

Abstract
Motivation: In ordinary regression, imposition of a lasso penalty makes continuous model selection straightforward. Lasso penalized regression is particularly advantageous when the number of predictors far exceeds the number of observations.
Method: The present article evaluates the performance of lasso penalized logistic regression in case–control disease gene mapping with a large number of SNPs (single nucleotide polymorphisms) predictors. The strength of the lasso penalty can be tuned to select a predetermined number of the most relevant SNPs and other predictors. For a given value of the tuning constant, the penalized likelihood is quickly maximized by cyclic coordinate ascent. Once the most potent marginal predictors are identified, their two-way and higher order interactions can also be examined by lasso penalized logistic regression.
Results: This strategy is tested on both simulated and real data. Our findings on coeliac disease replicate the previous SNP results and shed light on possible interactions among the SNPs.
Availability: The software discussed is available in Mendel 9.0 at the UCLA Human Genetics web site.
Contact: klange@ucla.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The recent successes in association mapping of disease genes have been propelled by logistic regression using cases and controls. In most ways this represents a step down from the computational complexities of linkage analysis performed on large pedigrees. The most novel feature of these genome-wide association studies is their sheer scale. Hundreds of thousands of SNPs (single nucleotide polymorphisms) are now being typed on samples involving thousands of individuals. This avalanche of data creates new problems in data storage, manipulation and analysis. Size does matter. For instance, with hundreds of thousands of predictors, the standard methods of multivariate regression break down. These methods involve matrix inversion or the solution of linear equations for a very large number of predictors p. Since these operations scale as p3, it is hardly surprising that geneticists have opted for univariate linear regression SNP by SNP. This simplification goes against the grain of most statisticians, who are trained to consider predictors in concert. In this article, we explore an intermediate strategy that permits fast computation while preserving the spirit of multivariate regression.
The lasso penalty is an effective device for continuous model selection, especially in problems where the number of predictors p far exceeds the number of observations n (Chen et al., 1998; Claerbout and Muir, 1973; Santosa and Symes, 1986; Taylor et al., 1979; Tibshirani, 1996). Several authors have explored lasso penalized ordinary regression (Daubechies et al., 2004; Friedman et al., 2007 Fu, 1998; Wu and Lange, 2008) in both the ℓ1 and ℓ2 settings. Let yi be the response for case i, xij the j-th predictor for case i, βj the regression coefficient corresponding to the j-th predictor and μ the intercept. For notational convenience also let θ=(μ, β1,…, βp)t and xi=(xi1,…, xip)t. In ordinary linear regression, the objective function is f(θ)=∑i=1n(yi−μ−xitβ)2. In ℓ1 regression one replaces squares by absolute values. Lasso penalized regression is implemented by minimizing the modified objective function
| (1) |
Note that the intercept μ is ignored in the lasso penalty λ ∑j=1p|βj|. The tuning constant λ controls the strength of the penalty, which shrinks each βj toward the origin and enforces sparse solutions. A ridge penalty λ∑j=1pβj2 also shrinks parameter estimates, but it is not as effective in actually forcing many estimates to vanish. This defect of the ridge penalty reflects the fact the |b| is much larger than b2 for small b.
Many diseases are believed to stem from the interaction of risk factors. This further complication can also be handled by lasso penalization if we proceed in two stages. In the first stage, we select the important marginal predictors; in the second stage, we look for interactions among the supported predictors. In both stages, we adjust the penalty constant to give a fixed number of supported predictors. In most genetic studies, researchers have a general idea of how many true predictors to expect. Our software encourages experimentation and asks the user to decide on the right balance between model completeness and quick computation.
This article, like most papers, has its antecedents. In particular, Shi et al. (2006, 2007, 2008); Uh et al. (2007) and Park and Hastie (2008) make substantial progress in adapting the lasso to logistic regression and to the discovery of interactions. Malo et al. (2008) apply ridge regression to distinguish causative from non-causative SNPs in a small region. Schwender and Ickstadt (2008) and Kooperberg and Ruczinski (2005) identify interactions using logic regression. These and other relevant papers are reviewed by Liang and Kelemen (2008). We focus on a coordinate descent algorithm because it appears to be the fastest available. Competing algorithms for lasso penalized logistic regression include non-negative quadratic programming (Sha et al., 2007), quadratic approximations (Lee et al., 2006) and interior point methods (Koh et al., 2007). Friedman et al. (2008) compared coordinate descent with several competing algorithms and concluded that it performs the best.
The specific contributions made in this article include (i) the consistent use of the lasso penalty for both marginal and interaction predictors, (ii) selection of the tuning constant to give a fixed number of predictors, (iii) application of cyclic coordinate ascent in maximizing the lasso penalized loglikelihood, (iv) rigorous pre-selection of a working set of predictors and (v) application of false discovery rates for global significance. Our overall strategy combines fast computing with good recovery of the dominant predictors.
In the remainder of the article, Section 2 fleshes out our statistical approach to data. In particular, it covers the lasso penalized logistic model, selection of the tuning constant, cyclic coordinate ascent and assessment of significance for both marginal and interaction predictors. The procedures are summarized as follows:
prescreening by a score criterion (Section 2.6);
selection of the tuning parameters λ for a fixed number of predictors by bracketing and golden section search (Section 2.2);
parameter estimation via cyclic coordinate descent (Section 2.5);
significance assessment based on leave-one-out (LOO) indices (Section 2.3) and false discovery rate (FDR) (Section 2.7);
lasso identification and quantification of interactions among previously selected features (Section 2.4).
Section 3 evaluates the method on simulated data. Section 4 applies the method to real data on coeliac disease. Finally, Section 5 summarizes the advantages and limitations of lasso penalized logistic regression in association testing, puts our specific findings into the larger context of current research and mentions the availability of relevant software.
