

Abstract

A regio- and stereoselective isomerization of allenamides is described, leading to preparations of de novo 2-amido-dienes and a tandem isomerization–6π-electron electrocyclic ring-closure.
Synthesis of conjugated dienes via an allene isomerization, while a thermodynamically favored process, is not trivial kinetically. The required 1,3-H-shift constitutes a four-electron [2π + 2σ] process that would call for an antarafacial approach if proceeding through a concerted and anti-Hückel [or Möbius] transition state.1,2 Although impossible in an allylic system, it is relatively more feasible for an allenic system because of the presence of orthogonally oriented p-orbitals of the sp-hybridized central allenic carbon [Scheme 1]. The orthogonal p-orbital at C3 [in blue] introduces a formal phase change required for an anti-Hückel transition state, or formally allows a six-electron [2π +2σ + 2π] process when the second set of allenic π-electrons becomes involved. Nevertheless, the calculated2a ΔEact value remains high at 77.7 kcal mol−1 and consequently, concerted or not, most thermal isomerizations of allenes take place at high temperatures,3,4 thereby rendering it difficult to control E/Z ratios of the resulting dienes. There are more practical approaches would involve stepwise processes promoted by acid, base, or metal, but their examples are limited and the level of stereo- and regiochemical control need to be improved.3,5
Scheme 1.

Allene Isomerizations.
Given that most dienes can be prepared from an array of stereoselective transformations, synthesizing conjugated dienes from structurally more challenging allenes through a kinetically demanding and stereochemically undistinguished isomerization does not appear to be a logical first choice. However, our efforts with the chemistry of allenamides6 allowed us to envision a much greater potential in constructing amido-dienes through isomerizing allenamides7–9 because there are no consistent approaches for synthesizing amido-dienes.10–12 Of the two major methods for preparing amido-dienes,10 the one involving acid-mediated condensations suffers from functional group tolerance with the metal-mediated amidative cross-coupling13,14 suffering from limited access to halo-dienes [Scheme 1]. In contrast, substituted allenamides are quite accessible through α-alkylations of parent allenamide15,16 or amidative cross-couplings of allenyl halides.17 Their isomerizations can prove to be an invaluable entry to amido-dienes. We communicate here a regio- and stereoselective isomerization of allenamides in the synthesis of 2-amido-dienes and a tandem isomerization–6π-electron electrocyclic ring-closure.
Screening through various thermal conditions [entries 1–7 in Table 1] including several solvents distinctly revealed that isomerization of achiral allenamide 1 was the most effective at 115 °C in CH3CN [sealed tube], leading to 2-amido-diene 218 in 78% isolated yield and 16:1 ratio [entry 4] in favor of the E-geometry [assigned later]. While there appears to be a solvent effect on the E/Z ratio [entries 5–7], we found that with the exception of HNTf2 and PTSA [entries 8–9], a range of Brønsted acids were equally effective and more facile at RT in providing 2-amido-diene 2 with excellent E/Z ratio [entries 10–13].
Table 1.
Thermal vs. Acidic Conditions.
 | ||||||
|---|---|---|---|---|---|---|
| entry | solvent | acid [10 mol %] | temp [°C] | time [h] | yield [%]a,b | E:Zc |
| 1 | CH3CN | - | 25 | 16 | 0 | -d |
| 2 | CH3CN | - | 55 | 16 | 51 | ≥20:1 |
| 3 | CH3CN | - | 85 | 16 | 88 | ≥20:1 |
| 4 | CH3CN | - | 115 | 16 | 91 [78] | 16:1 |
| 5 | THF | - | 115 | 16 | 51 | 9:1 |
| 6 | ClCH2CH2Cl | - | 115 | 16 | 79 | 7:1 |
| 7 | Tol | - | 150 | 16 | 55 | 4:1 |
| 8 | CH2Cl2 | HNTf2 | 25 | 5 min | 0 | -e |
| 9 | CH2Cl2 | PTSA | 25 | 1 | 66 | 2:1 |
| 10 | CH2Cl2 | 4-NO2PhCO2H | 25 | 16 | 81 | 15;1 |
| 11 | CH2Cl2 | PhCO2H | 25 | 16 | 85 [55] | 18:1 |
| 12 | CH2Cl2 | PPTS | 25 | 16 | 77 | 15:1 |
| 13 | CH2Cl2 | CSA | 25 | 10 min | 95 [74] | 18:1 |
NMR yields.
