mRNA, no (none of 18) GnRH neurons were Lepr mRNA positive. Collectively, these results show that leptin does not act directly on GnRH neurons in rats and mice. Leptin appears to regulate GnRH function via forebrain neurons that are afferent to GnRH because forebrain neuronal LEPR deletion caused infertility. The location and phenotype of these leptin-responsive neurons remains to be elucidated.
Leptin does not act directly on GnRH neurons, which do not contain leptin receptor mRNA; rather, leptin regulates fertility via neurons afferent to GnRH.
Neuroendocrine regulation of mammalian fertility is governed by episodic release of the hypothalamic peptide GnRH, which acts in a hypophysiotropic manner to drive the anterior pituitary gonadotropes to secrete LH and FSH in a pulsatile fashion. These hormones promote male and female gonadal function (1,2). Environmental regulation of GnRH neuronal activity determines the reproductive status of the individual (1). For example, during periods of food restriction, GnRH and LH secretion are reduced and infertility may occur (3,4). This state is also characterized by a low circulating concentration of the adipose-derived hormone leptin (5,6). Leptin communicates the availability of oxidizable fuels to the central nervous system and causes reduced appetite and increased energy expenditure (7) via actions on proopiomelanocortin (POMC) (8,9) and neuropeptide Y (10) neurons in the hypothalamic arcuate nucleus.
In addition to metabolic regulation, leptin is required for reproductive function. Animals lacking functional leptin, or its receptor, show marked suppression in pulsatile LH secretion and are infertile (11,12,13,14). Infertility can be reversed by treatment with exogenous leptin (15). Additionally, leptin treatment overcomes the reduction in LH pulse frequency caused by short-term fasting in rats and primates (6,16). However, refeeding fasted animals restores GnRH and LH pulses before leptin levels rise (17,18). Therefore, there are multiple pathways involved between nutritional status and reproductive function. Indeed, others have shown metabolic fuels such as glucose and fatty acids are important for maintenance of the reproductive system (19,20).
Whereas leptin receptor (LEPR) isoforms are widely expressed in the brain and periphery (21,22,23), intracerebroventricular (i.c.v.) leptin injections are sufficient to overcome both the appetite and reproductive effects of leptin deficiency (24,25), whereas central infusion of leptin antiserum mimics the effects of fasting (26). However, in one experiment in which LEPRs were selectively deleted from brain neurons using conditional transgenics, 70–80% of the mice remained fertile (27). The LEPR-B isoform is strongly expressed in the hypothalamus and other regions of the brain (28). Most lines of evidence indicate GnRH neurons do not express LEPRs, but this remains to be conclusively shown. No LEPRs have been detected on GnRH neurons in vivo, as assessed in rats by immunofluorescence (29) and in monkeys by in situ hybridization (16). RT-PCR analysis revealed expression of Lepr mRNA in GnRH-expressing (GT1–7 and NLT) cell lines (23,30). Moreover, inhibition of Janus kinase-2, responsible for the leptin-dependent phosphorylation of signal transducer and activator of transcription (STAT)-3, within GnRH neurons abolishes the effects of leptin on GnRH neuronal activity (31). Furthermore, leptin can stimulate GnRH secretion from cultured rat hypothalamic tissue, even when dispersed into single cells (32), suggesting direct actions of leptin on GnRH neurons.
To resolve the question of whether leptin can directly regulate GnRH neuronal function in vivo, we used three different approaches in the present study. Immunohistochemical detection of leptin-induced phosphorylated STAT (pSTAT)-3 expression is a widely used technique for identifying leptin-responsive cells (33,34). Therefore, pSTAT3 immunohistochemistry was used to assay leptin signaling in GnRH neurons in female and male rats. Second, we used CRE-loxP mediated conditional transgenics to remove LEPR either from all forebrain neurons or specifically from GnRH neurons and examined the fertility of these mutant mice. Finally, we used single-cell RT-PCR to examine for the presence or absence of Lepr mRNA in GnRH neurons.
Materials and Methods
Animals
Male and female Sprague Dawley rats and transgenic mice (C57BL/6J background strain) were obtained from the University of Otago animal breeding facility. Rodents were housed under conditions of controlled lighting (lights on from 0600 to 1800 h) and temperature (22 ± 1 C), and had free access to standard rodent chow and water. The University of Otago Animal Ethics Committee approved all animal experimental protocols.
Experiment 1: measurement of leptin-induced pSTAT3 colocalization in GnRH neurons
Male and diestrous female rats (aged 9–12 wk and weighing 250–350 g) were fitted with 22-gauge i.c.v. cannulae under anesthesia to permit leptin injection into a lateral ventricle as previously described (34). The placement of the guide cannula in the lateral ventricle was confirmed by monitoring water intake after 10 ng of angiotensin was administered i.c.v. A positive response was taken as greater than 5 ml water consumed within 30 min. After at least a 3-d recovery period, rats were fasted overnight to reduce the basal circulating leptin concentration. The following morning rats received a single i.c.v. injection of 4 μg recombinant mouse leptin (National Hormone and Peptide Program, Torrance, CA) or 2 μl artificial cerebrospinal fluid using an injection cannula prepared to protrude 1 mm beyond the guide cannula. The rats were perfused 30 min later for brain collection and pSTAT3 + GnRH dual immunohistochemical staining (all groups: n = 5, except leptin-treated males: n = 3).
Coronal (40 μm thick) sections throughout the septal-preoptic area and hypothalamus were cut from each brain on a sliding microtome with a freezing stage to provide four sets of consecutive sections (160 μm apart). One set of sections was dual labeled for GnRH and pSTAT3 by first incubating in polyclonal rabbit anti-pSTAT3 primary antibody (Tyr705; Cell Signaling Technology, Danvers, MA;1:1000 dilution) followed by biotinylated goat antirabbit IgG secondary antibody (1:500; Vector Laboratories Inc., Burlingame, CA) and then incubation in a nickel-enhanced diaminobenzidine solution to visualize pSTAT3 immunoreactivity (blue-black nuclear staining). After this, remaining peroxidase activity was removed by washing and sections incubated in a polyclonal rabbit anti-GnRH primary antibody (1:6000 rabbit anti-GnRH; SW1 kindly donated by Susan Wray, National Institute of Neurological Disorders and Stroke, Bethesda, MD), followed by biotinylated goat antirabbit IgG secondary antibody as before. GnRH immunoreactivity was visualized using diaminobenzidine without nickel to generate a brown precipitate. All GnRH neurons in three sections per animal, which included the medial septum and region around the organum vasculosum of the lamina terminalis (OVLT), were counted (Figs. 17–18 in Ref. 35). Additionally, total numbers of pSTAT3-immunoreactive cells were counted within the anteroventral periventricular nucleus (AVPV; Fig. 18 in Ref. 35) in two sections per animal. Omission of either primary antibody resulted in a complete absence of staining.