2 METHODS
2.1 Lasso penalized logistic regression
In case–control studies, the dichotomous response variable yi is typically coded as 1 for cases and 0 for controls. By analogy to ordinary linear regression, in linear logistic regression we write the probability pi=Pr(yi=1) of case i given the predictor vector xi as
| (2) |
The parameter vector θ=(μ, β1,…, βp)t is usually estimated by maximizing the loglikelihood
| (3) |
To encourage sparse solutions, we subtract a lasso penalty from the loglikelihood as just suggested. For the purposes of this article, we consider only additive models where the range of the predictors xij is restricted to the three values −1, 0 and 1, corresponding to the three SNPs genotypes 1/1, 1/2 and 2/2, respectively. A dominant model can be achieved by collapsing the genotypes 1/1 and 1/2, and a recessive model can be achieved by collapsing genotypes 1/2 and 2/2. In both models the assigned quantitative values are −1 and 1. In our experience, the set of markers entering the model is relatively insensitive to the genetic model assumptions. We recommend standardizing all non-SNP quantitative predictors to have mean 0 and variance 1.
2.2 Selection of the tuning constant λ
For a given value of the tuning constant λ, maximizing the penalized loglikelihood singles out a certain number of predictors with non-zero regression coefficients. Let r(λ) denote the number of predictors selected. If we reduce λ and relax the penalty, then more predictors can enter the model. Although minor exceptions occasionally occur, r(λ) is basically a decreasing function of λ with jumps of size 1. Hence, once a predictor enters the model, it usually remains in the model as λ decreases. Although a predictor's order of entry tends to be correlated with its marginal significance, violations of this rule of thumb occur with correlated predictors. For every integer s≤p, we assume that there is an interval Is on which r(λ)=s. One can quickly find a point in Is by a combination of bracketing and bisection. In bracketing, we start with a guess λ. If r(λ)=s, we are done. If r(λ)<s and a∈(0, 1), then there is a positive integer j such that r(aj λ)≥s. If r(λ)>s and b>1, then there is a positive integer k such that r(bk λ)≤s. In practice, we set a=1/2 and b=2 and take the smallest integer j or k yielding the second bracketing point. Once we have a bracketing interval [λl, λu], we employ bisection. This involves testing the midpoint λm=1/2(λl+λu). There are three possibilities: if r(λm)=s, we are done; if r(λm)<s, we replace λu by λm; and if r(λm)>s, we replace λl by λm. In either of the latter two cases, we bisect again and continue. As soon as we hit a point in Is, we halt the process.
The primary danger in bracketing is visiting a λ with r(λ) very large. To limit the damage from a poor choice of λ, we abort optimization of the objective function whenever the search process encounters too many non-zero predictors. Since predictors can enter and leave the model repeatedly prior to convergence, this check is delayed for several iterations, say 10. As a further safeguard, we set the maximum number of non-zero predictors allowed well above the desired number of predictors s. In practice we use s+10.
In simpler settings, cross-validation is used to find the best value of λ. Recall that in k-fold cross-validation, one divides the data into k equal batches (subsamples) and estimates parameters k times, leaving one batch out per time. The testing error for each omitted batch is computed using the estimates derived from the remaining batches, and the cross-validation curve c(λ) is computed by averaging testing error across the k batches. The curve c(λ) can be quite ragged, and many values of λ must be tried to find its minimum. To avoid this time-consuming process, we let the desired number of predictors drive statistical analysis. In actual gene mapping studies, geneticists would be thrilled to map even 5 or 10 genes. In our coeliac disease example, it is necessary to consider a larger number of predictors to uncover the full biological truth.
2.3 Assessing significance
When SNPs are tested one by one, it is easy to assign a P-value to a SNP by conducting a likelihood ratio test. If we ignore non-genetic predictors such as age, sex and diet, then the only relevant parameters are the intercept μ and the slope β of the SNP. The null hypothesis β=0 can be tested by maximizing the loglikelihood under the null and alternative hypotheses and forming twice the difference in maximum loglikelihoods. This statistic is asymptotically distributed as a χ2-distribution with 1 degree of freedom. Collectively, the P-values must be corrected for multiple testing, either by a Bonferroni correction or some version of a FDR correction. The latter choice is more appropriate when we anticipate a fairly large number of true positives. We will say more about FDR corrections later. A more compelling concern is that proceeding SNP by SNP omits the impact of other SNPs. Most statisticians prefer to assess significance in the context of multiple linear regression rather than simple linear regression. They resist this natural impulse in association studies because of the computational barriers and the mismatch between numbers of observations and predictors.
In our multivariate setting, we compare the standard SNP by SNP P-values with alternative P-values generated by considering the s selected predictors as a whole. Once we have selected the s model predictors, we discard the non-selected predictors and re-estimate parameters for the selected predictors with λ=0. Since s is small, say 10 to 20 in our numerical studies, re-estimation is now a fully determined problem. We then undertake s further rounds of estimation, omitting each of the selected predictors in turn. These actions put us into position to conduct likelihood ratio tests by leaving one predictor out at a time. It is tempting to assign P-values by comparing the resulting likelihood ratio statistics to the percentile points of a χ2-distribution with 1 degree of freedom. This is invalid because it neglects the complex selection procedure for defining the reduced model in the first place. Nonetheless, these LOO P-values are helpful in assessing the correlations between the retained predictors in the reduced model. To avoid confusion, we will refer to the LOO P-values as LOO indices. The contrast between the univariate P-values and the LOO indices is instructive. Although both of these measures are defective and should not be taken too seriously, they are defective in different ways and together give a better idea of the truth.
2.4 Interaction effects
As mentioned previously, we advocate testing for interactions after identifying main effects. This strategy is prompted by the sobering number of interactions possible. With p predictors, there are 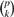 k-way interactions, and 2p interactions in all. With hundreds of thousands of SNPs, it is impossible even to examine all two-way interactions. These problems disappear once we focus on a handful of interesting marginal predictors. However, our commitment to a two-stage strategy brings in its wake certain technical problems.
k-way interactions, and 2p interactions in all. With hundreds of thousands of SNPs, it is impossible even to examine all two-way interactions. These problems disappear once we focus on a handful of interesting marginal predictors. However, our commitment to a two-stage strategy brings in its wake certain technical problems.