Isolated yields in the bracket.
Determined by 1H-NMR.
Allenamide 1 was recovered.
Allenamide 1 decomposed.
Generality of this α-isomerization could be established as shown in Table 2. Key features are: (1) An array of chiral allenamides 5–7 could be employed to construct de novo 2-amido-dienes 8–10 with comparable yields and E/Z ratios under thermal [higher temperature at 135 °C] or acidic conditions [entries 2–11]; (2) unsubstituted 2-amido-dienes 8d and 9c could also be accessed in good yields [see R = H in entries 7 and 9]; (3) allenamide 11 containing an acyclic amide is also feasible for the isomerization; and (4) a single-crystal X-ray structure of 10b was attained to unambiguously assign the E-configuration [Figure 1].
Table 2.
Isomerization of Allenamides at the α-Position.
| entry | allenamides | conditions [time]a | dienes | yield [%]b | E:Zc | ||
|---|---|---|---|---|---|---|---|
| 1 | 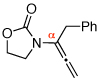 |
3 | 115 °C, 16 h | 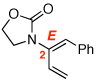 |
4 | 71 | 6:1 |
| 2 | 5a: R = n-Pr | 135 °C, 6 h | 8a | 77 | ≥20:1 | ||
| 3 | 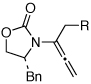 |
5a: R = n-Pr | CSA, 4 hd | 8a | 87 | ≥20:1 | |
| 4 | 5b: R = Ph | 135 °C, 16 h | 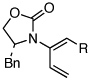 |
8b | 74 | ≥20:1 | |
| 5 | 5b: R = Ph | CSA, 2 h | 8b | 83 | ≥20:1 | ||
| 6 | 5c: R = 2Nap | 135 °C, 16 he | 8c | 73 | ≥50:1 | ||
| 7 | 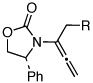 |
5d: R = H | 135 °C, 16 h | 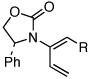 |
8d | 69 | - |
| 7 | 6a: R = n-Pr | CSA, 10 min | 9a | 82 | ≥50:1 | ||
| 8 | 6b: R = Ph | CSA, 10 min | 9b | 76 | ≥50:1 | ||
| 9 | 6c: R = H | 135 °C, 16 h | 9c | 69 | - | ||
| 10 | 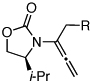 |
7a: R = n-Pr | 135 °C, 16 h | 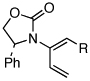 |
10a | 62 | ≥50:1 |
| 11 | 7b: R = Ph | 135 °C, 16 h | 10b | 82 | ≥50:1 | ||
| 12 | 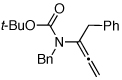 |
11 | 135 °C, 16 h | 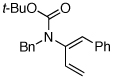 |
12 | 45 | ≥20:1 |
| 13 | 11 | CSA,f 2 h | 12 | 61 | ≥20:1 | ||
Unless otherwise noted, CH3CN was the solvent for thermal conditions, and CH2Cl2 was the solvent when using 10 mol % of CSA at rt. For all reactions, concn = 0.10 M.
Isolated yields.
Determined by 1H-NMR.
Temp started at −78 °C.
ClCH2CH2Cl was used.
4 Å MS was used.
Figure 1.

X-Ray Structure of 2-Amido-Diene 10b.
Although our main interest resides in identifying a useful protocol for synthesizing 2-amido-dienes given its greater scarcity,10–12,19,20 we examined isomerizations of allenamides from the γ-position en route to more well-known 1-amido-dienes.21 As shown in Table 3, isomerizations of two types of γ-substituted allenamides, those with a cyclohexylidene group [see 13–16 in entries 1–13], and those with an isopropylidene group [see 17–19 in entries 14–19] led to 1-amido-dienes 20–26 exclusively as E-enamides [assigned based on the trans-olefinic proton coupling constant].
Table 3.
