

Abstract
Motivation: The modeling of conservation patterns in genomic DNA has become increasingly popular for a number of bioinformatic applications. While several systems developed to date incorporate context-dependence in their substitution models, the impact on computational complexity and generalization ability of the resulting higher order models invites the question of whether simpler approaches to context modeling might permit appreciable reductions in model complexity and computational cost, without sacrificing prediction accuracy.
Results: We formulate several alternative methods for context modeling based on windowed Bayesian networks, and compare their effects on both accuracy and computational complexity for the task of discriminating functionally distinct segments in vertebrate DNA. Our results show that substantial reductions in the complexity of both the model and the associated inference algorithm can be achieved without reducing predictive accuracy.
Contact: bmajoros@duke.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Models of molecular evolution have proven useful in performing a variety of tasks, including phylogeny reconstruction (Felsenstein, 1981), RNA secondary structure prediction (Gulko and Haussler, 1996), gene finding (Pedersen and Hein, 2003; Siepel and Haussler, 2004a), motif discovery (Moses et al., 2004; Siddharthan et al., 2005) and others. In the case of DNA analysis, the accurate modeling of substitution rates can in some cases facilitate effective discrimination between genomic elements of differing functions by uncovering the different selective pressures shaping those elements. These substitution models typically consist of a phylogeny and one or more matrices describing substitution propensities between different residues in the genomic sequences of related taxa.
While the phylogeny components of these models control for the non-independence (due to commonality of descent) of residues observed at homologous sites in different genomes (i.e. within individual columns of a multi-species alignment), they do not take into account possible dependencies between sites (i.e. between columns), though such dependencies certainly exist (Averof et al., 2000; Smith et al., 2003; Whelan and Goldman, 2004). In the case of short-range dependencies (i.e. extending linearly along a several-nucleotide stretch of DNA), the effect of such dependencies on substitution rates has been referred to as context-dependence. Context-dependence in substitution rates between orthologous sites may conceivably result from a number of distinct but potentially interacting phenomena, including context-dependent mutation, context-dependent selection, multiple compensatory changes at nearby sites, or perhaps other effects. For the purpose of identifying functional elements in extant genomes, however, a productive first step is to model only the sum result of these various causal phenomena.
Practical systems utilizing context-dependent models have been described previously [Siepel and Haussler, 2004b (SH04); Gross and Brent, 2005 (GB05)], which specify rates of substitution between entire n-mers (i.e. oligomers of length n). By utilizing a Markov assumption (a form of conditional independence), these solutions are able to decompose the conditional likelihood according to individual columns in the alignment. As with existing codon models [e.g. Goldman and Yang, 1994 (GY94)], however, these approaches tend to be both parameter rich (a potential liability when training data are limited, due to the possibility of overfitting) and computationally intensive, with both the number of parameters and the computational cost growing exponentially with the size of the modeled context (n). For these reasons, n has often been limited in practice to n < 3.
In this article, we show how a more general decomposition of the likelihood is possible under an alternative set of conditional independence assumptions, allowing one to vary the dependency structure and the number of parameters in the model to better suit the available data. Our formulation is based on the theory of Bayesian networks, a well-established probabilistic modeling framework which is both flexible and theoretically rigorous (Pearl, 1988). Although previous authors have cast the problem in terms of Bayesian networks (Gross and Brent, 2005) and other graphical models (Jojic et al., 2004; McAuliffe et al., 2004), our treatment is more general in that we show how a greater range of network topologies may be utilized to improve the robustness of the resulting model and/or improve its computational complexity. We show how significant reductions in numbers of parameters may be achieved through the use of sparse dependency graphs and conditional probability distributions on substitution events. This reduction in complexity renders the modeling of longer contexts more feasible than before.
2 METHODS
2.1 Notation
We denote by A the multiple-sequence alignment which is provided as a fixed input to our models; A is a 2D matrix in which the rows correspond to taxa and the columns (or ‘sites’) correspond to homologous nucleotide positions (or gaps) in the aligned genomes. Although an input alignment will contain only sequences from extant taxa (denoted ℒ, for ‘leaves’), we may augment the alignment with additional rows for ancestral species (denoted 𝒜); the set of all taxa to be included in our model is thus 𝒯 = 𝒜∪ ℒ. We assume that the phylogeny 𝒫 relating these taxa has a known topology, though the branch lengths β may be unknown; as described later, we will be rooting the tree so as to place a chosen ‘target’ genome at the root. Note that the definition of ℒ and 𝒜 will not be affected by a re-rooting of the tree, so that after re-rooting, the root of the tree may be an element of 𝒜 or ℒ.
Given a taxon v∈𝒯, Aiv will denote the character state (i.e. residue) belonging in the i-th column of the alignment and corresponding to taxon v; in those cases where v∈𝒜, Aiv is unobservable and must be either inferred (through maximum likelihood or some other means) or eliminated via marginalization. We assume 1-based coordinates, so that the columns of A range from 1 to ℓ, where ℓ is the length of the alignment. Ai will denote a single column of A, whereas A[i,j] will denote the closed interval [i,j] consisting of columns {Ai,…,Aj}. Av denotes a single row of the alignment; A[i,j]v is an interval within that row. If V⊆𝒯 is a set of taxa, then A V denotes the alignment which results when all other taxa are omitted from A.
Since we consider only DNA substitution in this article, residues in an alignment will be drawn from the alphabet α = {A,C,G,T}; the additional symbols ‘−’ (denoting a gap) and ‘.’ (denoting an unaligned position) are considered separate from α (i.e. they are treated as missing data). A ‘higher order’ alphabet αn may be derived from α by taking all n-mers over the base alphabet: αn = {x1,x2, …,xn|xi∈α, 1⩽i⩽n}. Concatenation is denoted w + x, where w and x may be sequences or individual symbols. A model θ = (𝒫,β,ψ) will consist of a phylogeny 𝒫, a set of branch lengths β for that phylogeny and a substitution model ψ.