First, there is the combinatorial question of how to generate all subsets of {1,…, r} up to a given size. Fortunately, good algorithms for this task already exist. Minor changes to the NEXKSB code in Nijenhuis and Wilf (1978) permit one to generate one subset after another, with smaller subsets coming before larger subsets. Thus, when the number of predictors r retained from stage one is too large to generate all subsets, one can easily visit all lower order interactions and bypass higher order interactions. Second, there is the problem of storing the interaction predictors. We finesse this problem by computing interaction products on the fly. Third, there is the question of how to integrate SNP predictors with other predictors such as sex, age and environmental exposures. Since this is largely a programming problem, we omit further discussion of it. Fourth, our interactions do not involve any self-interactions. Inclusion of self-interactions would force us to pass from subsets to multisets. For SNPs the potential gain seems worth less than the bother. Other predictors such as age have a richer range of values, so it may be useful to add predictors such as age squared, age cubed and so forth to the original list of predictors. Finally, there are the problems of model selection and hypothesis testing for the interaction effects. Here, again it seems reasonable to rely on lasso penalized estimation and LOO indices.
2.5 Cyclic coordinate ascent algorithm
In linear logistic regression, maximum likelihood estimates are usually found by the scoring algorithm. This requires the score and observed information
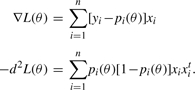 |
(4) |
of the loglikelihood (3). Because scoring coincides with Newton's method, it is fast and reliable, and most statisticians would agree that it is the method of choice for low-dimensional problems. Its Achilles heel is the need to invert the observed information at each iteration. If we add to this drawback the complication of dealing with the non-differentiable lasso penalty, then it becomes abundantly clear that competing algorithms should be considered in association analysis.
The oldest and simplest alternative, coordinate ascent, updates one parameter one at a time. Coordinate ascent comes in two flavors, cyclic and greedy (Wu and Lange, 2008). In cyclic coordinate ascent, each parameter is updated in turn; in greedy coordinate ascent, the parameter leading to the greatest increase in the objective function is updated. Although greedy coordinate ascent makes faster initial progress in logistic regression, it suffers from excess overhead. For this reason we will confine our attention to cyclic coordinate ascent.
Although the logistic loglikelihood (3) is non-linear, it has the compensating property of concavity. Concavity fortunately carries over to the lasso penalized loglikelihood
because the sum of two concave functions is concave. The objective function g(θ) is non-differentiable, but it does possess a directional derivative along each forward or backward coordinate direction. For instance, if uj is the coordinate direction along which βj varies, then
and for vj=−uj
When a function such as L(θ) is differentiable, its directional derivative along uj coincides with its ordinary partial derivative, and its directional derivative along v=−uj coincides with the negative of its ordinary partial derivative.
To update a single parameter of the objective function g(θ), we use one-dimensional scoring. This works well for the intercept parameter μ because there is no lasso penalty. For a slope parameter βj, the lasso penalty intervenes, and particular care must be exercised near the origin. In fact, it simplifies matters to start scoring at the origin. Here, we test the directional derivatives dujg(θ) and dvjg(θ). If both are non-positive, then g(θ) cannot be increased by moving away from the origin. This claim follows from the concavity of g(θ). If one of the directional derivatives dujg(θ) and dvjg(θ) is positive and the other is non-positive, then progress can be made along the corresponding arm of g(θ), and scoring is commenced until convergence is achieved along that arm. Concavity rules out the possibility that both directional derivatives are positive. A simple sketch of a concave function will convince the reader of this assertion.
In practice, we start all parameters at the origin. In overdetermined problems, the vast majority of slopes βj are permanently parked there. Only those with considerable evidence in their favor can overcome the pressure of the lasso pushing them toward the origin. Even those that escape this pressure can be forced back to the origin as other more potent predictors enter the model. It is clearly computationally beneficial to organize parameter updates by tracking the linear predictor μ+xitβ of each case. These start at 0, and when a single component of θ is updated, it is trivial to update the linear predictors.
2.6 The score criterion and efficient computations
In Section 2.2, we demonstrated that the lasso penalty can be tuned to select a predetermined number of the most relevant SNPs. Once the value of the tuning constant λ is fixed, the penalized likelihood is quickly maximized by cyclic coordinate ascent to give us the desired number of non-zero coefficients. However, since we face a very large number of SNP predictors, it would be much more efficient if we could start our search procedure by focusing on a substantially smaller set of features that are more likely to be associated with the response. We accomplish this by a ‘swindle’ that screens the predictors according to a simple score criterion.
The score equations of the loglikelihood (4) for linear logistic regression define part of the Karush–Kuhn–Tucker (KKT) conditions (Lange, 2004)
| (5) |
| (6) |
for optimality in the penalized model. Here pi(λ) is the fitted probability for observation i, fit using the indicated value of λ. For very large λ, all the βj are estimated as zero, and the only non-trivial parameter is the intercept μ, which is unpenalized. If p0 is the overall proportion of cases in the data, then the intercept is estimated as 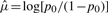 for large λ.
for large λ.
We accordingly define the following initial absolute score:
| (7) |
for each predictor. Note that aj determines the standard score statistic for testing the null model βj=0 with μ fixed at 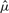 . The first predictor to enter the lasso penalized model as λ decreases is the predictor with the largest value of aj.
. The first predictor to enter the lasso penalized model as λ decreases is the predictor with the largest value of aj.
These considerations suggest a screening device for models with large numbers of SNPs. Because we insist on tuning the lasso penalty to select just a handful of predictors, the final absolute scores are apt to correlate strongly with the precomputed absolute scores. Thus, if we desire s predictors, we take k to be a reasonably large multiple of s, say k=10s, sort the aj, and extract the k predictors with the largest values of aj. Call this subset Sk. We now subject Sk to our estimation procedure and choose a value λk to give us exactly k predictors. The selected predictors satisfy the KKT conditions (5) and (6). If the predictors omitted from Sk also satisfy the KKT condition (6), then we have found the global minimum for the given value λk and stop. If one of the omitted predictors fails the KKT condition (6), we replace k by 2k, say, and repeat the process. Eventually, the KKT conditions are satisfied by all predictors. Since the KKT conditions are sufficient as well as necessary for a global maximum, this process legalizes the swindle. Often the value 10s works. When it does not, usually just a few doublings suffice. For example, if the desired number of predictors is s=10, in stage one we fit a model with 100 predictors. When stage two is needed, we fit a model with 200 predictors, and so forth. If there are hundred of thousands of SNPs, our swindle saves an enormous amount of computing with no loss in rigor.