GnRH neurons were counted as pSTAT3 positive if they had a distinct blue-black-stained nucleus surrounded by brown cytoplasmic staining, by an operator blind to the treatment groups.
Experiment 2a: effects of neuron-specific LEPR knockout on fertility
To generate a neuron-specific LEPR knockout mouse, homozygous female Lepr flox mice (Leprfl/fl; loxP sites flanking Lepr coding exon 17, a region that encodes a Janus kinase docking site required for STAT3 signaling) (36) were bred to male CamKIIα-iCre BAC (referred to as CamKIIα-Cre) mice (37). CamKIIα is expressed around birth in almost all forebrain neurons but not glial cells (38,39). The resulting male Leprfl/+,CamKIIα-Cre mice were backcrossed to female Leprfl/fl mice to generate Leprfl/fl,CamKIIα-Cre conditional knockout mice (referred to as neuron-specific LEPR knockout mice). Leprfl/fl littermates served as controls. Transgenic mice were identified by PCR analysis of genomic DNA isolated from tail biopsies using the following primer sets and annealing temperatures: CamKIIα-Cre identification, GGT TCT CCG TTT GCA CTC AGG A, CCT GTT GTT CAG CTT GCA CCA G and CTG CAT GCA CGG GAC AGC TCT (62 C); Leprfl identification, AAT GAA AAA GTT GTT TTG GGA CGA and CAG GCT TGA GAA CAT GAA CAC AAC AAC (59 C); and to detect Lepr excision, AAT GAA AAA GTT GTT TTG GGA CGA and CTG ATT TGA TAG ATG GTC TTG AG (59 C).
For fertility assessment in males and females, Leprfl/fl (females: n = 9, males: n = 7) and neuron-specific LEPR knockout (females: n = 12, males: n = 7), mice were monitored for puberty onset and subsequent fertility. Females were examined for vaginal opening from 25 d of age and vaginal smears collected from the day of vaginal opening for determination of first estrus occurrence by cytological examination. At 10 wk of age, females were housed in groups of three in a split cage that allowed pheromonal and visual but not physical contact with a mature wild-type male (40), and daily vaginal cytology was again conducted for 10 d to examine estrous cyclicity. Immediately after this, all females were individually paired with mature wild-type males for 120 d and the frequency and size of litters produced recorded. The experimental male mice were paired with mature wild-type females from 38 d of age and the frequency and size of litters produced recorded for 110 d. Date of puberty onset in males was determined by backdating 21 d (gestation length) from the date of birth of the first litter.
To test whether neuronal leptin signaling is required for the preovulatory GnRH/LH surge response to high concentrations of estradiol, additional adult Leprfl/fl (n = 7) and neuron-specific LEPR knockout (n = 5) female mice were subjected to a preovulatory surge induction model as described previously (41). Briefly, mice were ovariectomized and had an estradiol-filled silicone rubber capsule, 1.0 mm internal, 2.1 mm external diameter (Dow Corning, MI), filled with medical-grade silicone rubber adhesive (Dow Corning) containing 17β-estradiol (0.1 mg/ml adhesive; Sigma-Aldrich, St. Louis, MO) implanted sc. Each mouse was given a 1 cm long capsule (containing 1 μg estradiol) per 20 g body weight. Six days after ovariectomy, mice received a sc injection of estradiol benzoate (1 μg/20 g body weight; Sigma-Aldrich) at 0900 h to induce an LH surge. At 1800 h on the following day, the surge peak time (41), animals were perfused for brain collection and c-Fos + GnRH dual-immunohistochemical staining. A trunk blood sample was collected, the plasma harvested, and stored at −20 C for LH RIA.
Coronal (30 μm thick) sections throughout the septal-preoptic area and hypothalamus were cut as above to provide three sets of consecutive sections (90 μm apart). One set of sections was dual labeled for GnRH and c-Fos using a similar protocol to experiment 1, except that the primary antibodies were rabbit anti c-Fos (AB5; Calbiochem,San Diego, CA; 1:20,000 dilution) and mouse monoclonal anti-GnRH (HU4H, a kind gift from Henry Urbanski, Oregon Health Sciences University, Portland, OR; 1:10,000 dilution). The c-Fos antibody does not cross-react with Fos-related antigens or FosB. The monoclonal GnRH antibody was followed with biotinylated goat antimouse IgG antibody (1:500; Vector Laboratories). Omission of any of the primary antibodies resulted in a complete absence of staining. GnRH neurons in three sections per animal, which included the medial septum and region around the OVLT, were counted by an operator blind to the treatment groups (Figs. 25–28 in Ref. 42).
Serum LH concentrations were measured in 25 μl plasma by RIA. Values are expressed in terms of the rat standard NIDDK-rat LH-RP-3. The tracer, iodinated hormone (NIDDK-rat LH-I-10) was used, and the primary antiserum was NIDDK-rabbit antirat LH-S11 (final dilution 1:500,000). The sensitivity of the assay (95% confidence interval at 0 ng/ml on the standard curve) was 0.2 ng/ml. For a serum pool falling in the middle of the standard curve, the intraassay coefficient of variation was 16%. All samples were analyzed in a single assay.
Experiment 2b: effects of GnRH-specific knockout on fertility
Breeding female Leprfl/fl mice to male GnRH-Cre mice generated GnRH neuron-specific LEPR knockout mice (41). The GnRH-Cre line has been shown to selectively express cAMP response element (CRE) recombinase in 97% of all GnRH neurons (41). The resulting male Leprfl/+,GnRH-Cre mice were backcrossed to female Leprfl/fl mice to generate Leprfl/fl,GnRH-Cre conditional knockout mice (referred to as GnRH-specific LEPR knockout mice). Leprfl/fl littermates served as controls. PCR primers for identification of GnRH-Cre DNA from tail biopsies were CCT GGA AAA TGC TTC TGT CCG and CAG GGT GTT ATA AGC AAT CCC (55 C).
