

Abstract
Motivation: Pathway and gene set-based approaches for the analysis of gene expression profiling experiments have become increasingly popular for addressing problems associated with individual gene analysis. Since most genes are not differently expressed, existing gene set tests, which consider all the genes within a gene set, are subject to considerable noise and power loss, a concern exacerbated in studies in which the degree of differential expression is moderate for truly differentially expressed genes. For a significantly differentially expressed pathway, it is also of substantial interest to select important genes that drive the differential expression of the pathway.
Methods: We develop a unified framework to jointly test the significance of a pathway and to select a subset of genes that drive the significant pathway effect. To achieve dimension reduction and gene selection, we decompose each gene pathway into a single score by using a regularized form of linear discriminant analysis, called sparse linear discriminant analysis (sLDA). Testing for the significance of the pathway effect proceeds via permutation of the sLDA score. The sLDA-based test is compared with competing approaches with simulations and two applications: a study on the effect of metal fume exposure on immune response and a study of gene expression profiles among Type II Diabetes patients.
Results: Our results show that sLDA-based testing provides a powerful approach to test for the significance of a differentially expressed pathway and gene selection.
Availability: An implementation of the proposed sLDA-based pathway test in the R statistical computing environment is available at http://www.hsph.harvard.edu/∼mwu/software/
Contact: xlin@hsph.harvard.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Traditional high-level analysis of gene expression microarrays involves individual gene analysis: for each gene a statistic (e.g. t-statistic) and an associated P-value are computed to measure the difference in expression level between RNA samples from subjects with different diseases, experimental conditions or exposures. To account for multiple comparisons, procedures controlling the family-wise error rate or false discovery rate (FDR) (Benjamini and Hochberg, 1995) are performed and genes that survive the correction are considered differentially expressed. This usual mode of analysis has been found to have several limitations. In particular, individual gene analysis is often too conservative due to the need to control for a large number of multiple comparisons and correlation among genes, and results are subject to poor interpretability and reproducibility (Subramanian et al., 2005).
An alternative approach is to incorporate prior biological information. Specifically, it is known that biological phenomena occur through the concerted expression of multiple genes. Thus, we can use our prior knowledge of what genes belong to various pathways to focus our analysis on groups of functionally related genes called gene sets. We can, operationally, use the term gene sets interchangeably with gene pathway despite important differences. The logic behind this type of analysis is that several functionally related genes demonstrating moderate differences between experimental conditions may be more important than a single, possibly spurious, highly significant gene. Instead of considering individual genes, the pathway approach treats the gene set as a single unit to be tested. This approach is becoming increasingly popular as it addresses various issues associated with individual gene analysis and provides more directly interpretable and reproducible results.
A few methods focusing on analysis of entire gene sets and pathways have been previously proposed. The most commonly used approaches are based on overrepresentation analysis (Draghici et al., 2003) and gene set enrichment analysis (GSEA) (Mootha et al., 2003). Both of these methods are found to suffer from methodological problems, and may provide misleading and confusing results (Goeman and Buhlmann, 2007). Alternative approaches are available, but most were developed in experimental contexts where the signal is very high. However, in many practical settings, only a small number of genes within a pathway are likely to have differential expression. Since the existing gene set tests place weights on all the genes within a gene set, they may be subject to considerable noise and power loss due to contamination by many null genes. This is particularly a concern for studies in which the degree of the change in expression is relatively low for most truly differentially expressed genes, e.g. studies considering milder exposures or interstitial fluids rather than primary tissue sources.
In this article, we propose a new method for pathway-based gene expression analysis. Our method summarizes each gene set with a composite expression value computed as a linear combination of all the constituent genes' expression values. The optimal weights for the linear combination can be estimated using linear discriminant analysis (LDA), which identifies weights that allow for optimal separation between two groups. However, many genes in a differentially regulated pathway are expected to have no effect and the estimated LDA weights for these null genes are small but non-zero. This implies that the use of the regular LDA weights is likely to introduce substantial noises accumulated from the small weights of these null genes which could result in considerable power loss and mask true signals, especially when signals are moderate. Therefore, it is desirable to use a data-driven method to eliminate such noisy genes when constructing the composite expression. Reduced noise will increase the power of the test and allow one to identify important genes that drive the pathway effect.