Of course, the swindle sets Sk may contain highly correlated features with redundant information. This turns out to be the case with the HLA SNPs in our coeliac example. Fortunately, most of the redundant features are discarded by the lasso penalty. Our numerical results, for instance those displayed in Table 1, confirm that the swindle dramatically speeds up computation while preserving model selection results.
Table 1.
Simulation results based on 50 random samples
| (p, n) | ρ (s1, s2) | λ1 | Ktrue,1 | λ2 | Ktrue,2 | Time no | Time |
|---|---|---|---|---|---|---|---|
| swindle | swindle | ||||||
| (5000, 500) | 0.0 (10, 10) | 29.43 (1.50) | 5.00 (0.00) | 29.64 (1.90) | 5.84 (0.67) | 1.36 (0.34) | 0.68 (0.11) |
| (5000, 500) | 0.0 (10, 20) | 29.43 (1.50) | 5.00 (0.00) | 10.86 (1.71) | 6.98 (0.14) | 2.18 (0.39) | 1.57 (0.26) |
| (5000, 500) | 0.0 (20, 10) | 25.46 (1.06) | 5.00 (0.00) | 30.06 (1.65) | 5.84 (0.67) | 2.67 (0.40) | 1.10 (0.17) |
| (5000, 500) | 0.0 (20, 20) | 25.46 (1.06) | 5.00 (0.00) | 25.49 (1.25) | 6.24 (0.65) | 2.75 (0.36) | 2.17 (0.39) |
| (5000, 500) | 0.8 (10, 10) | 19.51 (1.94) | 5.00 (0.00) | 17.62 (3.24) | 5.04 (0.20) | 3.06 (0.52) | 1.76 (0.46) |
| (5000, 500) | 0.8 (10, 20) | 19.51 (1.94) | 5.00 (0.00) | 6.16 (1.12) | 6.58 (0.57) | 5.91 (5.74) | 4.61 (5.63) |
| (5000, 500) | 0.8 (20, 10) | 16.40 (1.50) | 5.00 (0.00) | 19.79 (2.01) | 5.04 (0.20) | 6.40 (2.94) | 3.08 (0.94) |
| (5000, 500) | 0.8 (20, 20) | 16.40 (1.50) | 5.00 (0.00) | 16.28 (1.65) | 5.12 (0.38) | 6.50 (4.32) | 5.14 (3.33) |
| (50 000, 2000) | 0.0 (10, 20) | 67.39 (2.21) | 5.00 (0.00) | 21.83 (3.18) | 7.00 (0.00) | 39.17 (11.45) | 10.09 (10.81) |
| (50 000, 2000) | 0.8 (10, 20) | 45.99 (2.12) | 5.00 (0.00) | 15.09 (2.39) | 7.00 (0.00) | 102.31 (33.92) | 14.59 (10.37) |
| (100 000, 2000) | 0.0 (10, 20) | 69.77 (2.13) | 5.00 (0.00) | 23.62 (3.24) | 7.00 (0.00) | 110.24 (22.59) | 8.94 (11.27) |
| (100 000, 2000) | 0.8 (10, 20) | 47.71 (2.30) | 5.00 (0.00) | 14.66 (2.54) | 7.00 (0.00) | 197.20 (53.17) | 10.81 (1.69) |
2.7 Computation of FDR
The score swindle also has implications for the assessment of the FDR for the univariate P-values. We will not pursue these delicate connections here because in practice most geneticists demand that all univariate tests be done. Fortunately, it takes just a few minutes of computing time to carry out the univariate logistic regressions encountered in a modern association study. Even substituting likelihood ratio tests for score tests does not change this fact.
In the Simes procedure highlighted by Benjamini and Hochberg (1995) in their analysis of FDR, there are n null hypotheses H1,…, Hn and n corresponding P-values P1,…, Pn. The latter are replaced by their order statistics P(1),…, P(n). If for a given α≥0, we choose the largest integer j such that P(i)≤(i/n)α for all i≤j, then we can reject the hypotheses H(1),…, H(j) at an FDR of α or better. This procedure is justified in theory when the tests are independent or positively correlated. In the presence of linkage equilibrium, association tests are independent; in the presence of linkage disequilibrium, they are positively correlated. For a more detailed discussion of the multiple testing issues in SNP studies, see Nyholt (2004).
3 ANALYSIS OF SIMULATED DATA
To evaluate the performance of lasso penalized regression in association testing, we focus on underdetermined problems where the number of predictors p far exceeds the number of observations n. Our simulation model
| (8) |
involves both marginal effects and two-way interactions. For ease of simulation, we assume that each predictor vector xi is derived from a realization of a multivariate normal vector Yi whose marginals are standard normal and whose covariances are
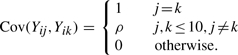 |
Thus, only the first 10 predictors are correlated. To mimic a SNP with equal allele frequencies, we set xij equal to −1, 0 or 1 according to whether Yij<−c, −c≤Yij≤c, or Yij>c. The cutoff −c is the first quartile of a standard normal distribution. In every simulation, we set μ=1, βj=1 for 1≤j≤5, and βj=0 for j>5. We also set ηkl=0 except for the special cases η12=η34=0.5. These substantial effect sizes allow us to discern signal from noise in fairly small samples.
To ameliorate the shrinkage of the non-zero estimates for a particular λ, we always re-estimate the selected parameters in the final model, omitting the non-selected parameters and the lasso penalty. This yields better parameter estimates for testing purposes. We compute LOO indices as mentioned earlier and contrast them to univariate P-values based on estimating the impact of each predictor without reference to the other predictors.