Isomerization of Allenamides at the γ-Position.
| entry | allenamides | conditions [time]a | dienes | yield [%]b,c | ||
|---|---|---|---|---|---|---|
| 1 | 13a: R = (R)-Ph | 135 °C, 16 h | 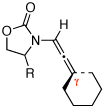 |
20a | ≤10d | |
| 2 | 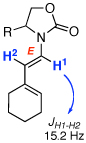 |
PTSA, 10 min | 95 | |||
| 3 | CSA, 10 min | 89 | ||||
| 4 | 13b: R = (R)-Bn | 135 °C, 16 h | 20b | 25e | ||
| 5 | PTSA, 5 min | 96 | ||||
| 6 | CSA, 10 min | 95 | ||||
| 7 | 13c: R = (S)-i-Pr | 135 °C, 16 he | 20c | 50f | ||
| 8 | PTSA, 10 min | 88 | ||||
| 9 | 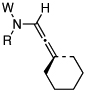 |
14: W = Ac; R = Ph | 175 °C,g 24 h | 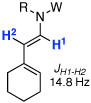 |
21 | 95 |
| 10 | CSA,h 10 min | 90 | ||||
| 11 | 15: W = Ts; R = Bn | 175 °C,g 24 h | 22 | 96 | ||
| 12 | CSA,h 10 min | 97 | ||||
| 13 | 16: W = PhCH2CH2CO R = Ph | 175 °C,g 24 h | 23 | 98 | ||
| 14 | 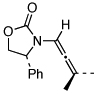 |
17 | 135 °C, 16 h | 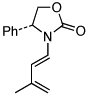 |
24 | 95 |
| 15 | 17 | PTSA, 5 min | 24 | -i | ||
| 16 | 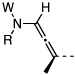 |
18: W = Ac; R = Ph | 175 °C,g 24 h | 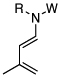 |
25 | 46j |
| 17 | CSA,h 10 min | 97 | ||||
| 18 | 19: W = Ts; R = Bn | 135 °C, 48 h | 26 | 77j | ||
| 19 | CSA,h 10 min | 99 | ||||
Unless otherwise noted, CH3CN was the solvent for thermal conditions, and CH2Cl2 was the solvent when using 10 mol % of PTSA or CSA at rt. In all reactions, concn was 0.10 M.
Isolated yields.
Only E isomers were observed.
90% Starting allenamide recovered.
70% Starting allenamide recovered.
44% Starting allenamide recovered.
Toluene was the solvent.
4 Å MS was used.
Decomposition.
NMR yields.
A keen observation here for the γ-isomerization is that acidic conditions appear to be more effective in general with the exception of 17 [entry 15]. In addition, thermal isomerizations at the γ-position required higher temperatures and/or longer reaction times than those of α-isomerizations. This difference prompted us to explore a possible regioselective isomerization. As shown in Scheme 2, when heating allenamides 27a and 27b, containing both α- and γ-substituents, at 135 °C in CH3CN, isomerizations occurred exclusively at the α-position, leading to 2-amido-dienes 28a and 28b22 in 71% and 94% yields, respectively, all in favor of the E-enamide [assigned by NOE18]. Isomerization of allenamide 27c took place at RT when in contact with silica gel but again α-isomerization was favored. This regioselective isomerization are both mechanistically intriguing23 and should be great synthetic value in constructing highly substituted 2-amido-dienes.
Scheme 2.

Regioselective α-Isomerizations.
The E-selectivity23 attained from α-isomerization provides an excellent platform for the following important pericyclic transformation. As shown in Scheme 3, isomerization of α-allylated allenamide 29 under acidic conditions afforded 3-amido-triene 30 in 86% yield. With the E-selectivity, triene 30 is perfectly suited for a thermal 6π-electron electrocyclic ring-closure24 to give cyclic diene 31. Although only in 35% yield,25 examples of cyclic 2-amido-dienes such as 31 are more rare.26 Allenamide 32a provided a good example of synthesizing cyclic 2-amido-diene 34a via acid-promoted α-isomerization followed by ring-closure. Allenamide 32b demonstrated that the thermal isomerization could be arrested with the gem-dimethyl group in triene 33b impeding the ring-closure. Unfortunatedly, attempted ring-closure of 32b at 200 °C led to an unidentified product instead of 34b.
Scheme 3.

3-Amido-Trienes and Pericyclic Ring-Closure.
At last, this process could be rendered in tandem under thermal conditions to access cyclic 2-amido-dienes 34a, 37, and 38 in good overall yields directly from respective allenamides 32a, 35, and 36 [Scheme 4]. It is noteworthy that these 6π-electron pericyclic ring-closures mostly took place at 135 °C, which implies an accelerated process. This feature is consistent with related ring-closures of 1,3,5-hexatrienes bearing a C3-donating group.27,28
Scheme 4.