We propose to use sparse LDA (sLDA) to achieve the dual goals of testing for the significance of a pathway and gene selection. The sLDA regularizes the usual LDA loss function by adding an L1 constraint on the weights. The L1 constraint causes some of the weights for the discriminant direction to be estimated as exactly zero (Tibshirani, 1996), thereby allowing for simultaneous estimation of an optimal set of sparse weights that permits a high degree of separation of two groups and selection of important genes. We propose permutation test of the sLDA score to test for the significance of the pathway effect. We compare the sLDA-based test to competing approaches with two applications: a study on the effect of metal fume exposure on immune response and a study of gene expression profiles among Type II Diabetes patients. The key advantage of this method is that it provides a unified framework to simultaneously test for the significance of a pathway with improved power and select a subset of genes in the pathway that drive differential expressions of the pathway. We find that sLDA-based testing provides a powerful approach for pathway-based gene expression analysis.
2 METHODS
Pathway-based analysis borrows information from different but correlated genes within the same pathway and hence provides results with improved reproducibility and increased power, especially when individual gene effects are moderate. Testing the significance of a gene pathway proceeds with a two-step procedure: (i) compute a statistic that measures the degree of overall differential expressions of genes within a pathway between the two groups and (ii) evaluate the statistical significance of the observed statistic. To accomplish the first step, we identify a sparse set of weights using sLDA and use the estimated weights to calculate the composite expression for the pathway. The degree of the sLDA-based composite pathway expression can be compared using a two-sample t-statistic. We can use permutation to generate the P-value for evaluating whether the pathway is significantly differentially expressed. Gene selection occurs since some weights used in computing the composite expression score are estimated as exactly zero and hence the gene does not contribute. In this section, we describe each step of the testing procedure in detail and then give the overall testing algorithm.
2.1 Two-group sLDA
The defining feature of our approach is the application of sLDA, which is a regularized form of LDA. LDA was originally proposed by Fisher (1936) as a means for finding the linear combination of the predictors that maximizes the between class variance relative to the within class variance, the discriminant direction. LDA estimates the discriminant direction w by maximizing the Rayleigh quotient:
| (1) |
where Sb is the between group covariance matrix and Sw is the within group (pooled) covariance of the gene expression values. sLDA differs from LDA in that sLDA finds w by solving (1) subject to an additional L1-constraint on w. Using an L1-constraint ensures that some wj will be estimated as exactly zero and the corresponding genes will not contribute to the discriminant direction and the composite expression value.
In this section, we will consider the computation of w via sLDA. First, however, we note that in the two-class setting, Sb is of rank 1 so (1) may be simplified to
where we define L(w)=w′Sww and 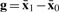 with
with 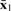 and
and  given as the vectors of mean gene expression values corresponding to the two groups, respectively. We will use this notation throughout.
given as the vectors of mean gene expression values corresponding to the two groups, respectively. We will use this notation throughout.
As discussed earlier, genes in the gene set that are null merely introduce extra noise. Filtration of these genes by variable selection improves the power of the test, especially when the number of noise predictors is large. To accomplish this, we place an L1-constraint on the vector w and define the sLDA solution as
| (2) |
for a fixed τ. The value of τ controls the degree of sparsity; when τ is small, some of the wj will be estimated as exactly zero.
Although (2) may be found by standard quadratic programming (QP) solvers for each value of τ, the high computational cost of permutation renders QP impractical. We show in Wu et al. (2008) that (2) belongs to a class of problems that have piecewise linear solution paths for w as a function of τ and develop an efficient algorithm to find the entire regularized solution path.
A final w is computed using the selected value of τ. In general, τ may be selected by maximizing the cross-validated (CV) Rayleigh quotient, but in our setting, we will choose to instead minimize the criterion:
where  is the estimate for w given a value of τ and rτ is the number of non-zero components of
is the estimate for w given a value of τ and rτ is the number of non-zero components of 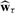 . This criterion is similar to the Bayesian information criterion (BIC) (Schwarz, 1978). τ may be selected by computing BICτ across a range of τ and selecting the τ that minimizes the BICτ. CV is a possible alternative, but since we are using permutation to compute the P-value, the additional computational expense is undesireable. Moreover, given the limited sample size in most gene expression profiling studies, CV is likely to be unstable since the outcome is discrete and the number of genes exceeds the number of samples (Ahn et al., 2007).