Given a list of variables 𝒱 = (v1, …,vm) and a list of values X = (x1, …,xm), 𝒱∼X will denote the putative assignment v1 = x1, v2 =,x2, …vm =xm, so that P(𝒱 ∼X) denotes the probability of the variables in 𝒱 taking their respective values from X. The operator δ(a,b) denotes the Kronecker delta function, which evaluates to 1 if a = b, and 0 otherwise; δn(a,b) for strings a and b evaluates to 1 if the first n letters of a and b match, and 0 otherwise. The operator ∧ denotes logical conjunction.
C(u) will denote the children of u in some tree-structured directed graphical model (such as a phylogeny or a tree-structured Bayesian network). Given random variables u and v, and residues x and y, P(v =y|u=x,λ) will denote the probability that v assumes the value y, given that u assumes the value x and also conditional on the predicate λ; we may use the abbreviated notation P(v|u,λ) when y and x are unambiguous (such as when v and u are associated with specific positions in an alignment). In directed tree models, the edges are assumed to point away from the root. Edges are denoted u → v for parent u and child v. When we consider Bayesian networks defined on a multiple-sequence alignment, an edge uj → vk will correspond to residues Aju and Akv. Alternatively, when each of the variables in a network have been associated with specific positions in an alignment, we may denote by AV the residues in A associated with the variables in the set v; this will permit us to utilize predicates of the form: V∼AV. In such situations, we may also use either of the abbreviations P(V) or P(AV) to mean P(V∼AV).
The Supplementary Data contain a glossary of terms and several figures which provide a conceptual overview of our methods (Supplementary Figs S1, S2 and S3).
2.2 Bayesian networks for substitution modeling
Substitution models, both context-dependent and context-independent, may be represented via Bayesian networks—probability distributions represented by weighted, directed acyclic graphs (Friedman, 2004; Heckerman, 1999; Pearl, 1988). In a Bayesian network, directed edges represent dependencies between nodes. A key advantage of this formalism is that it readily permits the exploration of alternative dependency structures exhibiting different tradeoffs between model complexity and predictive accuracy. In the case of context-dependent substitution models, this means that one is able to observe the effect on model fit and overtraining tendencies as edges are added to or removed from the network; by using, e.g. an appropriately regularized cross-validation strategy one may thus search for the network with the greatest predictive accuracy when applied to a particular modeling task.
Formally, we define a Bayesian network as a tuple 𝒢 = (𝒱, α, ℰ, P) in which 𝒱 is a set of vertices or variables taking values from the finite alphabet α, ℰ⊆ 𝒱 × 𝒱 is a set of directed edges forming an acyclic graph, and P(v|V) is a conditional probability function defined for each vertex v∈𝒱 conditional on sets of vertices V⊆𝒱. Each edge u→v in ℰ denotes a dependence relation between u and v in which the value assumed by variable u is taken to directly influence the probability of v assuming particular values. Formally, if ρ(v) = {u|(u→v)∈ ℰ} is the set of vertices directly influencing v according to ℰ, then:
| (1) |
where σ(v) is the set consisting of v and all the vertices which v can influence, either directly or transitively:
| (2) |
Thus, we say that v is conditionally independent of the set 𝒱-σ(v)-ρ(v), given the values of ρ(v). Given the set of conditional independence assumptions encoded by a Bayesian network, one can utilize Equation (1) to compute the likelihood that the variables 𝒱 = (v1,…,vm) of the network will take their respective values from the list X = (x1,…,xm)∈ αm, as follows:
| (3) |
In this case, we say that all the variables are observable, since they have been assigned putative values. When only a proper subset of variables W⊆𝒱 are assigned values, the remaining variables U = 𝒱−W are termed unobservables. Denote the unobservables in the network by U = (u1,…,uk). To evaluate the likelihood of a putative assignment W∼X to the observables, we can marginalize by summation over all possible combinations of values for the unobservables:
| (4) |
where P(W∼XU∼Y) is given by Equation (3). When the network is a tree—in which case ∀v∈ 𝒱 |ρ(v)|⩽ 1—the likelihood given by Equation (4) admits an efficient factorization given by:
| (5) |
for root variable r and recursive function Lu:
| (6) |
which is precisely Felsenstein's ‘pruning’ algorithm for phylogeny likelihood on ungapped alignments (Felsenstein, 1981), and is also known as the sum-product algorithm on trees (Kschischang et al., 2001; Lauritzen and Spiegelhalter, 1988). This recursion may be efficiently computed via dynamic programming (Durbin et al., 1998), in which case the time complexity is O(|α|2|𝒱 |), though a temporary matrix of size O(|α||𝒱 |) is also required to store intermediate values. Because we will be considering enhancements to the formulation of Equation (6), we refer to this base version as ℱ0
Modeling DNA substitution patterns at a single site with a Bayesian network is simple in the absence of context effects: the network is given by the known (rooted) phylogeny of the species present in a multiple-sequence alignment, with individual variables (i.e. vertices) corresponding to taxa in the phylogeny (of which only the leaves, representing extant species, are observable). The values of the observables are given by the residues in the alignment; variables that would normally be observable may be treated as unobservables when the corresponding position in the alignment contains a gap (i.e. ‘-’ or ‘.’). Felsenstein's algorithm gives an efficient means of computing the likelihood of a single column of the alignment, as given by Equations (5) and (6). To compute the likelihood of the entire alignment, we note that the lack of context effects warrants an independence assumption between columns:
| (7) |
for alignment A of length ℓ; we assume the alignment has been augmented with empty rows (i.e. all gaps) corresponding to unobserved taxa. We now have a Bayesian network consisting of ℓ disjoint trees, in which each variable uj corresponds to the residue at site j in species u (i.e. uj is the variable to which the residue Aju is assigned).