We analyzed the simulated data in two stages. In stage one, we considered only main effects and selected s1 predictors. In stage two, we discarded the non-selected predictors and sought s2 marginal effects or interactions among the selected predictors. The sensible choice s2≥s1 permits all predictors singled out in stage one to remain in contention as marginal effects in stage two. Because virtually all association studies yield only a handful of predictors that can be replicated, we took s1 and s2 small and considered the specific pairs (s1, s2)=(10, 10), (10, 20), (20, 10), (20, 20). Table 1 summarizes our results over 50 random replicates for various choices of the number of predictors p, the number of subjects n and the correlation coefficient ρ. Table 1 reports the average values of the tuning constants λ1 and λ2, the average number of true predictors Ktrue,1 and Ktrue,2 found and the average computing times in seconds. The subscripts 1 and 2 refer to the first and second stages. The standard error of each average appears in parentheses.
The last two columns of Table 1 summarize computing times with and without our computational swindle. Forgoing the swindle inflates all times in Table 1. For p=5000 the differences are not too noticeable, but for p=100000 it takes 10 to 20 times longer to reach the lasso solution without the swindle.
The results Table 1 for the choice (s1, s2)=(10, 20) appear best. In general, we recommend using a substantially larger s2 than s1. Performance degrades as we pass from uncorrelated to highly correlated predictors. More iterations are needed for convergence, and the fraction of true predictors captured falls. With a large enough sample size, performance is perfect. Table 1 in our submitted Supplementary Materials displays our results for a single representative sample with p=50 000, n=2000, ρ=0 and (s1, s2)=(10, 20). At stage one, all five true predictors are correctly selected with impressive univariate P-values and LOO indices. At stage two, all five main effects and both interaction effects are selected. In both instances, the univariate P-values and LOO indices of the true predictors are much smaller than the corresponding values for the false predictors.
It is also instructive to consider what happens in the simulated data with p=5000, n=500 and ρ=0 when the stage one tuning constant λ1 varies. Figure 1 plots six things as a function of λ1: (i) the number of predictors selected at stage one, (ii) the number of predictors selected at stage two, (iii) the number of true predictors selected at stage one, (iv) the number of true predictors selected at stage two, (v) the FDR at stage one and (vi) the FDR at stage two. In stage two, we set the tuning constant λ2=25. In counting true predictors, we consider only marginal predictors at stage one and marginal plus interaction predictors at stage two. When we know the true predictors, estimating FDR is trivial, and the Simes procedure can be ignored. Inspection of the six plots shows that all true predictors are recovered for a fairly broad range of λ1 values. As λ1 decreases, more predictors enter the model, and FDR increases.
Fig. 1.

Plots of the stage one penalty constant λ1 versus the number of selected predictors, the number of true predictors and FDR. The stage two penalty constant λ2=25.
4 ANALYSIS OF COELIAC DATA
4.1 Data description
In the British coeliac data of van Heel et al. (2007), p=310,637 SNPs are typed on n=2200 subjects (938 males and 1262 females). Controls outnumber cases 1422 to 778. Across the sample, an impressive 99.875% of all genotypes are assigned; no individual has more than 10% missing data. We impute missing genotypes at a SNP by the method sketched in Ayers and Lange (2008). Only 32 SNPs show a minor allele frequency below 1%; these are dropped from further analysis.
4.2 Simulation study based on coeliac data
We also tested our method by conducting a simulation study based on the coeliac data. Here in model (8), we took μ=−3, βj=1 for gender, rs3737728 (SNP2), rs9651273 (SNP4) and rs4970362 (SNP9), and βj=0 for the remaining SNPs. We also set ηkl=2 for the interaction of gender and rs3934834 (SNP1) and the interaction of SNP4 and SNP9; all other ηkl we set to 0. Notice that SNP1 has no marginal effect even though it interacts with gender in determining the response. The lower right-hand block of the correlation matrix
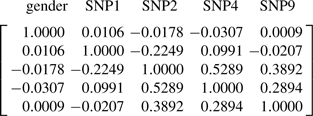 |
indicates fairly strong linkage disequilibrium among the three marginally important SNPs. Table 2 summarizes Fisher's exact test for Hardy–Weinberg equilibrium on the four SNPs (Lazzeroni and Lange, 1998). A total of 10 000 random tables were sampled to approximate P-values at each SNP.
Table 2.
Fisher's exact test for Hardy–Weinberg equilibrium on the 2200 coeliac cases and controls
| Locus | Est. | Expected | Observed | ||
|---|---|---|---|---|---|
| name | P-value | Range | homozygotes | homozygotes | |
| s3934834 | 1.0000 | ||||
| s3737728 | 0.7101 | ± | 0.0090743 | 1303.39 | 1296 |
| s9651273 | 0.6445 | ± | 0.0095733 | 1304.25 | 1294 |
| s4970362 | 0.7412 | ± | 0.0087595 | 1189.35 | 1197 |
Following our previous plan of analysis, we varied the numbers of predictors (s1, s2) in the model. The best results summarized in Table 3 reflect the sensible choice (s1, s2)=(10, 20). At stage one, all four true predictors are correctly selected. In stage two all four main effects are selected, and both interaction effects are selected for the vast majority of the 50 random replicates.
Table 3.
Results for 50 random replicates using the coeliac genotypes
| (s1, s2) | λ1 | Ktrue,1 | λ2 | Ktrue,2 | Time |
|---|---|---|---|---|---|
| Additive model | |||||
| (10, 10) | 48.78 | 4.00 | 52.19 | 4.46 | 45.33 |
| (1.57) | (0.00) | (4.11) | (0.50) | (13.48) | |
| (10, 20) | 48.78 | 4.00 | 18.24 | 5.70 | 66.36 |
| (1.57) | (0.00) | (4.05) | (0.61) | (12.82) | |
| (20, 10) | 45.10 | 4.00 | 53.93 | 4.44 | 74.22 |
| (1.24) | (0.00) | (4.00) | (0.50) | (29.64) | |
| (20, 20) | 45.10 | 4.00 | 45.11 | 4.54 | 137.16 |
| (1.24) | (0.00) | (1.63) | (0.50) | (51.70) | |
| Dominant model | |||||
| (10, 20) | 85.18 | 3.96 | 26.68 | 5.70 | 182.98 |
| (4.57) | (0.20) | (5.69) | (0.54) | (20.62) | |
| Recessive model | |||||
| (10, 20) | 62.53 | 3.00 | 20.76 | 3.14 | 83.76 |
| (3.93) | (0.00) | (8.32) | (0.35) | (66.12) | |
Our success with the additive model was partially replicated when we simulated under dominant and recessive models. In the dominant model, we score a SNP predictor as 1 if the number of minor alleles is 1 or 2; otherwise we score it as −1. In the recessive model, we score a SNP predictor as 1 if the number of minor alleles is 2; otherwise we score it as −1. The last two rows of Table 3 report our analysis results for the dominant and recessive models. The results under the dominant model are nearly as good as those under the additive model. Since the numbers of predictor values equal to 1 and −1 are better balanced under the dominant model, it is hardly surprising that the recessive model does worse.