. This criterion is similar to the Bayesian information criterion (BIC) (Schwarz, 1978). τ may be selected by computing BICτ across a range of τ and selecting the τ that minimizes the BICτ. CV is a possible alternative, but since we are using permutation to compute the P-value, the additional computational expense is undesireable. Moreover, given the limited sample size in most gene expression profiling studies, CV is likely to be unstable since the outcome is discrete and the number of genes exceeds the number of samples (Ahn et al., 2007).
Although we have proposed a straightforward formulation of the sparse LDA problem, we note that Fung and Ng (2007) also attempted to address the sLDA problem, though with a significantly different approach. Their approach is based on the method of Feng et al. (2003) which, instead of regularizing the Rayleigh quotient, changes the problem into a simple linear regression and then adds an L1 and an L2 penalty to achieve sparsity. The relationship between maximizing the Rayleigh quotient (LDA) and linear regression is well known, but as soon as penalties are added, then the problems become different. Although this is motivated by LDA and achieves sparsity, it is unclear whether it can still obtain the optimality guaranteed by LDA.
2.1.1 Additional L2-constraint
In the linear regression setting, it was shown that addition of an L2 (ridge) penalty improved prediction and variable selection in cases where predictors are highly correlated (Zou and Hastie, 2005). We can also add an L2 penalty to (2) as a Lagrangian term. In this case, the discriminant directions given by
where ϑ is a Lagrangian term corresponding to an additional L2-constraint. For each fixed ϑ, we add ϑ to the diagonal terms of Sw, and as in the sLDA case, we can use QP to compute 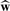 with the modified Sw. Empirically, however, the power appears somewhat robust to the specific value of ϑ (data not shown). Including ϑ appears to stabilize the algorithm, so we set ϑ=2log(p)/n when we apply sLDA to pathway testing.
with the modified Sw. Empirically, however, the power appears somewhat robust to the specific value of ϑ (data not shown). Including ϑ appears to stabilize the algorithm, so we set ϑ=2log(p)/n when we apply sLDA to pathway testing.
One may see that if a large value of ϑ is applied, then the regularized within class covariance matrix essentially mimics the identity matrix and the procedure approaches the shrunken centroid method (Tibshirani et al., 2002).
2.2 sLDA-based pathway testing
Throughout this section, we assume that we are interested in comparing gene expression profiles for exactly two groups. In order to test a pathway for differential activity, we can decompose the testing procedure into two steps: (i) summarize the pathway's differential activity with a single relevant statistic; and (ii) determine whether the computed statistic is statistically significant.
Our proposed sLDA-based testing approach begins by reducing each gene set to a composite expression value computed as a linear combination of the constituent genes. Let X be an n × p matrix of gene expression values with (i, j) component equal to the gene expression value of the j-th gene in the gene set for the i-th subject (array), such that n is the number of arrays (samples) and p is the number of genes in the gene set. Then for the i-th subject, set the composite expression value zi=w′Xi, where w is a vector of weights for each gene in the gene set computed using sLDA. The differential activity is then summarized by T, the t-statistic comparing the zi for cases versus controls (or exposed with non-exposed subjects). Note that this value is equal to the square root of the Rayleigh quotient:  .
.
sLDA is a supervised approach so using a parametric P-value for T, i.e. comparing T to a classical/usual t-distribution, would give biased results. As an alternative, we propose to use permutation to evaluate significance. Specifically, we consider the use of the following procedure:
Algorithm 1. —
sLDA-based Pathway test
Estimate the pathway statistic T by: (i) find sparse w via sLDA; (ii) estimate z=w′X; (iii) estimate T.
Permute the class labels, and repeat Step 1 with the permuted data to compute T*.
Repeat Step 2 B times to obtain {T*(b), b=1, …, B}, for some large number B. A new τ must be re-selected for each permutation.
If the pathway is differentially expressed, examine the individual wj to identify important driver genes.
The last step of Algorithm 1 is a direct result of using sLDA to estimate w, where some weights are estimated exactly as 0 by sLDA. Genes such that wj=0 do not contribute to the estimation of z or T. In other words, only non-zero wj contribute to a differentially expressed pathway's significant result. We consider those genes with non-zero or large wj ‘important’ or ‘informative’ in driving differential pathway activity.