Note that in adopting the Bayesian network formulation for context modeling we follow Siepel and Haussler (2004b) in committing only to an empirical model of local dependencies in the alignment, rather than to an underlying process of evolutionary change at the level of individual generations. Process-based models [e.g. Hwang and Green, 2004 (HG04); Pedersen and Hein, 2003] are forced to admit that local dependencies within one generation may give rise over evolutionary time to long-range dependencies between taxa, and require sampling based methods (such as Markov chain Monte Carlo—MCMC) to properly capture the resulting long-range dependencies, due to the exorbitant computational costs which would otherwise be incurred. Thus, although in evaluating P(v = y|u = x) we make use of the construction P(t) = eQt often associated with continuous-time Markov models described by an instantaneous rate matrix Q and branch length t, we utilize this construction merely for the purposes of parameter tying—i.e. so that the same Q can be shared across all branches of the phylogeny.
As stated previously, it is convenient to re-root the phylogeny so that one of the extant taxa occupies the root; this will generally allow the removal of a single unobservable from the network (Gross and Brent, 2005), and is also convenient when the purpose of evolution modeling is to inform the prediction of functional elements in a single target genome (e.g. gene finding with a phyloHMM), so that the target genome is the most natural choice for the root. Re-rooting of the phylogeny is always possible when using a reversible substitution matrix (Felsenstein, 1981). Our interest will therefore be the computation of the conditional likelihood P(A𝒱−R|AR), for R = {r(i)|1⩽i⩽ℓ} the set of root vertices r(i) in all columns i:
| (8) |
Incorporation of context effects into the network involves the addition of edges connecting trees from different columns—e.g. ui → vj for taxa u and v, and columns i and j. We will initially require these additional edges to respect the original phylogeny, so that, e.g. if ui → vj is added to the network to represent context effects between columns i and j, then either u → v must be present in the original phylogeny (with u as the parent of v), or u =v; we will relax this requirement later when we consider models having only observable contexts. It should be clear that the conditional independence assumptions imposed by the phylogeny are well-justified (in the absence of lateral DNA transfer) by evolutionary principles.
Unfortunately, the network when augmented in this way is no longer a collection of trees, so that it no longer suffices to apply Felsenstein's algorithm to each column independently. In the next section, we describe two solutions to this problem. First, however, we formalize the notion of a network template. Let 𝒢t = (𝒱t,α,ℰt,Pt) be a Bayesian network defined on an abstract alignment with some small number n of columns, and let A be the full n-column alignment for which we wish to evaluate the likelihood, n ≪ N. Let 𝒯 be the set of taxa in the phylogeny, and define Vt = {uiz|1≤i≤n,z∈T}. We can use 𝒢t as a template in order to construct a full Bayesian network 𝒢 = (𝒱,α,ℰP) for A, by instantiating the template once for each column in A. Formally, define V = {viz|1≤i≤N, z∈T}. For each uiz→unw in ℰt, and each column j>n in A, add to ℰ an edge vj−n+iz for columns j≤n add only those edges vj−n+iz for which ℰt has an edge uiz → unw such that n−i < j. Finally, define P(vjz|ρ(vjz)) = Pt (unz |ρ(unz)) for all j>n, or P(vjz |ρ (vjz)) = Pt (unz |ρ (unz)∩ {uiz |n-i<j}) for j≤n. We say that the template 𝒢t is of order n−1. Supplementary Figure S4 illustrates template instantiation.
2.3 Observable versus unobservable contexts
Given a context-dependent network 𝒢 = (𝒱,α,ℰ,P) instantiated on an alignment A from some template 𝒢t, we say that contexts in 𝒢 are fully observable if, for every edge zi → wj ∈ ℰ such that i ≠ j, the residue Aiz associated with variable zi is present in the alignment (i.e. it is not a gap or an unaligned position). Likelihood evaluation for networks with fully observable contexts can be carried out using a simple extension of Felsenstein's algorithm:
| (9) |
where context(v) is the joint event describing the observed context: ρ(v)−{u}∼Aρ (v)−{u}.
When contexts are not fully observable, Felsenstein's single-site algorithm will not suffice, since there will be context variables for which the value is unknown; Felsenstein's algorithm marginalizes only over those unobservables in the current column of the alignment. Although the general sum–product algorithm can still be applied on the full network 𝒢 (Kschischang et al., 2001),, in the most general case the dense web of dependencies in the full network will prevent efficient factorization, so that summations must be evaluated over all columns simultaneously, resulting in a time complexity of O(|α|2ℓ|𝒯|) and space requirements on the order of O(|α|ℓ|𝒯|)—i.e. exponential in the length ℓ of the alignment. The MCMC approaches of Jensen and Pedersen (2000), Hwang and Green (2004) and Arndt and Hwa (2005) circumvent this computational difficulty via sampling.
An alternative solution is to utilize a windowing scheme, in which a fixed-length window of width n+1, for some small n, is superimposed over each interval of the alignment, with the computation of the column likelihoods within the window taking into account only those dependencies falling within the window. Under such a discipline, we need only perform summations over the (n+1)-mers inhabiting each taxon within the window (rather than over the ℓ-mers making up an entire row in the alignment), so that for reasonable values of n (say, 1≤n≤5) the inference problem is rendered tractable, though not extremely fast—O(ℓ |α|2n+2|𝒯|). The cost of such an approach is a willingness to assume that the variables within the sliding window are conditionally independent of variables outside the window. Such an assumption is not unreasonable so long as actual context effects do not extend over distances longer than n columns. Note that while variables close to the edge of the window will be most severely affected by the conditional independence assumption, these same variables will be re-evaluated at each of the other window positions as the window is moved (in 1,bp steps) along the length of the alignment. As long as n is made sufficiently large to cover the longest inter-column dependence in the Bayesian network, all dependencies in the network will contribute to the computation of the likelihood.
Formally, let 𝒢 = (𝒱,α,ℰ,P) be a network instantiated on an alignment A from an n-th order template 𝒢t—i.e. such that the longest inter-column dependency spans n+1 columns. Let u→v ∈𝒫 be an edge in the phylogeny 𝒫. Then we can approximate the probability P(A[i,j]v|A[i,j]u of observing the substitution of the (n+1)-mer A[i,j]u in ancestor u by A[i,j]v in descendant v as follows:
| (10) |
for vk the variable in 𝒱 associated with cell Akv in the alignment. We can then compute the (conditional) likelihood of an interval [ij] of the alignment via Lrn(A[i,j]r, for the observable root genome r and a generalized Felsenstein recurrence Lun defined on complete (n+1)-mers x:
| (11) |
We refer to this version of Felsenstein's algorithm as ℱNMER; it permits a dynamic-programming implementation identical to the one for ℱ0 except for the use of the higher order alphabet αn+1.