4.3 Results of real data analysis
Replicating earlier results with antigenic markers, van Heel et al. (2007) find overwhelming evidence for association in the human leukocyte antigen (HLA) region of chromosome 6. SNP rs2187668 in the first intron of HLA-DQA1 has the strongest association, followed by SNPs rs9357152 and rs9275141 within or adjacent to HLA-DQB1. van Heel et al. also identify a more weakly associated region on chromosome 4 centered on SNPs rs13119723 and rs6822844 in the KIAA1109-TENR-IL2-IL21 linkage disequilibrium block. Their results are reproduced in our supplementary Table 2. The P-values listed in the table are univariate P-values taking one SNP at a time.
We now examine several models with different numbers of desired predictors. Since the grand mean μ always enters the model first, we omit it from further discussion. In model 0 with one predictor mandated, SNP rs2187668 on chromosome 6 HLA region is selected. This SNP has the smallest univariate P-value (9.48 × 10−191) among all the 310 605 SNPs tested. In model 1 with five predictors mandated, we identify four HLA SNPs in addition to rs2187668. In model 2 with 10 predictors mandated, once again we recover only HLA SNPs from chromosome 6; these results are summarized in Table 3 of our submitted Supplementary Materials. Univariate P-values appear in column 4 and LOO indices in column 5 of the table. It is striking how different the univariate P-values and LOO indices are for these SNPs. This phenomenon is just another manifestation of the high linkage disequilibrium among the SNPs. The estimated FDRs for the selected SNPs are all much smaller than 0.01. In model 3 with 50 predictors mandated, we finally see predictors outside the HLA region. Table 4 records the non-HLA predictors identified. Here, univariate P-values differ less from LOO indices because the SNPs are largely uncorrelated.
Table 4.
The non-HLA predictors found under model 3 with 50 mandated predictors
| Position | Univariate | LOO | |||
|---|---|---|---|---|---|
| SNP | Chr | in BP | P-value | index | Estimate |
| gender | 2.77489e-25 | 9.20120e-18 | 0.61074 | ||
| rs1888176 | 1 | 63298344 | 0.001561 | 0.000234 | 0.36746 |
| rs13397583 | 2 | 23459535 | 1.60268e-05 | 0.001518 | 0.32605 |
| rs6735141 | 2 | 142468480 | 0.000684 | 2.84818e-05 | 0.44583 |
| rs1836577 | 3 | 5776886 | 0.000955 | 0.000119 | −0.38543 |
| rs6762743 | 3 | 180494694 | 8.41159e-07 | 3.04012e-06 | 0.50470 |
| rs1559810 | 3 | 189607048 | 6.24178e-05 | 0.000587 | −0.36208 |
| rs991316 | 4 | 100541468 | 0.000286 | 5.86295e-05 | 0.38997 |
| rs12642902 | 4 | 123727951 | 4.07547e-05 | 2.46611e-06 | 0.48330 |
| rs153462 | 5 | 150585263 | 0.001544 | 8.49478e-05 | 0.42474 |
| rs13357969 | 5 | 150731750 | 8.89698e-05 | 0.000153 | 0.38409 |
| rs916786 | 7 | 109841758 | 0.000804 | 0.000174 | 0.37033 |
| rs736191 | 8 | 99264380 | 0.000261 | 0.000493 | 0.35143 |
| rs10505604 | 8 | 134096770 | 4.38312e-06 | 3.47134e-05 | −0.46746 |
| rs1499447 | 8 | 138051471 | 3.90991e-11 | 6.99196e-05 | 0.42571 |
| rs1901633 | 10 | 4800561 | 0.000474 | 0.007989 | 0.28059 |
| rs1064891 | 10 | 6316580 | 3.41871e-06 | 0.089192 | −0.35675 |
| rs1539234 | 10 | 6316749 | 5.33772e-06 | 0.843098 | 0.03108 |
| rs10501723 | 11 | 89922680 | 8.56749e-05 | 0.004005 | 0.28470 |
| rs7320671 | 13 | 19407203 | 0.001174 | 1.64655e-05 | −0.43494 |
| rs2879414 | 18 | 47962958 | 0.000272 | 2.78969e-05 | −0.41487 |
| rs10503018 | 18 | 53326747 | 0.000247 | 0.000925 | −0.36819 |
| rs2836985 | 21 | 39623039 | 0.000149 | 0.014373 | 0.24478 |
| rs6517581 | 21 | 40276738 | 0.001141 | 0.000959 | 0.33649 |
| rs5764419 | 22 | 42291261 | 0.000411 | 0.003744 | 0.28829 |
| rs2283693 | X | 9625063 | 0.000248 | 0.000731 | 0.28610 |
| rs5934725 | X | 9885994 | 0.000757 | 0.010604 | −0.22593 |
| rs4335267 | X | 44940333 | 0.000848 | 0.012583 | 0.21747 |
We find similarities and differences between the van Heel et al. (2007) results and our results. Almost all of the SNPs in Table 4 with univariate P-values below 10−4 are singled out by van Heel et al. (2007). The one exception is SNP rs1499447 on chromosome 8, which they dismiss because of irregularities in genotyping. We find different SNPs in the KIAA1109-TENR-IL2-IL21 block on chromosome 4. This is the region that replicates well in their Dutch and Irish samples. Our failure to identify the same SNPs in the KIAA1109-TENR-IL2-IL21 block is hardly a disaster; the region and ultimately the underlying gene are more important than the individual SNPs. It is noteworthy that among the 1000 most significant SNPs listed by van Heel et al. (2007), 979 are in the HLA region. Since SNPs in the HLA region on chromosome 6 are highly correlated with coeliac disease, model 4 with 10 mandated predictors removes the HLA SNPs, with the aim of finding associated SNPs outside the HLA region. Table 4 in our submitted Supplementary Materials now picks up SNPs on chromosomes 9, 11, 14 and 18 that do not appear in Table 4. Removing all chromosome 6 SNPs rather than just HLA SNPs leads to virtually the same results as displayed in Supplementary Table 4.