3 RESULTS
3.1 Metal particulate exposure data
Our research was motivated by a gene-environment study evaluating whether metal particulate exposure causes systemic inflammation and whether evidence of this could be found in gene expression profiling of peripheral blood. Briefly, the study was conducted as follows: after a wash-out period of at least 5 days, nine healthy, non-smoking subjects were exposed to metal fumes and airborne particulate matter (≈5 h) from shielded metal arc welding, gas tungsten arc welding and plasma arc cutting at a welding apprentice school. On the same day, seven other subjects were assigned as controls and performed bookwork and office tasks at an office in the same welding school. All subjects wore monitors to measure exposure to fine particulate matter (particulate matter with a mass median aerodynamic diameter ≤2.5 μm, PM2.5). Cases were found to have a median PM2.5 exposure of 0.948 mg/m3, while the median PM2.5 for controls was 0.021 mg/m3. For all subjects, complete blood samples were collected at baseline (at the beginning of the day) and post-exposure (6 h later). Gene expression profiling of each collected blood sample was performed using Affymetrix Human Genome U133A GeneChips with 22 215 probe sets. Following preprocessing using the dChip software (Li and Wong, 2001) and filtration of unexpressed genes, 5543 genes (probes) were available for analysis. For each probe set on each subject, the (log) baseline expression level was subtracted from the (log) post-exposure expression level.
A traditional individual gene analysis using two-sample t-tests was initially attempted to identify genes which showed a different degree of change from pre- to post-exposure between welders and controls. However, after controlling for the FDR, no genes were significantly differentially expressed. This result was not surprising because our experimental conditions involved an environmental exposure rather than a stronger disease phenotype and because we used blood rather than a primary tissue.
3.2 The candidate pathway approach with application to the metal particulate exposure data: pathway significance test and gene selection
When specific biology-driven hypotheses are of interest, as was the case in the motivating study, analysis of candidate pathways rather than a large-scale screen of many pathways may be more effective and powerful. For the metal particulate data, 35 gene sets involving biological processes related to inflammation and immune response were distilled from the gene ontology (GO) database (Ashburner et al., 2000). Each gene set from the GO database is a group of genes known to have common function. After filtering the gene sets to remove genes on the basis of electronic annotation information, we applied sLDA to each gene set. We performed 1000 permutations to generate the P-value for significance of each gene set. Of the 35 pathways, 15 pathways were differentially expressed at the nominal α=0.05 level. Controlling for the FDR at 5%, 13 pathways were found to be significantly differentially expressed. The significant pathways, the number of genes in each pathway, the number of selected informative genes and the corresponding sLDA-based test P-values are given in Table 1. Among the 15 pathways presented in Table 1, the number of genes per pathway varies from 4 to 154. The number of selected informative genes per pathway by sLDA varies from 2 to 8, suggesting that sLDA has a strong ability to filter out a large number of noisy genes and select a subset of informative genes that drive the pathway effects. A total of 39 unique genes were selected among the 15 pathways as informative for the exposure effects on the pathway expressions. This provides a parsimonious list of genes for possible further analyses.
Table 1.
The 15 significant differentially expressed gene pathways using sLDA at the nominal α=0.05 level (FDR < 0.11) for the metal particulate exposure data
| Pathway | No. of genes | No. of selected genes | P-value | Q-value |
|---|---|---|---|---|
| Response to external biotic stimulus | 153 | 7 | <0.001 | 0.02 |
| Response to pest, pathogen or parasite | 149 | 7 | 0.001 | 0.02 |
| Inflammatory response | 54 | 5 | 0.003 | 0.04 |
| Activation of MAPK activity | 7 | 5 | 0.005 | 0.04 |
| Response to biotic stimulus | 198 | 7 | 0.005 | 0.04 |
| Taxis | 32 | 3 | 0.006 | 0.04 |
| Response to external stimulus | 56 | 4 | 0.007 | 0.04 |
| Chemotaxis | 32 | 3 | 0.008 | 0.04 |
| Oxygen and reactive oxygen species metabolism | 5 | 2 | 0.011 | 0.04 |
| Superoxide metabolism | 5 | 2 | 0.011 | 0.04 |
| Immune response | 69 | 6 | 0.018 | 0.05 |
| Monocyte differentiation | 4 | 3 | 0.018 | 0.05 |
| Positive regulation of I-kappaB kinase/NF-kappaB cascade | 24 | 8 | 0.020 | 0.05 |
| DNA damage response, signal transduction | 5 | 5 | 0.035 | 0.09 |
| Response to oxidative stress | 13 | 4 | 0.048 | 0.11 |
For comparison purposes, we also applied several other gene pathway methods to the metal particulate data. Specifically, we used the global test (Goeman et al., 2004), the singular value decomposition (SVD) approach (Tomfohr et al., 2005), SigPath (Tian et al., 2005) and GSEA (Mootha et al., 2003), to test the 35 pathways related to the immune response process. The global test, SVD and SigPath failed to identify any pathways as significantly differentially expressed at the nominal α=0.05 level or the FDR=0.20 level. GSEA, which tests a competitive null hypothesis, identified only the activation of MAPK activity pathway as differentially expressed at the nominal level (P=0.047), but this was no longer significant after controlling the FDR at 20%. These approaches were developed under the classical microarray setting and appear to require stronger effects to be detected than sLDA. Further, they do not perform gene selection. Hence, accumulation of the noises from a large number of null genes are likely to mask the effects when the pathway effects are moderate. Overrepresentation analysis was not applied due to major methodological issues that suggest the null hypothesis is of entirely tangential interest to the investigators (Goeman and Buhlmann, 2007).