As noted previously (Gross and Brent, 2005; Siepel and Haussler, 2004b), (n+1)-mer substitution probabilities such as those given by Equation (11) can be converted into conditional, single-column probabilities via:
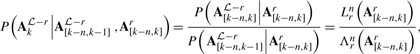 |
(12) |
for k>n, where Λun is a modified version of Lun which marginalizes over the final symbol in the leaf (n+1)-mers:
| (13) |
The likelihood of the entire alignment may then be estimated by employing an n-th order Markov assumption (Supplementary Fig. S2 illustrates these steps):
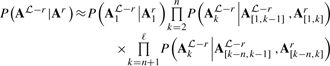 |
(14) |
Such an assumption can be justified by noting that context-dependence is, by definition, a local effect. As with Markov chains, conditioning may be performed on columns to the right (instead of to the left) of the current column; furthermore, conditioning simultaneously on both left and right contexts is also possible as long as cycles are not induced in the dependency structure.
2.4 Joint versus conditional models
By generalizing the Markov assumption of Equation (14) to apply to the ancestral taxon as well as the descendant, we can decompose the substitution probability P(v = y|u = x) on (n+1)-mers into a product of conditional probabilities in which a single-nucleotide substitution is conditioned on a pair of n-mers (one from the ancestor and one from the descendant):
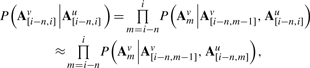 |
(15) |
where the second line invokes an additional conditional independence assumption (illustrated in Supplementary Fig. S3). For contexts of length n we say the substitution is of n-th order.
Setting λ = (ui-n, …,um−1)∼A[i−n,m−1]u ∧ (vi-n,…,vm−1)∼A[i−n,m−1]v, the final term in Equation(15) may be rewritten:
| (16) |
where P(Amv|Amu,λ) may be represented by any standard 4×4 substitution matrix (e.g. GTR, HKY, etc.) drawn from a collection of matrices indexed by A[i−n,m−1]u+A[i−n,m−1]v. We refer to such a collection of matrices as a conditional substitution model, as compared to the joint substitution model comprising a single 4n+1×4n+1 substitution matrix on entire (n+1)-mers.
Examples of joint substitution models are SH04 and GB05, mentioned previously, as well as the codon model GY94. To appreciate the reduction in parameters achieved by employing a conditional substitution model rather than a joint model, note that in the case of a dual-context (i.e. taking context from both the immediate taxon and its parent), general reversible model, the conditional scheme requires only 6× 42n free parameters (the coefficient 6 arising from the number of parameters in the GTR ‘core’ matrix), as opposed to (42n+2−4n+1)/2 for the joint model. Table 1 lists the numbers of parameters required for various model configurations.
Table 1.
Numbers of parameters required by models of orders 1–5
| Order | REV+JOINT | REV+JOINT+INDEP | GTR+COND+DUAL | GTR+COND+SINGLE | HKY+COND+SINGLE | FEL+COND+SINGLE |
|---|---|---|---|---|---|---|
| 1 | 120 | 48 | 96 | 24 | 8 | 4 |
| 2 | 2016 | 288 | 1536 | 96 | 32 | 16 |
| 3 | 32 640 | 1536 | 24 576 | 384 | 128 | 64 |
| 4 | 523 000 | 7680 | 393 216 | 1536 | 512 | 256 |
| 5 | 8×106 | 36 864 | 6×106 | 6144 | 2048 | 1024 |
GTR: reversible 4×4 model (six parameters + equilibrium frequencies) of Tavaré (1986); REV: general reversible model on n-mers of any size; JOINT: utilizes a single joint matrix; COND: utilizes a collection of conditional matrices; DUAL: assumes dual contexts; SINGLE: assumes single contexts; INDEP: assumes multiple changes in an n-mer are conditionally independent; HKY: Hasegawa et al. (1985) model (two parameters + equilibrium frequencies); FEL: Felsenstein's (1981) model (one parameter + equilibrium frequencies).
Many joint models (e.g. GY94, SH04 and HG04) avoid an explosion of parameters by assuming conditional independence between substitution events (denoted+INDEP in the table) within an n−mer—i.e. instantaneous rate Qx,y = 0 for any two n-mers x and y differing in more than one position.
Conditional substitution models afford much additional convenience due to their use of standard 4 × 4 nucleotide matrices, particularly in terms of software re-use and parameter estimation (Supplementary Methods). In particular, much existing software for modeling of molecular evolution may be fairly easily modified to model context effects in this way.
2.5 Single versus dual contexts
We now consider several specific network topologies. The reference model for all of our further descriptions will be the full n-th order model described above in relation to Equations (11–14), which we call the ‘whole hog’ model (abbreviated HOG). Under this model, Aiv is assumed to directly depend on the parent residue in the current column (i) as well as on the residues of the current taxon v and its parent taxon u in all of the preceding n columns (i.e. the n-th order Markov assumption applied to a pair of taxa), giving rise to the following Bayesian network description:
| (17) |
for all v∈ 𝒯−{r}. Because we take context from two taxa we say that we are using dual contexts. Due to the use of dual contexts, evaluation of the window likelihood via ℱNMER induces a time complexity of O(42n+2|𝒯|) for a single window, since for each branch in the phylogeny we require iteration over all possible pairs of (n+1)-mers. Thus, for even moderate values of n, HOG incurs a large computational cost due to the nested summations resulting from the recursion, and also due to the large number of parameters (especially in the case of a general reversible matrix: see REV+JOINT in Table 1). In addition, a dynamic-programming matrix of size O(4 affn+1|𝒯|) is required during inference.