Puberty onset and fertility was assessed in Leprfl/fl (females, n = 7; males n = 7) and GnRH-specific LEPR knockout mice (females: n = 6, males: n = 6) mice as in experiment 2a.
Experiment 3: single-cell collection and RT-PCR for Lepr
Single-cell RT-PCR analysis of Lepr mRNA presence in GnRH neurons was undertaken as reported previously (43,44). For consistency, C57BL/6J diestrous female mice expressing green fluorescent protein (GFP) under the control of the GnRH promoter (GnRH-GFP mice) (45) were used to collect coronal brain slices (200 μm thick) containing the medial septum and preoptic area of the hypothalamus. To harvest fluorescent neurons, slices were placed into a recording chamber and mounted onto an upright microscope (BX51; Olympus, Tokyo, Japan) fitted with differential interference contrast optics. GnRH neurons were identified as vertically oriented GFP-expressing bipolar cells located in the medial septum and rostral preoptic area adjacent to the OVLT.
Patch electrodes used to harvest cell cytoplasm were pulled on a Flaming-Brown P-97 puller (Sutter Instruments, Novato, CA) to a tip resistance of 4–5 mΩ. Electrodes were filled with about 8 μl of sterile internal solution [140 mm KCl, 10 mm EGTA, 1 mm MgCl2, 1 mm CaCl2, 10 mm HEPES (pH 7.3) with KOH] to a resistance of 8 mΩ. Fluorescent cells from the medial septum and region around the OVLT were located then approached with a patch pipette under positive pressure. When the cell of interest was contacted, a high-resistance seal was made with the cell body via negative pressure (1 GΩ) and the cell cytoplasm, but not the nucleus, was harvested under visual control. To control for contamination by foreign debris, we conducted mock harvests, in which the pipette was lowered into the slice and contact made with a labeled cell, but no cell contents were removed.
As a positive control for Lepr mRNA within individual cells, the cytoplasmic contents of POMC cells were collected (8). To generate POMC-GFP mice, POMC-Cre mice were mated with mice homozygous for a CRE-dependent GFP reporter cassette [B6;129-Gt(ROSA) 26Sortm2Sho/J] (46). The resulting offspring showed GFP expression specifically in cells matching the location of POMC neurons in the ventral portion of the arcuate nucleus. Coronal brain slices containing the arcuate nucleus were used to collect these cells as described for GnRH neurons.
Immediately after harvesting, cytoplasmic contents were expelled from the collection pipette into reverse transcriptase priming solution [5 μm Oligo(dT)20, 2.5 ng/μl random hexamers, 1 mm deoxynucleotide triphosphates] and heated to 65 C for 5 min. After briefly cooling on ice, buffer [55 mm Tris-HCl, 80 mm KCl, 3 mm MgCl2 (pH 8.3)], 6 mm dithiothreitol, and 4 U/μl of ribonuclease inhibitor (RNaseOUT, Invitrogen, Carlsbad, CA) were added to the mixture and incubated at room temperature for 5 min before the addition of 20 U/μl of reverse transcriptase (SuperScript III; Invitrogen). Samples were incubated at 50 C for 50 min and the enzyme inactivated at 70 C for 15 min. Resulting cDNA was stored at −20 C until multiplex nested PCR was performed the following day as reported previously (43,47). A 100-μl first round PCR [0.2 mm deoxynucleotide triphosphates, 10 mm Tris-HCl, 50 mm KCl, 1.5 mm MgCl2 (pH 8.3)] was performed on 5 μl of the reverse transcriptase product from each individual cell using 2.5 U of Taq DNA polymerase (Roche Diagnostics, Mannheim, Germany). The following negative controls were included: 1) cell contents processed in parallel but without reverse transcriptase, 2) pipette solution from mock harvests, and 3) water in place of template. Primers for GnRH, POMC, and leptin receptor spanned at least one intron/exon boundary and could therefore be used to detect the presence of contaminating genomic DNA. GnRH or POMC and leptin receptor oligonucleotide primer pairs at 10 pmol each were pooled in the same PCR (see Table 1), performed as follows: 94 C (3 min), 59 C (2 min), 72 C (3 min), and then 35 cycles of 94 C (40 sec), 59 C (1 min), and 72 C (1 min). A final extension step of 72 C for 5 min was used to complete the reaction. Second-round PCR using 1 μl of the first round product was performed as above (see Table 1 for inner primers pairs) to detect GnRH, POMC, and LEPR in separate 20-μl reactions for an additional 35 cycles. Primers for LEPR were designed to detect all isoforms of the receptor that are generated by alternative splicing (48). Resulting amplicons (GnRH = 213 bp, POMC = 173 bp, Lepr = 214 bp) were resolved on a 2% agarose gel containing ethidium bromide and visualized on a gel documentation system (Ultra.LUM, Inc., Carson, CA).
Table 1.
Primers sequences (5′–3′) used in nested single-cell RT-PCR experiments
| Gene name | Accession no. | Sequence | Amplicon (bp) |
|---|---|---|---|
| GnRH | NM_008145 | ||
| 1st round | F, tgctccagccagcactggtcc | 262 | |
| R, caatgttatactcgggtgttgtgg | |||
| 2nd round | F, cactggtcctatgggttgcgc | 213 | |
| R, agtgcatatacatcttcttctgcc | |||
| POMC | NM_008895 | ||
| 1st round | F, cgacggaagagaaaagaggtt | 357 | |
| R, ttttcagtcaggggctgttc | |||
| 2nd round | F, catctttgtccccagagagc | 173 | |
| R, gcaccagctccacacatcta | |||
| Lepra | NM_146146 | ||
| 1st round | F, ctggatgaaaggggacttga | 537 | |
| R, ctcttgctcctcacctgga | |||
| 2nd round | F, agtcttcggggatgtgaatg | 214 | |
| R, tttggctgtcccaagaaatc |
F, Forward; R, reverse.