A Tandem α-Isomerization–Pericyclic Ring-Closure.
We have described here a regio- and stereoselective isomerization of allenamides, leading to preparations of de novo 2-amido-dienes and a tandem isomerization–6π-electron electrocyclic ring-closure. Studies involving applications of these dienes and this new tandem process as well as mechanistic understanding of this allene-isomerization are underway.
Supplementary Material
Experimental procedures as well as NMR spectra, characterizations, and X-ray structural files are available for all new compounds and free of charge via Internet http://pubs.acs.org.
Acknowledgement
Authors thank NIH [GM066055] for support and Dr. Victor Young [University of Minnesota] for X-ray structural analysis.
References
- 1.Woodward RB, Hoffmann R. Angew. Chem. Int. Ed. 1969;8:781. [Google Scholar]
- 2.(a) Jensen F. J. Am. Chem. Soc. 1995;117:7487. [Google Scholar]; (b) Lewis DK, Cochran JC, Hossenlopp J, Miller D, Sweeney T. J. Phys. Chem. 1981;85:1787. [Google Scholar]
- 3.For general reviews on allenes, see: Krause N, Hashmi ASK. Modern Allene Chemistry. Vol 1 and 2. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2004.
- 4.For some examples, see: Meier H, Schmitt M. Tetrahedron Lett. 1989:5873. Crandall JK, Paulson DR. J. Am. Chem. Soc. 1966;88:4302. Bloch R, Perchec PL, Conia J-M. Angew. Chem. Int. Ed. 1970;9:798. Jones M, Hendrick ME, Hardie JA. J. Org. Chem. 1971;36:3061. Patrick TB, Haynie EC, Probat WJ. Tetrahedron Lett. 1971;27:423. Lehrich F, Hopf H. Tetrahedron Lett. 1987;28:2697.
- 5.For some examples, see: Tsuboi S, Masuda T, Takeda A. J. Org. Chem. 1982;47:4478. Peng W, Zhu S. Tetrahedron. 2003;59:4641. Al-Masum M, Yamamoto Y. J. Am. Chem. Soc. 1998;120:3809. Bibas H, Koch R, Wentrup C. J. Org. Chem. 1998;63:2619. doi: 10.1021/jo972137m. Jacobs TL, Johnson RN. J. Am. Chem. Soc. 1960;82:6397. Buzas AK, Istrate FM, Gagosz F. Org. Lett. 2007;9:985. doi: 10.1021/ol063031t. Trost BM, Kazmaier U. J. Am. Chem. Soc. 1992;114:7933. Guo C, Lu X. J. Chem. Soc. Perkin Trans. 1993;1:1921.
- 6.For a review on allenamide chemistry, see: Hsung RP, Wei L-L, Xiong H. Acc. Chem. Res. 2003;36:773. doi: 10.1021/ar030029i.
- 7.For an earlier example of allenamide isomerizations, see: Overman LE, Clizbe LA, Freerks RL, Marlowe CK. J. Am. Chem. Soc. 1981;103:2807.
- 8.For a study that likely involved an allenamine intermediate, see. Farmer ML, Billups WE, Greenlee RB, Kurtz AN. J. Org. Chem. 1966;31:2885.
- 9.For an allenamide isomerization via Grubb’s catalyst, see: Kinderman SS, van Maarseveen JH, Schoemaker HE, Hiemstra H, Rutjes FPJT. Org. Lett. 2001;3:2045. doi: 10.1021/ol016013e. (b) Also see reference 17a.
- 10.For a review on the synthesis of enamides, see: Tracey MR, Hsung RP, Antoline J, Kurtz KCM, Shen L, Slafer BW, Zhang Y. In: Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Weinreb Steve M., editor. Chapter 21.4. Georg Thieme Verlag KG; 2005.
- 11.For reviews on chemistry of dienamides, see: Overman LE. Acc. Chem. Res. 1980;13:218. Petrzilka M. Synthesis. 1981:753. Campbell AL, Lenz GR. Synthesis. 1987:421.