Because of the high computational cost of the full HOG model, it is worthwhile to consider simpler networks in which some subset of dependencies in the HOG model is omitted. The first alternative model which we consider occurs under the assumption that the descendant context is likely to be identical to the ancestral context most of the time and can therefore be ignored; we call this model ACO (ancestral contexts only—see Fig. 1). We expect this assumption to hold best in the case of relatively low substitution rates, or when context dependence is not overly strong. Since this model utilizes single (rather than dual) contexts, it requires considerably fewer parameters than HOG (6 × 4n instead of 6 × 4 2n in the case of general reversible models), though the inference procedure has the same time complexity.
Fig. 1.

ROC curves for TRCO+GTR (orders 0–2) and SH04 (first order) on ENCODE data. y-axis: sensitivity; x-axis: false-positive rate. n: order of model.
ACO can be formally characterized via its Bayesian network description:
| (18) |
for u the parent of v in 𝒫. Inference in ACO again uses the generalized Felsenstein recurrence ℱNMER, except that the (n+1)-mer substitution term of Equation (11) is replaced with the one below:
| (19) |
We denote this modified inference procedure ℱACO. Even though the ACO model utilizes only a single context, the inference procedure must still sum over all possible pairs of (n+1)-mers, so that the time complexity is identical to that of ℱNMER.
The summation over (n+1)-mers in ℱACO can be eliminated by assuming that unobserved contexts are either identical to the root context or similar enough to it that the root context (which is observable) can be used in their place—a somewhat stronger assumption than that used in ACO regarding the low rate of substitution in the context sequence. Under this assumption, the summation term in ℱACO may be modified to sum over single nucleotides rather than (n+1)-mers, resulting in a significant reduction in time complexity for inference. We refer to the resulting model as TRCO (transitive root contexts only) and the associated inference algorithm as ℱTRCO. The Bayesian network for TRCO is described by:
| (20) |
for root r. The inference algorithm ℱTRCO for this model may be derived directly from Equation (9) since contexts are observable:
| (21) |
where λ = (ri−n, …,ri−1)∼A[i−n,i−1]r. In contrast to Equation (12), only one invocation of this recursion is necessary to obtain the conditional probability of a column: Lrn(Air).
A further simplification of TRCO is achieved by ignoring contexts altogether, except in the evaluation of substitutions between the root and its immediate descendants:
| (22) |
where Mx,yn,v(λ) denotes the (x,y) entry of the n-th order substitution matrix between v and its parent taxon, which is conditional on context λ; Mx,y0,v is the corresponding entry from the zeroth-order matrix, which has no context. This model we refer to as RCO (root contexts only).
As a final alternative model, we consider the use of Fitch's maximum parsimony algorithm (Fitch, 1971) for reconstruction of ancestral states, followed by ACO applied to the resulting, augmented alignment. The resulting model we denote MP (maximum parsimony) and its inference algorithm ℱMP:
| (23) |
for the current column i and context predicate λ = (ui−n, …,ui−1)∼ A[i−n,i−1]u. Because the ancestral sequences are inferred prior to the computation of the likelihood, there are no unobservables in the network. Thus, ℱMP need not perform any summation, rendering the algorithm extremely fast. Fitch's parsimony algorithm is itself very fast, since asymptotically it has O(|𝒯|) time complexity and most of the operations are simple memory accesses and bit operations. Like the other alternatives to HOG considered above, we expect MP to be most useful when substitution rates are low, so that the ancestral state reconstruction would be most similar to the actual ancestral states. Note that once the ancestral states have been reconstructed we may apply any of the foregoing models (e.g. HOG, ACO, RCO and TRCO) or other conceivable models, though we explicitly consider only ACO here.
3 RESULTS
3.1 Experiments
We compared both joint and conditional models of various orders on the task of modeling context-dependent substitution rates at third codon positions of human protein-coding exons, which are known to exhibit strong context effects due to the degeneracy structure of codons in their mapping to amino acids (Percudani, 2001). The conditional models included ACO, RCO, TRCO and MP models of first order, each with a core GTR matrix for evaluation of the P(Amv|Amu,λ) term. A first-order SH04 model served as an arbitrary representative of current n-mer-based approaches—i.e. REV+JOINT+INDEP (reversible, joint and assuming conditional independence between substitution events within an n-mer). The effects of different context lengths were investigated by also testing models of zeroth (GTR) and second (TRCO+GTR) orders. For all of these we estimated the model parameters from third codon positions of internal exons; as a null model we re-estimated all parameters from period-3 positions in introns (choosing one of three frames arbitrarily). Denoting the resulting foreground and background models as θfg and θbg, respectively, we then evaluated the discriminative power of each model on a held-out set of coding exons and introns via the following likelihood ratio rule:
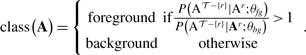 |
Under a uniform prior, such a rule is equivalent to a Bayes' classifier; we therefore included equal numbers of positive and negative test cases in each test set. Note that such a classifier is far simpler than a full parsing model such as a ‘PhyloHMM’ (Siepel and Haussler, 2004a); indeed, each of the substitution models we consider may be utilized in one or more individual states within a full-blown PhyloHMM, though in this work we focus only on the substitution models. Results were averaged via 5-fold cross-validation. Receiver operating characteristics (ROC) curves were also obtained; the area under the ROC curve (AUC) for each model was also computed, to allow comparison of classification accuracy at varying levels of sensitivity.
Genomic features were taken from random genes in the ENCODE regions (ENCODE Project Consortium, 2004); gene annotations were taken from the October 2005 set of VEGA ‘known genes’ (Harrow et al., 2006). Elements from a single gene were included either in the training partition or the test partition, but not both. MAVID (Bray and Pachter, 2004) alignments of human (hg17), mouse (mm5), rat (rn3), dog (canFam1) and chicken (galGal2) were used, with human selected as the target (root) genome. Each training set in the 5-fold cross-validation consisted of 200 elements of each type, totaling 329 kbp of human sequence on average, or roughly 1.6 Mb on average across five species.