Primers are designed to detect all forms of the receptor.
Statistical analysis
Significance (P < 0.05) was tested using nonparametric Mann-Whitney U tests or one-way ANOVA, with repeated measures analysis for body weight data as indicated in the text. This was followed, where appropriate, by the Bonferroni t test for post hoc analysis to determine where significant effects occurred. Results are presented as mean ± sem.
Results
Experiment 1: leptin-induced pSTAT3 expression in GnRH neurons
As expected, i.c.v. leptin treatment induced a marked increase in nuclear pSTAT3 immunoreactivity in the arcuate nucleus and ventromedial hypothalamic nucleus (data not shown). Leptin also induced pSTAT3 immunoreactivity in cells within the rostral preoptic area, particularly the region around the OVLT, in close proximity to where many GnRH neurons were observed. However, there was a striking absence of pSTAT3 in GnRH neurons in both leptin-treated and control rats; no GnRH neurons were found to express pSTAT3 in females (53 ± 7 GnRH neurons examined/rat) and only one GnRH neuron expressed pSTAT3 in all of the male rats (96 ± 4 GnRH neurons examined/rat; Fig. 1, A and B).
Figure 1.

Top panels, Leptin treatment did not induce pSTAT3 in GnRH neurons. Bottom panels, Leptin treatment induced pSTAT3 in the AVPV of female rats. Representative examples of GnRH (cytoplasmic staining) and pSTAT3 (dark nuclear staining) immunoreactivity in rats treated i.c.v. with vehicle (A and C) or 4 μg leptin (B and D). No colocalization between GnRH and pSTAT3 was seen in male or female rats. Arrows denote pSTAT3 immunoreactivity in unidentified cells in the vicinity of GnRH neurons. Dashed lines indicate the boundaries of the AVPV. Scale bars, 20 μm (upper panels); 75 μm (lower panels). 3V, Third ventricle.
In the AVPV, a region known to provide stimulatory input to GnRH neurons during the preovulatory GnRH/LH surge (41), leptin treatment caused a pronounced increase in pSTAT3 immunoreactivity in female rats (88 ± 5 positive cells/section; n = 5, vs. controls 26 ± 4 positive cells/section; n = 5; P < 0.001). Representative examples are shown in Fig. 1, C and D. A similar but less pronounced trend existed in male rats that failed to reach statistical significance (leptin treated: 39 ± 15 positive cells/section; n = 3, compared with controls 15 ± 5 positive cells/section; n = 5; P = 0.08).
Experiment 2a: effects of neuron-specific LEPR knockout on fertility
Postweaning body weight gain in neuron-specific LEPR knockout mice was approximately double that of Leprfl/fl controls (repeated measures ANOVA: P < 0.001; Fig. 2, A and B, for females and males, respectively). Female control mice exhibited vaginal opening at 32.8 ± 0.6 d old, with first estrus at d 37.2 ± 0.9. In female neuron-specific LEPR knockout mice, the mean date of vaginal opening was delayed compared with controls (38.5 ± 1.1 d; P < 0.001), with no estrous smears observed by 47 d of age (Fig. 2C). Adult control mice exhibited normal cyclicity, whereas the neuron-specific LEPR knockout mice were acyclic, exhibiting persistent diestrus-like vaginal smears (Fig. 2D). Furthermore, female mice were unable to produce litters when paired with wild-type males; compared with control females that produced a litter every 25.8 ± 0.9 d (Fig. 2E). The infertility in neuron-specific LEPR knockout females was associated with significantly reduced fresh weights of reproductive organs obtained from additional mice at 3 months of age (paired ovaries: controls = 11.4 ± 1.1 mg, neuron-specific LEPR knockouts = 2.8 ± 1.1 mg; uterus: controls = 81.4 ± 4.1 mg, neuron-specific LEPR knockouts = 14.6 ± 5.6 mg; cervix: controls = 28.5 ± 2.5 mg, neuron-specific LEPR knockouts = 7.9 ± 0.6 mg; all P < 0.01).
Figure 2.

Profound infertility in neuron-specific LEPR knockout mice. Female (A) and male (B) neuron-specific LEPR knockout mice (open markers) were obese, exhibiting more than double the body weight gain of control littermates (black markers). C, Vaginal opening was delayed and estrus was not observed in young female neuron-specific LEPR knockout mice; male neuron-specific LEPR knockout mice had delayed or absent puberty. D, Absence of estrous cycles in neuron-specific LEPR knockout female mice, as observed by vaginal cytology over a 1-wk period (data normalized to 4 d estrous cycle length; P, proestrus; E, estrus; M, metestrus; D, diestrus). E, In contrast to control littermates, female neuron-specific LEPR knockout mice were unable to produce any litters, and male neuron-specific LEPR knockout mice produced almost no litters when paired with wild-type mates for 120 d. F, When subjected to a preovulatory-like surge induction protocol, female neuron-specific LEPR knockout mice did not exhibit an LH surge or activation (i.e. c-Fos coexpression) of GnRH neurons in response to estradiol benzoate injection. *, P < 0.001.
Neuron-specific LEPR knockout females were also unable generate an exogenous estradiol-induced LH surge. At the time of surge peak, Leprfl/fl control mice exhibited a robust increase in plasma LH concentration (4.7 ± 0.7 ng/ml) and the percentage of GnRH neurons coexpressing c-Fos (35 ± 6%), whereas in neuron-specific LEPR knockout mice, these parameters remained at very low levels (0.1 ± 0.1 ng/ml and 3 ± 1%; both P < 0.001; Fig. 2F).
Male Leprfl/fl control mice attained puberty at an average age of 45.3 ± 0.8 d old, and thereafter sired a litter every 28 ± 1.6 d. In marked contrast, only two of seven neuron-specific LEPR knockout males were able to sire litters (puberty onset was 56 and 66 d of age; P < 0.001 vs. control males). A single litter was sired by each of these males over the 110 d that they were paired with wild-type females. Litter size did not differ from that of control males (Fig. 2E). Paired fresh testes weights (measured in additional 4 month old males) were not significantly reduced in the neuron-specific LEPR knockout mice (controls = 209.4 ± 3.9 mg, neuron-specific LEPR knockouts = 193.5 ± 16.5 mg); however, in contrast to control mice, the testes were undescended, the epididymis appeared regressed and no spermatozoa were present in the epididymal tail.