We use the activation of MAPK activity pathway to illustrate the gene selection feature of our method. Seven genes were in the gene set and also expressed on our chip. Five of the seven were selected using sLDA. Since their sLDA weights were estimated as non-zero and they contributed to the composite pathway expression score, these five genes were potentially important in driving the significant test result. Two genes were considered noise and removed in calculating the composite pathway expression score, as their sLDA weights were estimated as zero. The five selected genes and their sLDA weights are given in Table 2. For comparison, we also present the t-statistics gene analysis. Although only a single gene is individually differentially expressed, their linear combination is highly significant (P=0.005). This occurs because the genes are correlated (range=[−0.09, 0.85]), allowing sLDA to borrow information across genes.
Table 2.
The five genes (out of an original seven) in the activation of MAPK activity pathway selected as driving the significant pathway test result
| Gene | Gene Description | sLDA weights | t-statistic | P-value |
|---|---|---|---|---|
| CD81 | CD81 molecule | −0.511 | 1.498 | 0.156 |
| TRIB3 | Tribbles homolog 3 | 0.436 | −2.166 | 0.048 |
| ADRB2 | Adrenergic, beta-2-, receptor, surface | 0.194 | −0.217 | 0.831 |
| C5AR1 | Complement component 5a receptor 1 | 0.172 | −1.379 | 0.190 |
| FPR1 | Formyl peptide receptor 1 | 0.143 | −0.908 | 0.379 |
The two unselected genes, SHC1 and PIK3CB, had sLDA weights estimated as zero and were considered null genes. The t-statistic and P-values are from the original individual gene analysis.
3.3 Reanalysis of Type II Diabetes data
To explore the pathway significance test and gene selection properties of sLDA on a better studied phenotype than metal particulate exposure, we applied the sLDA-based testing procedure approach to a previously analyzed dataset that considered Type II Diabetes gene expression profiles. This dataset was presented in Mootha et al. (2003) and was originally analyzed using GSEA. We restricted our analysis to the subset of the data consisting of 17 patients with normal glucose tolerance and 18 patients with Type II Diabetes. The goal was to identify gene sets differentially expressed between normal and diabetic patients. After preprocessing as described in the original paper, we applied the sLDA-based pathway test to 124 of the 149 gene sets used in the original paper. Twenty-five gene sets were omitted after we limited the minimum number of probes per gene set to be four. The number of pathways deemed differentially expressed at the nominal level by sLDA and its competitors are given in Table 3. Our proposed method again identifies more gene sets as differentially expressed than the competitors.
Table 3.
Results from the analysis of 125 gene sets from the diabetes dataset using the sLDA-based test and four competitors.
| sLDA | Global Test | SigPath | SVD | GSEA | |
|---|---|---|---|---|---|
| sLDA | 9 | 1 | 1 | 0 | 1 |
| Global Test | 4 | 3 | 0 | 1 | |
| SigPath | 5 | 2 | 3 | ||
| SVD | 2 | 2 | |||
| GSEA | 4 |
Each cell gives the over-lapping number of gene sets called differentially expressed at the nominal 0.05 level by the methods shown in the corresponding column and row.