To test for interactions, we take the s1=50 predictors selected in model 3 and examine all marginal and two-way effects. The total number of predictors is 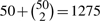 , and we keep s2=50 predictors in the model. Most of the 50 selected predictors have LOO indices close to one. Table 5 lists the marginal and interaction predictors with LOO indices less than 0.01. Several of these interactions are interesting. Given the predominance of female patients, the interaction between gender and one of the HLA SNPs is credible. The interactions between two HLA SNPs and SNPs on chromosomes 2, 3 and 8 are more surprising. It is particularly noteworthy that the univariate P-values for these three SNPs as marginal effects (Table 4) are far less impressive than their univariate P-values as interaction effects (Table 5).
, and we keep s2=50 predictors in the model. Most of the 50 selected predictors have LOO indices close to one. Table 5 lists the marginal and interaction predictors with LOO indices less than 0.01. Several of these interactions are interesting. Given the predominance of female patients, the interaction between gender and one of the HLA SNPs is credible. The interactions between two HLA SNPs and SNPs on chromosomes 2, 3 and 8 are more surprising. It is particularly noteworthy that the univariate P-values for these three SNPs as marginal effects (Table 4) are far less impressive than their univariate P-values as interaction effects (Table 5).
Table 5.
Strongest predictors under model 5 with all main effects and two-way interactions included
| SNP | Chr | Position in BP | Univariate P-value | LOO index | Estimate |
|---|---|---|---|---|---|
| gender | 2.77489e-25 | 0.00952 | 0.39359 | ||
| rs1888176 | 1 | 63298344 | 0.001561 | 0.001464 | 0.33090 |
| rs1836577 | 3 | 5776886 | 0.000955 | 8.69904e-05 | −0.38923 |
| rs1559810 | 3 | 189607048 | 6.24178e-05 | 9.71542e-05 | −0.40263 |
| rs991316 | 4 | 100541468 | 0.000286 | 0.000514 | 0.34159 |
| rs13357969 | 5 | 150731750 | 8.89698e-05 | 4.56935e-05 | 0.43473 |
| rs2187668 | 6 | 32713862 | 9.48302e-191 | 1.65234e-09 | −1.30307 |
| rs916786 | 7 | 109841758 | 0.000804 | 3.13293e-05 | 0.41684 |
| rs736191 | 8 | 99264380 | 0.000261 | 0.001485 | 0.33010 |
| rs10505604 | 8 | 134096770 | 4.38312e-06 | 0.001069 | −0.38067 |
| rs10501723 | 11 | 89922680 | 8.56749e-05 | 0.005633 | 0.28523 |
| rs2879414 | 18 | 47962958 | 0.000272 | 0.000125 | −0.38128 |
| rs10503018 | 18 | 53326747 | 0.000247 | 6.38471e-05 | −0.41328 |
| rs5934725 | 23 | 9885994 | 0.000757 | 0.004094 | −0.24651 |
| gender, rs2856997 | 6 | 1.45631e-19 | 0.00088 | 0.41372 | |
| gender, rs736191 | 8 | 1.42328e-06 | 0.008821 | 0.26378 | |
| rs6735141, rs9357152 | 2,6 | 2.29578e-22 | 0.000538 | 0.43816 | |
| rs6762743, rs9357152 | 3,6 | 1.11685e-26 | 9.16095e-06 | 0.55819 | |
| rs3129763, rs2187668 | 6,6 | 3.8393e-71 | 1.90299e-14 | −1.31326 | |
| rs2294478, rs1499447 | 6,8 | 3.89141e-27 | 0.008368 | 0.38291 |
Here, we take s1=50 and s2=50 and list an effect when its LOO index falls below 0.01.
5 DISCUSSION
Our analysis of simulated data demonstrates that lasso penalized regression is easily capable of identifying pertinent predictors in grossly underdetermined problems. Computational speed is impressive. If predictors are uncorrelated, then interaction effects can be found readily as well. As one might expect, correlations among important predictors degrade computational speed and the recognition of interactions. For very large datasets involving more than, say, 109 total SNP genotypes, data compression is mandatory. Repeated decompression of chunks of the data then slows computation. Our computational swindle circumvents this problem because all of the working predictors easily fit within memory.
The coeliac dataset of van Heel et al. (2007) is challenging for two reasons. First, the overwhelming HLA signal masks the weaker signals coming from other chromosome regions. Second, the HLA SNPs are in strong linkage disequilibrium and hence highly correlated. Linkage disequilibrium manifests itself as increased LOO indices and significant two-way interactions. Despite these handicaps, lasso penalized regression identifies several promising non-HLA regions and interaction effects. Our results for chromosome 4 differ slightly from those of van Heel et al. (2007) because we impute missing genotypes differently. Ayers and Lange (2008) introduce a new penalized method of haplotype frequency estimation that enforces parsimony and achieves both speed and accuracy. When phase can be deduced from relatives, this extra information can be included in estimation. Finally, it is noteworthy that van Heel et al. have validated the chromosome 4 association on two further datasets.
One can quibble with our method of picking candidate predictors for interaction modeling. An obvious alternative would be to look for two-way interactions between the top s predictors and all other predictors. This tactic requires little change in our numerical methods.