PCR analysis of experimental vs. control tissues showed that Cre-mediated flox excision of the Lepr gene was specific to brain tissue (supplemental Fig. 1 published on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org).
Experiment 2b: effects of GnRH-specific LEPR knockout on fertility
Post weaning body weight gain did not differ between GnRH-specific LEPR knockout mice and Leprfl/fl controls (Fig. 3A). In contrast to the profound infertility exhibited by neuron-specific LEPR knockout female mice, GnRH-specific LEPR knockout females exhibited normal fertility. Occurrence of vaginal opening and first estrus (Fig. 3B), estrous cyclicity (Fig. 3C), and litter frequency and size (Fig. 3D) did not differ from control values. A tendency for male GnRH-specific LEPR knockout mice to attain puberty later than control males (controls = 50.1 ± 1.8, GnRH-specific LEPR knockouts = 58.2 ± 4.9 d; Fig. 3B) did not reach statistical significance (P = 0.13), and subsequent fertility (litter frequency and size) did not differ between the two groups (Fig. 3D). Paired fresh testes weight at the end of the study also did not differ between the two groups (controls = 206.9 ± 12.1 mg, GnRH-specific LEPR knockouts = 199.0 ± 9.1 mg).
Figure 3.

Normal fertility in GnRH-specific LEPR knockout mice. A, GnRH-specific LEPR knockout mice (open markers) exhibited a similar rate of body weight gain to control littermates (black markers). B, Date of vaginal opening, first estrus, and male puberty onset was normal in young GnRH-specific LEPR knockout mice. C, Female GnRH-specific LEPR knockout mice also exhibited normal estrous cycles, as observed by vaginal cytology over a 12-d period (data normalized to 4 d estrous cycle length; P, proestrus; E, estrus; M, metestrus; D, diestrus). D, Male and female GnRH-specific LEPR knockout mice produced litters at the same frequency and size as control littermates.
Experiment 3: Lepr mRNA in individual GnRH neurons
A total of 28 GnRH cells were harvested from three adult female mice; 21 of these were screened for the presence of GnRH and LEPR transcripts. Eighteen of 21 GnRH-GFP harvested cells showed the presence of GnRH transcripts. None of the 21 GnRH-GFP neurons showed the presence of Lepr mRNA. Controls included no reverse transcriptase (n = 7), H2O in place of template (n = 3) and mock harvests (n = 3); none of these showed any product. As a positive control for the single-cell Lepr RT-PCR, POMC neurons were harvested and analyzed alongside GnRH cells in the nested PCR. In total, 14 POMC-GFP cells were collected from two adult female mice; 11 of these cells were screened for POMC and Lepr transcripts and three were used in no reverse transcriptase controls. Six of 11 were confirmed as POMC producing cells by nested RT-PCR and of these, four were positive for Lepr mRNA. Negative controls showed no product (Fig. 4).
Figure 4.
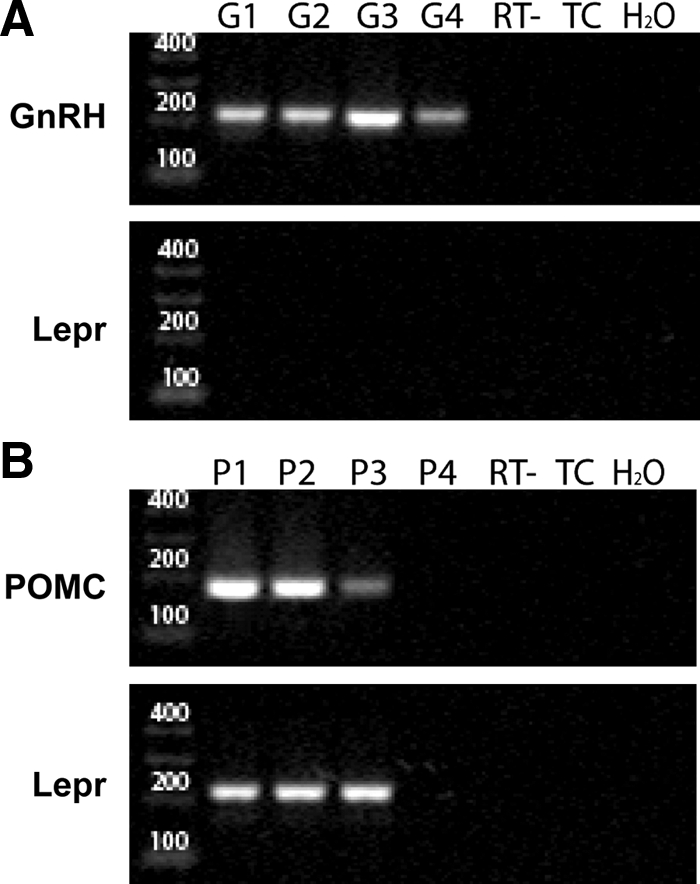
Agarose gel showing representative amplicons from two rounds of nested RT-PCR using cytoplasmic contents from individual GFP-labeled GnRH- or POMC-positive neurons. A, Cytoplasm from single GnRH-GFP neurons (lanes G1–G4) was harvested, subjected to reverse transcriptase (RT), and then nested PCR with gene-specific primers recognizing GnRH (A, upper panel) or Lepr (A, lower panel) mRNA. B, Cytoplasm from single POMC-GFP neurons of the arcuate nucleus (lanes P1–P4) was processed as above with primers recognizing POMC (B, upper panel) or Lepr (B, lower panel) mRNA. Negative controls (all panels), processed in parallel, included either reactions without reverse transcriptase (RT−), touch only control in which cell was touched but cytoplasmic contents not collected (TC), or water in place of template for the first round of amplification (H2O). Lepr transcripts were not detected in any GnRH neurons, whereas Lepr was present in 67% of POMC-positive neurons. Left lanes contain molecular weight markers, with sizes indicated in base pairs. The expected amplicon sizes are: GnRH, 213 bp; POMC, 173 bp; and Lepr, 214 bp.