- 12.For reviews on chemistry of 2-amino- or –amido-dienes: Krohn K. Angew. Chem. Int. Ed. Engl. 1993;32:1582. Enders D, Meyer O. Liebigs Ann. 1996:1023.
- 13.For reviews on Cu-mediated C-N and C-O bond formations, see: Lindley J. Tetrahedron. 1984;40:1433. Ley SV, Thomas AW. Angew. Chem. Int. Ed. 2003;42:5400. doi: 10.1002/anie.200300594. Dehli JR, Legros J, Bolm C. Chem. Commun. 2005:973. doi: 10.1039/b415954c. For reviews on Pd-catalyzed N-arylations of amines and amides, see: Hartwig JF. Angew. Chem. Int. Ed. 1998;37:2046. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. Wolfe JP, Wagaw S, Marcoux J-F, Buchwald S. Acc. Chem. Res. 1998;31:805.
- 14.For some examples, see: Klapper A, Huang X, Buchwald SL. J. Am. Chem. Soc. 2002;124:7421. doi: 10.1021/ja0260465. Han C, Shen R, Su S, Porco JA., Jr Org. Lett. 2004;6:27. doi: 10.1021/ol0360041. Shen R, Lin CT, Bowman EJ, Bowman BJ, Porco JA., Jr J. Am. Chem. Soc. 2003;125:7889. doi: 10.1021/ja0352350. (d) Also see reference 26b.
- 15.Xiong H, Hsung RP, Wei L-L, Berry CR, Mulder JA, Stockwell B. Org. Lett. 2000;2:2869. doi: 10.1021/ol000181+. [DOI] [PubMed] [Google Scholar]
- 16.For synthesis of parent allenamides, see: Wei L-L, Mulder JA, Xiong H, Zificsak CA, Douglas CJ, Hsung RP. Tetrahedron. 2001;57:459.
- 17.(a) Trost BM, Stiles DT. Org. Lett. 2005;7:2117. doi: 10.1021/ol050395x. [DOI] [PubMed] [Google Scholar]; (b) Shen L, Hsung RP, Zhang Y, Antoline JE, Zhang X. Org. Lett. 2005;7:3081. doi: 10.1021/ol051094q. [DOI] [PubMed] [Google Scholar]
- 18.See Supporting Information.
- 19.For leading examples of 2-amido-dienes, see: Ha JD, Kang CH, Belmore KA, Cha JK. J. Org. Chem. 1998;63:3810. González-Romero C, Bernal P, Jiménez F, Cruz MC, Fuentes-Benites A, Benavides A, Bautista R, Tamariz J. Pure Appl. Chem. 2007;79:181. and references cited therein. Movassaghi M, Hunt DK, Tjandra M. J. Am. Chem. Soc. 2006;128:8126. doi: 10.1021/ja0626180.
- 20.For some examples of 2-amino-dienes, see: Enders D, Meyer O, Raabe G. Synthesis. 1992:1242. Barluenga J, Canteli R–M, Flrez J, Garca-Granda S, Gutirrez-Rodrguez A, Martín E. J. Am. Chem. Soc. 1998;120:2514.
- 21.For examples, see: Terada A, Murata K. Bull. Chem. Soc. Jpn. 1967;40:1644. Overman LE, Clizbe LA. J. Am. Chem. Soc. 1976;98:2352. Oppolzer W, Bieber L, Francotte E. Tetrahedron Lett. 1979:4537. Smith AB, III, Wexler BA, Tu C-Y, Konopelski JP. J. Am. Chem. Soc. 1985;107:1308. Schlessinger RH, Pettus TRR, Springer JP, Hoogsteen K. J. Org. Chem. 1994;59:3246. Kozmin SA, Rawal VH. J. Am. Chem. Soc. 1997;119:7165. Huang Y, Iwama T, Rawal VH. J. Am. Chem. Soc. 2002;124:5950. doi: 10.1021/ja026088t. Robiette R, Cheboub-Benchaba K, Peeters D, Marchand-Brynaert J. J. Org. Chem. 2003;68:9809. doi: 10.1021/jo0302049.
- 22.There appears to be ~% of 1-amido-diene from γ-isomerization.
-
23.Without detailed studies, a rationale for lowering of the thermal activation barrier of 1,3-H-shift is the stabilization of the bi-radical intermediate provided by the nitrogen atom, assuming a radical intermediate is considered electron deficient. Based on the this model, this stabilization is direct when isomerizations take place at the α-position [see i], and "vinylogous" for isomerizations at the γ-position [see ii]. Thus, thermal isomerizations at the α-position were faster than at the γ-position.