For illustration, we examined the gene selection properties of sLDA by studying the individual genes found to be important in the carbon fixation pathway, which was statistically significant at the α=0.05 level (P=0.015). The previous study by Mootha et al. (2003) defined the carbon fixation pathway to contain 27 genes, of which 18 remained after the preprocessing procedure. Nine genes had non-zero weights in the estimated composite pathway expression score and were deemed by sLDA potentially important for the significant effect of the carbon fixation pathway on Type II Diabetes. These genes and their sLDA weights are provided in Table 4. The magnitudes of the weights give the relative importance of each gene. We also provide in Table 4 individual t-statistics and P-values for comparison purpose. Although only the two most heavily weighted genes are individually statistically significant, all nine genes have been previously postulated to play a role in diabetes (Hittel et al., 2005; Lemieux et al., 1984; Maniratanachote et al., 2005; Marcus and Hosey, 1980; Morral et al., 2007; Nakanishi et al., 2004; Oh et al., 2005; Park and Drake, 1982; Yang et al., 2002). Their joint effects drive the pathway to be significantly expressed.
Table 4.
The nine genes in the carbon fixation pathway selected from the original 28 genes by sLDA as potentially important for driving the significant pathway test
| Gene | Gene Description | sLDA weights | t-statistic | P-value |
|---|---|---|---|---|
| ME3 | Malic enzyme 3 | −0.415 | 2.098 | 0.044 |
| GOT2 | Glutamic-oxaloacetic transaminase 2 | 0.401 | −2.358 | 0.025 |
| FBP1 | Fructose-1,6-bisphosphatase 1 | −0.281 | 1.097 | 0.281 |
| ALDOA | Aldolase A | 0.264 | −1.505 | 0.142 |
| MDH2 | Malate dehydrogenase 2 | −0.171 | 1.063 | 0.296 |
| ALDOB | Aldolase B | −0.170 | 0.975 | 0.337 |
| ME1 | Malic enzyme 1 | 0.105 | −0.459 | 0.650 |
| PKM2 | Pyruvate kinase | 0.060 | −1.234 | 0.226 |
| ALDOC | Aldolase C | −0.041 | 0.832 | 0.411 |
The t-statistic and P-values are from the original individual gene analysis.
3.4 Simulation study
To compare the performance of sLDA-based testing to existing approaches under controlled settings, we conducted simulations to study the power of our proposed test.
For each configuration described below, we generated the gene expression values from a gene set containing p genes for n ‘cases’ and n ‘controls’. Each of the cases were generated from a multivariate normal distribution with mean μ(1) and covariance Σ, while each of the controls were simulated from a multivariate normal with mean μ(2) and covariance Σ.
Setting 1: we let n=10, p=100, and μ(2)=0. μ(1) was a vector with μ(1)1=μ(1)25=μ(1)75=μ(1)100=1, μ(1)10=μ(1)50=μ(1)90=−1, and all other components equal to zero. The covariance matrix, Σ was estimated using the empirical covariance between the first 100 genes in the ‘c0_133 probes’ gene set from the diabetes dataset.
Setting 2: this setting was identical to Setting 1 except we increased the sample size to n=15.
Setting 3: this setting was identical to Setting 1 except we increased the sample size to n=20.
Setting 4: We let n=10, p=50 : and μ(2)=0. μ(1) was a vector with μ(1)1=μ(1)20=μ(1)30=μ(1)49=1, μ(1)5=μ(1)45=−1 and all other components equal to zero. We allowed an autoregressive correlation structure such that Σi,j=0.85|i−j|.
Setting 5: this setting was identical to Setting 4 except we increased the sample size to n=20.
We also considered generating the data from a logistic regression model. For both of the following configurations, we generated n cases and n controls from the model: logit pi=x′iβ, where pi is the probability the i-th subject is a case and xi ∼ N(0, Σ) is the vector of expression values of genes in the gene set.
Setting 6: we set n=15 and p=100. β was a vector with β1=···=β5=1 and all other components equal to zero. We again allowed the same autoregressive correlation structure as in Setting 4.
Setting 7: this setting was identical to Setting 6 except we increased the same size to n=20.
For each of the settings, we ran 500 simulations. In each simulation, the data were generated as described and then sLDA-based testing, the global test, SVD and SigPath were applied to test for a differential expression between cases and controls. We also considered testing via two supervised dimension reduction techniques other than sLDA: (non-sparse) L2-constrained LDA (L2LDA) and supervised PCA (sPCA) (Bair et al., 2006). Testing using these two methods proceeded by substituting L2 LDA or sPCA for sLDA in Algorithm 1. For each setting and testing method, the power was estimated as the proportion of P-values less than α=0.05. The results are given in Table 5.
Table 5.