Discussion
The aim of this study was to determine whether leptin could act directly on GnRH neurons. We used three separate approaches, namely immunohistochemistry, conditional transgenic knockouts, and single-cell RT-PCR in living brain slices to address this question. Taken together, the data clearly show that GnRH neurons are unlikely to be direct targets for leptin in rodents.
The published literature is equivocal in determining whether leptin directly regulates GnRH neurons. STAT3 is considered to be the primary mediator of leptin’s actions in hypothalamic neurons, including appetite regulation via POMC and neuropeptide Y neurons (49). Hence, we used immunohistochemical detection of leptin-induced STAT3 phosphorylation as a functional indicator of leptin responsiveness in GnRH neurons. Leptin-induced pSTAT3 was readily detectable in the vicinity of GnRH neurons. Dual labeling, however, for pSTAT3 and GnRH in male and female adult rats showed no colocalization, indicating that leptin does not signal via this pathway in GnRH neurons. Interestingly, a significant induction of pSTAT3 expression was observed in the AVPV of females and a less pronounced trend noted in males. The sexually dimorphic AVPV contains neurons that are activated during the preovulatory surge (50). Additionally, neurons that reside in the AVPV (such as kisspeptin neurons) are capable of providing stimulatory input to GnRH neurons (51,52). Our pSTAT3 results raise the possibility that leptin may regulate GnRH neuronal function via AVPV afferents. Whereas our data suggest leptin does not act on GnRH neurons through pSTAT3, we cannot exclude the possibility that LEPR stimulates different signaling cascades, such as STAT1, STAT5, and ERK kinases (53). For example, leptin has recently been shown to induce STAT5 phosphorylation and translocation in hypothalamic neurons in vitro and in vivo (54,55). However, the STAT3 pathway is thought to be dominant (56) because neuronal STAT3 deletion recapitulates the infertility, obesity, diabetes, and thermal dysregulation of global leptin and LEPR knockout mice (57). In contrast, mice with neuronal STAT5 deletion remain completely fertile (unpublished observations from our laboratory). Thus, we believe that absence of pSTAT3 in GnRH neurons provides evidence to support our conclusion of indirect GnRH neuronal regulation by leptin. Future experiments should examine leptin signaling by other pathways in hypothalamic cells including GnRH neurons.
The role of neuronal expression of the LEPR in mediating effects of reproduction on fertility has been evaluated previously. McMinn et al. (27) used the CamKIIα promoter to drive CRE expression to generate a neuron-specific LEPR deleted mouse line. Contrary to our results, they found that fertility was largely unaffected, with 70–80% of male and female conditional knockout mice still able to produce offspring (27). However, the same group has also shown that when male and female db/db mice (which have a global Lepr mutation) were subjected to a neuron-specific Lepr knock-in, fertility was completely restored, showing that brain-specific leptin signaling is sufficient for reproductive function (58). Consistent with these latter data, in our study, male and female neuron-specific LEPR knockout mice were almost completely infertile, demonstrating the importance of central leptin actions for fertility. The infertility in our neuron-specific LEPR knockout mice was apparent, even when ovariectomized females were treated with high doses of estradiol to induce a preovulatory-like GnRH/LH surge. Unlike Leprfl/fl controls, neuron-specific LEPR knockout females were completely unable to exhibit GnRH neuronal activation or a consequent surge of plasma LH. The different levels of fertility in our mice compared with McMinn et al. may be due to differences in background strain (C57BL/6J in the present experiments and FVB in the aforementioned studies). FVB mice have a fecundity score almost double that of any other inbred mouse strain and are apparently fertile, even in absence of leptin signaling (59).
In contrast to the profound infertility observed in C57BL/6J neuron-specific LEPR knockout mice, GnRH-specific LEPR knockout mice exhibited normal fertility, supporting the concept that direct actions of leptin on GnRH neurons are not required for reproductive function. There are two possible interpretations as to why GnRH-specific LEPR knockouts showed relatively normal fertility: 1) GnRH neurons do not express LEPRs or 2) the GnRH-Cre cross-resulted in incomplete Lepr deletion within the GnRH neuron population. Previous studies examining the presence of LEPR in GnRH neurons have been inconclusive. Approximately 75% of GT1–7 GnRH-expressing cells contain LEPRs (30), whereas in vivo, Hâkansson et al. (29) reported few if any GnRH neurons with LEPRs in hypothalamic sections. Additionally, Finn et al. (16) did not find any GnRH-expressing cells clearly expressing Lepr using a dual GnRH/Lepr in situ hybridization. We reexamined this issue by looking for the presence of Lepr mRNA in GnRH neurons, using the highly sensitive technique of single-cell RT-PCR. To ensure the efficacy of single-cell RT-PCR to detect Lepr mRNA presence in single cells, we also examined POMC neurons as a known positive control (8). Whereas 75% of positive control (POMC) neurons contained Lepr mRNA, suggesting no technical difficulties in detecting Lepr in single cells, no GnRH neurons were positive for any of the Lepr mRNA splice variants. Although limitations on the number of cells able to be collected (18 GnRH neurons analyzed for Lepr) leave the possibility of some coexpression open, it is clear from these results that the vast majority of GnRH neurons do not express the LEPR.
Taken together, these data provide three lines of evidence to suggest that leptin action on fertility is mediated indirectly. Unlike other reproductive hormones such as estrogen (2) and prolactin (43), which appear to be able to interact with GnRH neurons both directly and indirectly, our results imply that the actions of leptin on the GnRH system occur exclusively via afferent interneurons. Although leptin does not bind to GnRH neurons to affect fertility, our results show that leptin actions on forebrain neurons essential for regulation of both energy balance and fertility.
Supplementary Material
Acknowledgments
The authors thank G. Schutz, C. Dulac, and S. Chua for supplying transgenic mice.
Footnotes
The New York Obesity Research Center funded the development of the floxed leptin receptor mouse (Grant 1PO1DK26687 to S.C.). This work was supported by The Health Research Council of New Zealand and New Zealand Lottery Health Research grants.
Disclosure Summary: The authors have nothing to declare.
First Published Online January 29, 2009
Abbreviations: AVPV, Anteroventral periventricular nucleus; CRE, cAMP response element; GFP, green fluorescent protein; i.c.v., intracerebroventricular; LEPR, leptin receptor; OVLT, organum vasculosum of the lamina terminalis; POMC, proopiomelanocortin; pSTAT, phosphorylated STAT; STAT, signal transducer and activator of transcription.