Phylogenies were constructed via a two-step process: first, we inferred tree topologies from training sets via neighbor-joining Saitou and Nei, 1987); given the fixed branching patterns produced by NJ, branch lengths were then estimated simultaneously with all other model parameters via maximum likelihood estimation (MLE) using quasi-Newton methods; identical meta-parameters (i.e. step-sizes and convergence thresholds) were used for all runs to ensure equal treatment of all model classes (Supplementary Methods). Computations were performed on a cluster of 80 Xeon processors running at 2.8 GHz.
Columns in the alignment for which a gap was present in the target sequence were deleted, as in Siepel and Haussler (2004b). Gaps in non-target genomes were treated as ‘missing information’, as in Whelan and Goldman (2004), by summing over all possible nucleotide states for those missing variables; better modeling of gaps is a current topic of research, but is not addressed by the present work.
A further set of cross-validation runs was performed on the ENCODE data to assess the effect of different types of core matrices within conditional models.
In order to verify that context dependence could be detected and exploited by our models in at least one other type of genomic element, we also applied our TRCO+GTR model at several orders (0–3) and SH04 at first order to the task of discriminating between experimentally validated regulatory elements (positive class) and ancestral repeats (negative class). The latter dataset included 1268 positive and 1268 negative examples from human (hg18), rhesus macaque (rheMac2), chimpanzee (panTro1), cow (bosTau2), dog (canFam2), mouse (mm8) and rat (rn4), totaling 1.1Mb of sequence from the positive class and 1.0Mb from the negative class; sequences were downloaded from the UCSC browser (Kent et al., 2002) using coordinates obtained from the recent study by Taylor et al. (2006).
3.2 Cross-validation results
Results on the ENCODE data are summarized in Table 2 (see also Supplementary Figs S5 and S6) and Figure 2. Among the first-order conditional models, TRCO+GTR produced the highest classification accuracy (77.8%), fully matching the accuracy of the n-mer model SH04 (77.4%), despite having many fewer parameters (31 versus 55) and requiring roughly one-fortieth the computational effort during training (10 CPU-hours versus 384 CPU-hours) as compared to the n-mer model. The only model to require less training time than TRCO+GTR was MP+GTR, which required 7h rather than 10, but produced a substantially lower classification accuracy (74.5%). Conversely, ACO+GTR was found to produce nearly the same accuracy as TRCO+GTR (77.3%) but required nearly as much time as the n-mer model for training (382 CPU-hours). Thus, among first-order models, TRCO was found to present the best tradeoff between model complexity and discriminative power.
Table 2.
Summary of 5-fold cross-validation results for ENCODE data
| Model | n | Accur (%) | SD | AUC | # parms | LLexon | LLintron | Ttrain | Tcol |
|---|---|---|---|---|---|---|---|---|---|
| GTR | 0 | 74.8 | 2.6 | .895 | 13 | −11 486 | −85 890 | 3.0 | 1.2×10-5 |
| SH04 | 1 | 77.4 | 2.4 | .918 | 55 | −11 392 | −85 550 | 384 | 2.1×10-4 |
| TRCO+GTR | 1 | 77.8 | 3.9 | .924 | 31 | −11 352 | −85 735 | 10 | 1.3×10-5 |
| ACO+GTR | 1 | 77.3 | 3.9 | .920 | 31 | −11 354 | −85 726 | 382 | 6.2×10-4 |
| RCO+GTR | 1 | 74.9 | 3.2 | .902 | 31 | −11 463 | −85 753 | 26 | 1.9×10-5 |
| MP+GTR | 1 | 74.5 | 2.1 | .859 | 31 | −12 520 | −112 213 | 7 | 1.4×10-5 |
| TRCO+GTR | 2 | 81.2 | 4.2 | .945 | 103 | −11 182 | −85 671 | 180 | 1.7×10-5 |
n: length of context (not including the current column); Accur: mean classification accuracy (percentage correctly classified test cases); SD: standard deviation of accuracy; AUC: area under ROC curve; #parms: number of parameters (including branch lengths); LLexon: mean log-likelihood of training exons; LLintron: mean log-likelihood of training introns; Ttrain: mean training time, in CPU-hours (elapsed time × number of CPUs); Tcol: mean time (seconds) required to evaluate a single column in test alignments.
Fig. 2.

ROC curves for TRCO+GTR (orders 0–2) and SH04 (first order) on ENCODE data. y-axis: sensitivity; x-axis: false-positive rate. n: order of model.
Clear differences in prediction accuracy were seen when comparing models of orders 0–2. The accuracy of the zeroth-order GTR model (74.8%) was substantially less than that of the best first-order model (TRCO+GTR) (77.8%), which was in turn substantially less than that of the second-order TRCO+GTR model (81.2%). These differences are starkly apparent in the ROC curves (Fig. 2), in which it can be seen that while the three orders are clearly separable by their ROC curves, the first-order curves for TRCO+GTR and SH04 are nearly inseparable. Thus, while these experiments had sufficient resolution to clearly separate models of different orders, no substantial difference could be detected between the NMER-based model and our conditional model.
Table 3 shows the effect of utilizing different core matrices within a zeroth- or first-order TRCO model. Results indicate that all five core matrices benefitted from the use of context, with the gain in accuracy generally being greater for the more complex models. Thus, the use of more complex core matrices does not obviate the need for context modeling. Conversely, context modeling did not eliminate the need for either explicit transition–transversion modeling (as in models K2P, HKY and GTR) or the specification of non-uniform equilibrium frequencies (as in FEL, HKY and GTR).
Table 3.