Comparison of the empirical power of sLDA and competing methods across seven simulation settings
| Setting | sLDA | L2LDA | sPCA | Global test | SVD | SigPath |
|---|---|---|---|---|---|---|
| 1 | 0.202 | 0.212 | 0.206 | 0.064 | 0.084 | 0.118 |
| 2 | 0.380 | 0.298 | 0.164 | 0.048 | 0.062 | 0.068 |
| 3 | 0.660 | 0.574 | 0.190 | 0.100 | 0.044 | 0.110 |
| 4 | 0.672 | 0.916 | 0.326 | 0.192 | 0.086 | 0.166 |
| 5 | 1.000 | 1.000 | 0.506 | 0.582 | 0.114 | 0.320 |
| 6 | 0.988 | 0.550 | 0.972 | 0.856 | 0.282 | 0.766 |
| 7 | 0.856 | 0.404 | 0.896 | 0.596 | 0.292 | 0.596 |
The results indicate that when the data were generated under a shifted-mean multivariate normal setup (Settings 1–5), sLDA and L2LDA had improved power over the competing approaches. sLDA was more powerful in the Settings 2 and 3 when the majority of genes did not contribute to differentiating cases from controls. In Settings 1, sLDA and L2LDA performed similarly since the signal was very low. Similarly, when the signal was high, both sLDA and L2 DA showed excellent power in Setting 5. In Setting 4, the degree of sparsity was lower and, as expected, in such a setting L2LDA outperformed sLDA. Under the logistic regression model (Settings 6 and 7), sLDA had higher power than L2LDA, the global test and SVD, but the supervised PCA approach was comparable to sLDA.
4 DISCUSSION
This article considers the use of sLDA for pathway-based analysis of gene expression profiling experiments. This method is particularly attractive in settings where the signal is moderate, i.e. a few genes are moderately differentially expressed while most show little change relative to the noisiness of the data. Our method simultaneously tests for differential pathway activity and selects informative genes within a pathway that drive the effects. By eliminating non-informative genes from our composite pathway expression score, we reduce noise and increase power. The same method can be applied to study proteomic and metabalomic pathways.
We illustrate the powerful results of sLDA for detecting pathway effects and gene selection within pathways using simulations and two data examples: the metal particulate exposure data and the Type II Diabetes data. Our results show that pathway analysis can be more powerful for detecting differential expression signals. Few genes selected within significantly differentially expressed pathways were called individually differentially expressed at the nominal α-level. By accounting for correlations among them, methods such as sLDA can detect the pathway genes composite effects, suggesting that marginal analyses of individual genes have limited power. Similarly, our simulations demonstrated that sLDA had improved power over several alternatives, particularly when the majority of genes are not differentially expressed.
An important aspect of our approach is that—as well as SVD, the global test and SigPath—it tests a self-contained null hypothesis. As noted in Goeman and Buhlmann (2007) and Tian et al. (2005), such a test considers the global null hypothesis. This is in contrast to GSEA which tests a competitive null hypothesis. The difference is that a self-contained null hypothesis is rejected if any of the genes in the gene set are truly differentially expressed whereas a competitive null hypothesis is rejected when the relative degree of differential expression of the genes in the gene set is higher when compared with other genes on the chip. Thus, because large pathways are more likely to contain some truly differentially expressed genes, self-contained tests are more likely to consider large pathways as truly differentially expressed. In practice, however, large pathways may not be more likely to be statistically significant because they may also contain more noise: if a small pathway contains a few differentially expressed genes and a larger pathway contains the same number of differentially expressed genes, the excess noise in the large pathway may decrease power. Further discussion on the differences in hypotheses may be found in Goeman and Buhlmann (2007), in which self-contained tests are advocated over competitive tests due to important issues in interpretability of results, loss of power and difficulty in adjusting for multiple comparisons.
The principal biological contribution of this work is in the analysis of the metal fume exposure data. These results are interesting from an environmental health perspective for two separate reasons. First, this study has demonstrated that use of peripheral blood, rather than primary tissue, is sufficient for studying changes in gene expression. This is promising since blood is readily obtainable and is one of the few options available when considering environmental exposures. Second, this work verifies the hypothesis that gene expression signatures indicate a systemic immune response to metal particulate exposure. All subjects appeared healthy after exposure and no obvious exposure effect could be discerned based on readily available phenotype. Nevertheless, at the molecular level, the body was responding as if it were in a distressed state. This suggests that in between healthy and diseased phenotypes, there exists an intermediate stage at which exposure effects may be seen only at the molecular level. Moreover, gene pathway expression appears to better capture the effect than individual genes. Therefore, molecular pathway responses in blood plasma may be a more sensitive method for assessing the effects of ambient air pollution or other environmental exposures.