References
- Freeman ME 2006 Neuroendocrine control of the ovarian cycle of the rat. In: Neill JD, ed. Physiology of reproduction. 3rd ed. San Diego: Elsevier; 2327–2388 [Google Scholar]
- Herbison AE 2006 Physiology of the GnRH neuronal network. In: Neill JD, ed. Knobil and Neill’s physiology of reproduction. San Diego: Academic Press; 1415–1482 [Google Scholar]
- Cameron JL, Nosbisch C 1991 Suppression of pulsatile luteinizing hormone and testosterone secretion during short term food restriction in the adult male rhesus monkey (Macaca mulatta). Endocrinology 128:1532–1540 [DOI] [PubMed] [Google Scholar]
- I'Anson H, Manning JM, Herbosa CG, Pelt J, Friedman CR, Wood RI, Bucholtz DC, Foster DL 2000 Central inhibition of gonadotropin-releasing hormone secretion in the growth-restricted hypogonadotropic female sheep. Endocrinology 141:520–527 [DOI] [PubMed] [Google Scholar]
- Hukshorn CJ, Menheere PP, Westerterp-Plantenga MS, Saris WH 2003 The effect of pegylated human recombinant leptin (PEG-OB) on neuroendocrine adaptations to semi-starvation in overweight men. Eur J Endocrinol 148:649–655 [DOI] [PubMed] [Google Scholar]
- Nagatani S, Guthikonda P, Thompson RC, Tsukamura H, Maeda KI, Foster DL 1998 Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology 67:370–376 [DOI] [PubMed] [Google Scholar]
- Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM 2001 Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest 108:1113–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua Jr SC, Elmquist JK, Lowell BB 2004 Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42:983–991 [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ 2001 Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411:480–484 [DOI] [PubMed] [Google Scholar]
- Erickson JC, Hollopeter G, Palmiter RD 1996 Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 274:1704–1707 [DOI] [PubMed] [Google Scholar]
- Barash IA, Cheung CC, Weigle DS, Ren H, Kabigting EB, Kuijper JL, Clifton DK, Steiner RA 1996 Leptin is a metabolic signal to the reproductive system. Endocrinology 137:3144–3147 [DOI] [PubMed] [Google Scholar]
- Chua Jr SC, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia L, Leibel RL 1996 Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 271:994–996 [DOI] [PubMed] [Google Scholar]
- Todd BJ, Ladyman SR, Grattan DR 2003 Suppression of pulsatile luteinizing hormone secretion but not luteinizing hormone surge in leptin resistant obese Zucker rats. J Neuroendocrinol 15:61–68 [DOI] [PubMed] [Google Scholar]
- Wauters M, Considine RV, Van Gaal LF 2000 Human leptin: from an adipocyte hormone to an endocrine mediator. Eur J Endocrinol 143:293–311 [DOI] [PubMed] [Google Scholar]
- Chehab FF, Lim ME, Lu R 1996 Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet 12:318–320 [DOI] [PubMed] [Google Scholar]
- Finn PD, Cunningham MJ, Pau KY, Spies HG, Clifton DK, Steiner RA 1998 The stimulatory effect of leptin on the neuroendocrine reproductive axis of the monkey. Endocrinology 139:4652–4662 [DOI] [PubMed] [Google Scholar]
- Bronson FH 1986 Food-restricted, prepubertal, female rats: rapid recovery of luteinizing hormone pulsing with excess food, and full recovery of pubertal development with gonadotropin-releasing hormone. Endocrinology 118:2483–2487 [DOI] [PubMed] [Google Scholar]
- Szymanski LA, Schneider JE, Friedman MI, Ji H, Kurose Y, Blache D, Rao A, Dunshea FR, Clarke IJ 2007 Changes in insulin, glucose and ketone bodies, but not leptin or body fat content precede restoration of luteinising hormone secretion in ewes. J Neuroendocrinol 19:449–460 [DOI] [PubMed] [Google Scholar]
- I'Anson H, Sundling LA, Roland SM, Ritter S 2003 Immunotoxic destruction of distinct catecholaminergic neuron populations disrupts the reproductive response to glucoprivation in female rats. Endocrinology 144:4325–4331 [DOI] [PubMed] [Google Scholar]
- Schneider JE, Goldman MD, Tang S, Bean B, Ji H, Friedman MI 1998 Leptin indirectly affects estrous cycles by increasing metabolic fuel oxidation. Horm Behav 33:217–228 [DOI] [PubMed] [Google Scholar]
- Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, Friedman JM 1997 Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci USA 94:7001–7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P 1996 Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett 387:113–116 [DOI] [PubMed] [Google Scholar]
- Zamorano PL, Mahesh VB, De Sevilla LM, Chorich LP, Bhat GK, Brann DW 1997 Expression and localization of the leptin receptor in endocrine and neuroendocrine tissues of the rat. Neuroendocrinology 65:223–228 [DOI] [PubMed] [Google Scholar]
- Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P 1995 Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269:546–549 [DOI] [PubMed] [Google Scholar]
- Watanobe H 2002 Leptin directly acts within the hypothalamus to stimulate gonadotropin-releasing hormone secretion in vivo in rats. J Physiol 545:255–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro E, Pinilla L, Seoane LM, Considine RV, Aguilar E, Casanueva FF, Dieguez C 1997 Influence of endogenous leptin tone on the estrous cycle and luteinizing hormone pulsatility in female rats. Neuroendocrinology 66:375–377 [DOI] [PubMed] [Google Scholar]
- McMinn JE, Liu SM, Liu H, Dragatsis I, Dietrich P, Ludwig T, Boozer CN, Chua Jr SC 2005 Neuronal deletion of Lepr elicits diabesity in mice without affecting cold tolerance or fertility. Am J Physiol Endocrinol Metab 289:E403–E411 [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI 1995 Identification and expression cloning of a leptin receptor, OB-R. Cell 83:1263–1271 [DOI] [PubMed] [Google Scholar]
- Hâkansson ML, Brown H, Ghilardi N, Skoda RC, Meister B 1998 Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci 18:559–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni P, Vettor R, Pagano C, Calcagno A, Beretta E, Messi E, Zanisi M, Martini L, Motta M 1999 Expression of a leptin receptor in immortalized gonadotropin-releasing hormone-secreting neurons. Endocrinology 140:1581–1585 [DOI] [PubMed] [Google Scholar]