Effect of core matrix type on accuracy of conditional model (TRCO) in 5-fold cross-validation experiments
| Model | acc(0) (%) | acc(1) (%) | Δ0,1 (%) | parms(0) | parms(1) | Y↔R | EQ |
|---|---|---|---|---|---|---|---|
| JC | 67.8 | 68.3 | 0.5 | 1 | 4 | no | no |
| FEL | 69.1 | 69.5 | 0.4 | 1 | 4 | no | yes |
| K2P | 73.6 | 74.9 | 1.3 | 2 | 8 | yes | no |
| HKY | 73.8 | 76.5 | 2.7 | 2 | 8 | yes | yes |
| GTR | 74.8 | 77.8 | 3.0 | 6 | 24 | yes | yes |
JC: Jukes and Cantor's (1969) model; K2P: Kimura's (1980) 2-parameter model; acc(n): classification accuracy for n-th order model; Δ0,1: acc(1)-acc(0); parms(n): number of free parameters (excluding equilibrium frequencies) in n-th order matrix; Y↔R: whether transition and transversion rates can be separately parameterized; EQ: whether non-uniform equilibrium frequences can be modeled.
Table 4 summarizes the results on the mammalian promoter dataset. Classification accuracy of the substitution model and a Markov chain applied to the root taxon (both singly and in combination) generally increased with increasing model order n. However, by third order there was no improvement in accuracy for the substitution model, and the Markov chain appeared to suffer from over-training. The first-order SH04 model performed less well than TRCO, even after the addition of the Markov chain; additional experiments utilizing an independently trained SH04 model are provided in the Supplementary Data.
Table 4.
Classification accuracy on mammalian promoter dataset
| Model | n | parms | acc | MC | acc+MC | Time |
|---|---|---|---|---|---|---|
| GTR | 0 | 17 | 72.9 | 81.0 | 86.0 | 6.6 |
| TRCO+GTR | 1 | 35 | 74.9 | 82.9 | 86.2 | 25.3 |
| TRCO+GTR | 2 | 107 | 76.2 | 85.8 | 88.3 | 161.3 |
| TRCO+GTR | 3 | 395 | 76.1 | 84.8 | 87.0 | 3910 |
| SH04 | 1 | 59 | 68.6 | 82.9 | 84.7 | 1146 |
n: order of substitution model;parms: number of free parameters (excluding equilibrium frequencies); acc: classification accuracy of substitution model alone; acc+MC: classification accuracy of combined substitution model and n-th order Markov chain applied to root taxon; MC: classification accuracy of Markov chain alone; time: training time in CPU-hours.
4 DISCUSSION
Context-dependent models based on substitutions of full n-mers, such as the various codon models derived from GY94 and the more general models SH04 and GB05, are computationally very expensive. Within the realm of purely phylogenetic applications, the GY94 model for triplet substitutions has seen relatively little use over the years, compared to single-nucleotide models of coding regions, due presumably to its high computational cost (Schadt and Lange, 2002; Shapiro et al., 2006; Whelan and Goldman, 2004).
Like the GY94 model, the more general SH04 model for n-mer substitutions employs a rate matrix in which simultaneous substitutions at multiple positions within an n-mer are not permitted. Although this constraint is to some degree relaxed when Q is compounded via P(t) = eQt, evaluation of joint probabilities for compound substitution events within an n-mer effectively treats these as conditionally independent events. Thus, while GY94 and SH04 both incur a high computational cost due to their use of dual contexts, even these models do not capture the full range of conceivable context-dependence over short distances—in particular, they are unable to fully utilize the information available in those dual contexts so as to capture effects such as compensatory changes at neighboring sites.
In contrast, for nongapped sequences the GB05 model is able to capture all possible context effects of a particular order, but requires significantly more parameters than even SH04, since separate matrices must be trained for each branch in the phylogeny. Reduction in numbers of parameters for n-mer-based models has been attempted by assuming strand symmetry (Jojic et al., 2004; Siepel and Haussler, 2004b); however, this is not an ideal assumption when modeling functional elements in DNA, since many such elements are not believed to be strand symmetric.
There has therefore remained a need for more practical context-dependent substitution models. While several recent studies have cast the problem in terms of graphical probability models, these have either ignored context-dependence (McAuliffe et al., 2004), considered only dual contexts (Gross and Brent, 2005), or considered only unobservable contexts (Jojic et al., 2004). Our investigation into the use of alternate dependency networks for single-nucleotide substitutions suggests that in the absence of rapid compensatory changes, context-dependent substitution modeling can be achieved with far fewer parameters and substantially lower computational costs while still permitting accurate discrimination between genomic elements under different selective pressures. Since the complexities of our single-context models are reduced at all orders compared to existing, n-mer-based models, the use of contexts of greater length than has been previously considered is now rendered feasible.
Within a Bayesian network framework, a potentially large number of different models may be considered, corresponding to the large number of possible networks for context-dependent nucleotide substitution. Although we have considered only a few possible networks, other topologies may prove useful, either generally or for specific modeling tasks; selection of optimal topologies for a given application via automated means is one interesting option for future investigation (e.g. Heckerman, 1999). The special case of observable contexts should be especially useful when combining large phylogenies and long contexts, in which the computational savings would be substantial.
Conditional substitution models potentially offer several additional advantages which we have not explored in this work; we briefly list a few of these. In the case of conditional models with fully observable contexts, an interpolation scheme such as those commonly used for variable-order Markov chains (Ohler et al., 1999; Salzberg et al., 1998) can be applied to mitigate the effects of sampling error for rare contexts. A conditional substitution model could, in principle, be conditioned on other random variables besides individual nucleotide identities—for example, one could condition on the local GC density computed over arbitrary window sizes (effectively capturing a much larger context but at a courser level), or on other intrinsic or extrinsic features of the DNA local to a given site (e.g. observed or predicted structural or chromatin-state features, etc.). The n-mer-based models could conceivably be so-conditioned as well, resulting in hybrid joint/conditional models; the latter may be particularly useful in capturing compensatory substitutions at adjacent sites while also conditioning on single-sequence (i.e. nondual) contexts at sites further away. Our simple conditional models may be especially useful in combination with indel (insertion/deletion) models, in which case an insertion or deletion within an n-mer would confuse an n-mer-based substitution model by introducing frameshifts between parent and child n-mers. Yet another possible avenue for future research is the use of degenerate alphabets to allow longer context modeling with fewer parameters—e.g. using the purine/pyrimidine alphabet {Y,R} for some or all of the context positions instead of the full {A,C,G,T}, similarly to the approach described by Taylor et al. (2006).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Adam Siepel and Sayan Mukherjee for useful discussions and advice.