Readers may want to compare our approach with the approach of Shi et al. (2006, 2007, 2008). One major difference is our application of cyclic coordinate ascent. A second major difference is that we always select a fixed number of predictors. These choices allow us to quickly process a very large numbers of SNPs or interactions among SNPs. The path following algorithm of Park and Hastie (2008) has the advantage of revealing the exact sequence in which predictors enter the model. Path following is more computationally demanding than simply finding the best r predictors, but note that their software [glmpath in R, Park and Hastie (2007)] can quickly post-process the best r predictors discovered.
We have featured univariate P-values and LOO indices in this article, but neither measure is ideal. Although FDR analysis is valuable, no one has said the last word on multiple testing (Balding, 2006, Kimmel and Shamir, 2006). For instance, some form of generalized cross-validation may ultimately prove useful. As a matter of principle, most geneticists would not accept a single study as definitive. All important findings are subject to replication. This attitude, whether justified or not, puts the onus on finding the most important SNPs rather than on declaring their global significance. Our approach to data analysis is motivated by this consideration. The software discussed here will be made available in the next release of Mendel.
Funding: USPHS (GM53275, MH59490 to K.L. in part).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Ayers KL, Lange K. Penalized estimation of haplotype frequencies. Bioinformatics. 2008;24:1596–1602. doi: 10.1093/bioinformatics/btn236. [DOI] [PubMed] [Google Scholar]
- Balding DJ. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006;7:781–791. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Chen SS, et al. Atomic decomposition by basis pursuit. SIAM J. Sci. Comput. 1998;20:33–61. [Google Scholar]
- Claerbout JF, Muir F. Robust modeling with erratic data. Geophysics. 1973;38:826–844. [Google Scholar]
- Daubechies I, et al. An iterative thresholding algorithm for linear inverse problems with a sparsity constraint. Commun. Pure Appl. Math. 2004;57:1413–1457. [Google Scholar]
- Friedman J, et al. Pathwise coordinate optimization. Ann. Appl. Stat. 2007;2:302–332. [Google Scholar]
- Friedman I, et al. Regularized Paths for Generalized Linear Models Via Coordinate Descent. Department of Statistics, Stanfard University; 2008. [Google Scholar]
- Fu WJ. Penalized regressions: the bridge versus the lasso. J. Comput. Graph. Stat. 1998;7:397–416. [Google Scholar]
- Kimmel G, Shamir R. A fast method for computing high-significance disease association in large population-based studies. Am. J. Hum. Genet. 2006;79:481–492. doi: 10.1086/507317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, et al. An interior-point method for large-scale l1-regularized logistic regression. J. Mach. Learn. Res. 2007;8:1519–1555. [Google Scholar]
- Kooperberg C, Ruczinski I. Identifying interacting SNPs using Monte Carlo logic regression. Genet. Epidemiol. 2005;28:157–170. doi: 10.1002/gepi.20042. [DOI] [PubMed] [Google Scholar]
- Lange K. Optimization. New York: Springer; 2004. [Google Scholar]
- Lazzeroni LC, Lange K. A conditional inference framework for extending the transmission/disequilibrium test. Hum. Hered. 1998;48:67–81. doi: 10.1159/000022784. [DOI] [PubMed] [Google Scholar]
- Lee S-L, et al. Efficient L1 regularized logistic regression. Proceedongs of the 21th National Conference on Artifical Intelligence (AAAI-06). 2006 Available at http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.64.1993. [Google Scholar]
- Liang Y, Kelemen A. Statistical advances and challenges for analyzing correlated high dimensional SNP data in genomic study for complex diseases. Stat. Surv. 2008;2:43–60. [Google Scholar]
- Malo N, et al. Accommodating linkage disequilibrium in genetic-association analyses via ridge regression. Am. J. Hum. Genet. 2008;82:375–385. doi: 10.1016/j.ajhg.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijenhuis A, Wilf HS. Combinatorial Algorithms for Computers and Calculators. 2. New York: Academic Press; 1978. [Google Scholar]
- Nyholt DR. A simple correction for multiple testing for SNPs in linkage disequilibrium with each other. Am. J. Human. Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MY, Hastie T. L1 Regularization Path for Generalized Linear Models and Cox Proportional Hazards Model. R package. 2007 Available at http://bm2.genes.nig.ac.jp/RGM2/pkg.php?p=glmpath. [Google Scholar]
- Park MY, Hastie T. Penalized logistic regression for detecting gene interactions. Biostatistics. 2008;9:30–50. doi: 10.1093/biostatistics/kxm010. [DOI] [PubMed] [Google Scholar]
- Santosa F, Symes WW. Linear inversion of band-limited reflection seimograms. SIAM J. Sci. Stat. Comput. 1986;7:1307–1330. [Google Scholar]
- Schwender H, Ickstadt K. Identification of SNP interactions using logic regression. Biostatistics. 2008;9:187–198. doi: 10.1093/biostatistics/kxm024. [DOI] [PubMed] [Google Scholar]
- Sha F, et al. Multiplicative Updates for L1-Regularized Linear and Logistic Regression, Lecture Notes in Computer Science. Springer; 2007. [Google Scholar]
- Shi W, et al. Technical Report 1131. Madison: University of Wisconsin; 2006. Lasso-Patternsearch Algorithm with Application to Ophthalmalogy Data. [Google Scholar]
- Shi W, et al. Detecting disease causing genes by LASSO-patternsearch algorithm. BMC Proc. 2007;1(Suppl. 1):S60. doi: 10.1186/1753-6561-1-s1-s60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, et al. Technical Report 1141. Madison: University of Wisconsin; 2008. LASSO-Patternsearch Algorithm with Applications to Ophthalmology and Genomic Data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor HL, et al. Deconvolution with the ℓ1norm. Geophysics. 1979;44:39–52. [Google Scholar]
- Tibshirani R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B. 1996;58:267–288. [Google Scholar]
- Uh H-W, et al. Model selection based on logistic regression in a highly correlated candidate gene region. BMC Proc. 2007;1(Suppl. 1):S114. doi: 10.1186/1753-6561-1-s1-s114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel D, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat. Genet. 2007;397:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, Lange K. Coordinate descent algorithms for lasso penalized regression. Ann. Appl. Stat. 2008;2:224–244. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.