- Sullivan SD, Moenter SM 2004 γ-Aminobutyric acid neurons integrate and rapidly transmit permissive and inhibitory metabolic cues to gonadotropin-releasing hormone neurons. Endocrinology 145:1194–1202 [DOI] [PubMed] [Google Scholar]
- Woller M, Tessmer S, Neff D, Nguema AA, Roo BV, Waechter-Brulla D 2001 Leptin stimulates gonadotropin releasing hormone release from cultured intact hemihypothalami and enzymatically dispersed neurons. Exp Biol Med 226:591–596 [DOI] [PubMed] [Google Scholar]
- Anderson KD, Lambert PD, Corcoran TL, Murray JD, Thabet KE, Yancopoulos GD, Wiegand SJ 2003 Activation of the hypothalamic arcuate nucleus predicts the anorectic actions of ciliary neurotrophic factor and leptin in intact and gold thioglucose-lesioned mice. J Neuroendocrinol 15:649–660 [DOI] [PubMed] [Google Scholar]
- Ladyman SR, Grattan DR 2004 Region-specific reduction in leptin-induced phosphorylation of signal transducer and activator of transcription-3 (STAT3) in the rat hypothalamus is associated with leptin resistance during pregnancy. Endocrinology 145:3704–3711 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C 2005 The rat brain atlas in stereotaxic coordinates. 5th ed. Sydney: Elsevier [Google Scholar]
- McMinn JE, Liu SM, Dragatsis I, Dietrich P, Ludwig T, Eiden S, Chua Jr SC 2004 An allelic series for the leptin receptor gene generated by CRE and FLP recombinase. Mamm Genome 15:677–685 [DOI] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, Schütz G 2001 A CamKIIα iCre BAC allows brain-specific gene inactivation. Genesis 31:37–42 [DOI] [PubMed] [Google Scholar]
- Burgin KE, Waxham MN, Rickling S, Westgate SA, Mobley WC, Kelly PT 1990 In situ hybridization histochemistry of Ca2+/calmodulin-dependent protein kinase in developing rat brain. J Neurosci 10:1788–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet CC, McGuinness TL, Greengard P 1984 Immunocytochemical localization of calcium/calmodulin-dependent protein kinase II in rat brain. Proc Natl Acad Sci USA 81:5604–5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clow OL, Hurst PR, Fleming JS 2002 Changes in the mouse ovarian surface epithelium with age and ovulation number. Mol Cell Endocrinol 191:105–111 [DOI] [PubMed] [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Grüne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE 2006 Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 52:271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ 2004 The mouse brain in stereotaxic coordinates. San Diego: Elsevier Academic Press [Google Scholar]
- Grattan DR, Jasoni CL, Liu X, Anderson GM, Herbison AE 2007 Prolactin regulation of gonadotropin-releasing hormone neurons to suppress luteinizing hormone secretion in mice. Endocrinology 148:4344–4351 [DOI] [PubMed] [Google Scholar]
- Jasoni CL, Todman MG, Han SK, Herbison AE 2005 Expression of mRNAs encoding receptors that mediate stress signals in gonadotropin-releasing hormone neurons of the mouse. Neuroendocrinology 82:320–328 [DOI] [PubMed] [Google Scholar]
- Spergel DJ, Krüth U, Hanley DF, Sprengel R, Seeburg PH 1999 GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J Neurosci 19:2037–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Chapdelaine A, Yang H, Orkin SH 2001 Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood 97:324–326 [DOI] [PubMed] [Google Scholar]
- Skynner MJ, Sim JA, Herbison AE 1999 Detection of estrogen receptor α and β messenger ribonucleic acids in adult gonadotropin-releasing hormone neurons. Endocrinology [Erratum (2001) 142:492–493] 140:5195–5201 [DOI] [PubMed] [Google Scholar]
- Chua Jr SC, Koutras IK, Han L, Liu SM, Kay J, Young SJ, Chung WK, Leibel RL 1997 Fine structure of the murine leptin receptor gene: splice site suppression is required to form two alternatively spliced transcripts. Genomics 45:264–270 [DOI] [PubMed] [Google Scholar]
- Håkansson ML, Meister B 1998 Transcription factor STAT3 in leptin target neurons of the rat hypothalamus. Neuroendocrinology 68:420–427 [DOI] [PubMed] [Google Scholar]
- Smith JT, Acohido BV, Clifton DK, Steiner RA 2006 KiSS-1 neurones are direct targets for leptin in the ob/ob mouse. J Neuroendocrinol 18:298–303 [DOI] [PubMed] [Google Scholar]
- Herbison AE 2008 Estrogen positive feedback to gonadotropin-releasing hormone (GnRH) neurons in the rodent: the case for the rostral periventricular area of the third ventricle (RP3V). Brain Res Rev 57:277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JT, Popa SM, Clifton DK, Hoffman GE, Steiner RA 2006 Kiss1 neurons in the forebrain as central processors for generating the preovulatory luteinizing hormone surge. J Neurosci 26:6687–6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekerman P, Zeidler J, Bamberg-Lemper S, Knobelspies H, Lavens D, Tavernier J, Joost HG, Becker W 2005 Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS J 272:109–119 [DOI] [PubMed] [Google Scholar]
- Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Münzberg H, Myers Jr MG 2007 The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J Biol Chem 282:31019–31027 [DOI] [PubMed] [Google Scholar]
- Mütze J, Roth J, Gerstberger R, Hübschle T 2007 Nuclear translocation of the transcription factor STAT5 in the rat brain after systemic leptin administration. Neurosci Lett 417:286–291 [DOI] [PubMed] [Google Scholar]
- Gao Q, Horvath TL 2008 Cross-talk between estrogen and leptin signaling in the hypothalamus. Am J Physiol Endocrinol Metab 294:E817–E826 [DOI] [PubMed] [Google Scholar]
- Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY 2004 Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci USA 101:4661–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua Jr SC 2005 Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest 115:3484–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LM 1995 Mouse genetics: concepts and applications. New York: Oxford University Press [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.