Funding: National Institures of Health (1R01-HG004065 to U.O.).
Conflict of Interest: none declared.
REFERENCES
- Arndt PF, Hwa T. Identification and measurement of neighbor-dependent nucleotide substitution processes. Bioinformatics. 2005;21:2322–2328. doi: 10.1093/bioinformatics/bti376. [DOI] [PubMed] [Google Scholar]
- Averof M, et al. Evidence for a high frequency of simultaneous double-nucleotide substitutions. Science. 2000;287:1283–1286. doi: 10.1126/science.287.5456.1283. [DOI] [PubMed] [Google Scholar]
- Bray N, Pachter L. MAVID: constrained ancestral alignment of multiple sequences. Genome Res. 2004;14:693–699. doi: 10.1101/gr.1960404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R, et al. Biological Sequence Analysis. Cambridge: Cambridge University Press; 1998. [Google Scholar]
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences. J. Mol. Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Fitch WM. Toward defining the course of evolution: minimum change for a specific tree topology. Syst. Zool. 1971;20:406–416. [Google Scholar]
- Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;303:799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Gross SS, Brent MR. Using multiple alignments to improve gene prediction. In: Miyano S, et al., editors. Lecture Notes in Computer Science. Vol. 3500. New York: Springer; 2005. pp. 374–388. [Google Scholar]
- Gulko B, Haussler D. Using multiple alignments and phylogenetic trees to detect RNA secondary structure. In: Hunter L, Klein T, editors. Biocomputing: Proceedings of the 1996 Pacific Symposium; World Scientific Publishing: Singapore; 1996. pp. 350–367. [PubMed] [Google Scholar]
- Harrow H, et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7(Suppl. 1):S4. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, et al. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Heckerman D. A tutorial on learning with Bayesian networks. In: Jordan MI, editor. Learning in Graphical Models. Cambridge, MA: MIT Press; 1999. pp. 301–354. [Google Scholar]
- Hwang DG, Green P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. PNAS. 2004;101:13994–14001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JL, Pedersen A-MK. Probabilistic models of DNA sequence evolution with context dependent rates of substitution. Adv. Appl. Prob. 2000;32:499–517. [Google Scholar]
- Jojic V, et al. Efficient approximations for learning phylogenetic HMM models from data. Bioinformatics. 2004;20:161–168. doi: 10.1093/bioinformatics/bth917. [DOI] [PubMed] [Google Scholar]
- Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. New York, NY: Academic Press; 1969. pp. 21–132. [Google Scholar]
- Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kschischang FR, et al. Factor graphs and the sum-product algorithm. IEEE Trans. Inf. Theory. 2001;47:498–519. [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Lauritzen SL, Spiegelhalter DJ. Local computations with probabilities on graphical structures and their application to expert systems. J. R. Statist. Soc. B. 1988;50:157–224. [Google Scholar]
- McAuliffe JD, et al. Multiple-sequence functional annotation and the generalized hidden Markov phylogeny. Bioinformatics. 2004;20:1850–1860. doi: 10.1093/bioinformatics/bth153. [DOI] [PubMed] [Google Scholar]
- Moses AM, et al. Phylogenetic motif detection by expectation-maximization on evolutionary mixtures. In: Altman RB, et al., editors. Biocomputing: Proceedings of the 2004 Pacific Symposium; Singapore: 2004. pp. 324–335. [DOI] [PubMed] [Google Scholar]
- Ohler U, et al. Interpolated Markov chains for eukaryotic promoter recognition. Bioinformatics. 1999;5:362–369. doi: 10.1093/bioinformatics/15.5.362. [DOI] [PubMed] [Google Scholar]
- Pearl J. Probabilistic Reasoning in Intelligent Systems. San Francisco: Morgan Kaufmann; 1988. [Google Scholar]
- Pedersen JS, Hein J. Gene finding with a hidden Markov model of genome structure and evolution. Bioinformatics. 2003;19:219–227. doi: 10.1093/bioinformatics/19.2.219. [DOI] [PubMed] [Google Scholar]
- Percudani R. Restricted wobble rules for eukaryotic genomes. Trends Genet. 2001;17:133–135. doi: 10.1016/s0168-9525(00)02208-3. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Salzberg SL, et al. Interpolated Markov models for eukaryotic gene finding. Genomics. 1998;59:24–31. doi: 10.1006/geno.1999.5854. [DOI] [PubMed] [Google Scholar]
- Schadt E, Lange K. Codon and rate variation models in molecular phylogeny. Mol. Biol. Evol. 2002;19:1534–1549. doi: 10.1093/oxfordjournals.molbev.a004216. [DOI] [PubMed] [Google Scholar]
- Shapiro B, et al. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 2006;23:7–9. doi: 10.1093/molbev/msj021. [DOI] [PubMed] [Google Scholar]
- Siddharthan R, et al. PhyloGibbs: a Gibbs sampling motif finder that incorporates phylogeny. PLoS Comp. Biol. 2005;1:e67. doi: 10.1371/journal.pcbi.0010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Combining phylogenetic and hidden Markov models in biosequence analysis. J. Comp. Biol. 2004a;11:413–428. doi: 10.1089/1066527041410472. [DOI] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol. 2004b;21:468–488. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- Smith NGC, et al. A low rate of simultaneous double-nucleotide mutations in primates. Mol. Biol. Evol. 2003;20:47–53. doi: 10.1093/molbev/msg003. [DOI] [PubMed] [Google Scholar]
- Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986;17:57–86. [Google Scholar]
- Taylor J, et al. ESPERR: learning strong and weak signals in genomic sequence alignments to identify functional elements. Genome Res. 2006;16:1596–1604. doi: 10.1101/gr.4537706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S, Goldman N. Estimating the frequency of events that cause multiple-nucleotide changes. Genetics. 2004;167:2027–2043. doi: 10.1534/genetics.103.023226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.