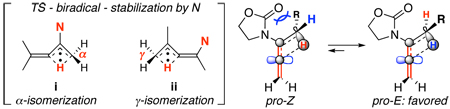 As one reviewer suggested, it is also possible that the nitrogen atom mediates a polarized transition sate in which an increasing charge density at the β-carbon could develop, leading to an N-acyl iminium ion-like character with the migrating hydrogen behaving more like a proton. This charged transition state instead of a neutral one should possess a lower thermal activation barrier for the 1,3-H-shift. Finally, a rationale for the E-selectivity from the thermal α-isomerization is that the pro-Z-TS experiences a greater allylic strain than the pro-E-TS during the 1,3-H-shift, although we cannot rule out equilibration from Z- to E-enamide after the initial isomerization.
As one reviewer suggested, it is also possible that the nitrogen atom mediates a polarized transition sate in which an increasing charge density at the β-carbon could develop, leading to an N-acyl iminium ion-like character with the migrating hydrogen behaving more like a proton. This charged transition state instead of a neutral one should possess a lower thermal activation barrier for the 1,3-H-shift. Finally, a rationale for the E-selectivity from the thermal α-isomerization is that the pro-Z-TS experiences a greater allylic strain than the pro-E-TS during the 1,3-H-shift, although we cannot rule out equilibration from Z- to E-enamide after the initial isomerization.
- 24.For reviews for pericyclic ring-closures, see: Marvell EN. Thermal Electrocyclic Reactions. New York: Academic Press; 1980. Okamura WH, de Lera AR. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Paquette LA, editors. Vol. 5. New York: Pergamon Press; 1991. pp. 699–750. For reviews on ring-closure in natural product synthesis, see: Pindur U, Schneider GH. Chem. Soc. Rev. 1994:409. Beaudry CM, Malerich JP, Trauner D. Chem. Rev. 2005;105:4757. doi: 10.1021/cr0406110.
- 25.When attempting the ring-closure at temperature ≥180 °C, diene 30 slowly isomerized to the respective 1-amido diene.
- 26.For examples, see: Martínez R, Jiménez-Vázquez HA, Delgado F, Tamariz J. Tetrahedron. 2003;59:481. Wallace DJ, Klauber DJ, Chen CY, Volante RP. Org. Chem. 2003;5:4749. doi: 10.1021/ol035959g. Wabnitz TC, Yu J-Q, Spencer JB. Chem. Eur. J. 2004;10:484. doi: 10.1002/chem.200305407.
- 27.For theoretical studies on substituent effects on electrocyclic ring-closure of 1,3,5-hexatrienes, see: Spangler CW, Jondahl TP, Spangler B. J. Org. Chem. 1973;38:2478. Guner VA, Houk KN, Davies IA. J. Org. Chem. 2004;69:8024. doi: 10.1021/jo048540s. Yu T-Q, Fu Y, Liu L, Guo GX. J. Org. Chem. 2006;71:6157. doi: 10.1021/jo060885i. Duncan JA, Calkins DEG, Chavarha M. J. Am. Chem. Soc. 2008;130:6740. doi: 10.1021/ja074402j.
- 28.For examples of accelerated ring-closures of 1,3,5-hexatrienes, see: Barluenga J, Merino I, Palacios F. Tetrahedron Lett. 1990;31:6713. Magomedov NA, Ruggiero PL, Tang Y. J. Am. Chem. Soc. 2004;126:1624. doi: 10.1021/ja0399066. Tessier PE, Nguyen N, Clay MD, Fallis AG.J. Am. Chem. Soc 2006128494616608316 Huntley RJ, Funk RL. Org. Lett. 2006;8:3403. doi: 10.1021/ol061259a. Yu T-Q, Fu Y, Liu L, Guo Q-X. J. Org. Chem. 2006;71:6157. doi: 10.1021/jo060885i. Sünnemann HW, Banwell MG, de Meijere A. Eur. J. Org. Chem. 2007:3879.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures as well as NMR spectra, characterizations, and X-ray structural files are available for all new compounds and free of charge via Internet http://pubs.acs.org.