Funding: National Cancer Institute (R37-CA-76404 and R01-CA-074386, in parts).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Ahn J, et al. The high-dimension, low-sample-size geometric representation holds under mild conditions. Biometrika. 2007;94:760. [Google Scholar]
- Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bair E, et al. Prediction by supervised principal components. J. Am. Stat. Assoc. 2006;101:119. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995;57:289–300. [Google Scholar]
- Draghici S, et al. Global functional profiling of gene expression. Genomics. 2003;81:98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- Feng J, et al. High dimensional feature selection for discriminant microarray data analysis. Adv. Data Mining Model. 2003;15:25–24. [Google Scholar]
- Fisher R. The use of multiple measurements in taxonomic problems. Ann. Eugen. 1936;7:179–188. [Google Scholar]
- Fung E, Ng M. On sparse Fisher discriminant method for microarray data analysis. Bioinformation. 2007;2:230. doi: 10.6026/97320630002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman J, Buhlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23:980. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- Goeman J, et al. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20:93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- Hittel D, et al. Proteome analysis of skeletal muscle from obese and morbidly obese women. Diabetes. 2005;54:1283. doi: 10.2337/diabetes.54.5.1283. [DOI] [PubMed] [Google Scholar]
- Lemieux G, et al. Renal enzymes during experimental diabetes mellitus in the rat. Role of insulin, carbohydrate metabolism, and ketoacidosis. Can. J. Physiol. Pharmacol. 1984;62:70–75. doi: 10.1139/y84-010. [DOI] [PubMed] [Google Scholar]
- Li C, Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:0032.1–0032.11. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniratanachote R, et al. Detection of autoantibody to aldolase B in sera from patients with troglitazone-induced liver dysfunction. Toxicology. 2005;216:15–23. doi: 10.1016/j.tox.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Marcus F, Hosey M. Purification and properties of liver fructose 1, 6-bisphosphatase from C57BL/KsJ normal and diabetic mice. J. Biol.Chem. 1980;255:2481–2486. [PubMed] [Google Scholar]
- Mootha V, et al. PGC-1 α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Morral N, et al. Effects of glucose metabolism on the regulation of genes of fatty acid synthesis and triglyceride secretion in the liver. J. Lipid. Res. 2007;48:1499. doi: 10.1194/jlr.M700090-JLR200. [DOI] [PubMed] [Google Scholar]
- Nakanishi N, et al. Serum γ-glutamyltransferase and risk of metabolic syndrome and type 2 diabetes in middle-aged Japanese men. Diabetes Care. 2004;27:1427–1432. doi: 10.2337/diacare.27.6.1427. [DOI] [PubMed] [Google Scholar]
- Oh H, et al. Identification of novel diagnostic marker candidates for diabetic retinopathy by serological proteome analysis. Invest. Ophtalmol. Vis. Sci. 2005;46:426–426. [Google Scholar]
- Park C, Drake R. Insulin mediates the stimulation of pyruvate kinase by a dual mechanism. Biochem. J. 1982;208:333–337. doi: 10.1042/bj2080333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Ann. Stat. 1978;6:461–464. [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, et al. Discovering statistically significant pathways in expression profiling studies. Proc. Natl Acad. Sci. USA. 2005;103:13544–13549. doi: 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, et al. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl Acad. Sci. USA. 2002;99:6567. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B. 1996;58:267–288. [Google Scholar]
- Tomfohr J, et al. Pathway level analysis of gene expression using singular value decomposition. BMC Bioinformatics. 2005;6:225. doi: 10.1186/1471-2105-6-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, et al. Technical report. Harvard University; 2008. Two-group classification via sparse linear discriminant analysis. Available at http://www.biostat.harvard.edu/∼mwu/Files/sLDA.pdf. [Google Scholar]
- Yang X, et al. Microarray profiling of skeletal muscle tissues from equally obese, non-diabetic insulin-sensitive and insulin-resistant Pima Indians. Diabetologia. 2002;45:1584–1593. doi: 10.1007/s00125-002-0905-7. [DOI] [PubMed] [Google Scholar]
- Zou H, Hastie T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005;67:301–320. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.