

Abstract
Purpose
To review the scientific literature supporting the participation of caveolin-1 in the pathogenesis of tissue fibrosis and that modulation of the caveolin-1 pathway may represent a novel treatment for systemic sclerosis (SSc) and other fibrotic diseases.
Recent Findings
Caveolin-1 plays an important role in the regulation of transforming growth factor β (TGF-β) signaling owing to its participation in TGF-β receptor (TβR) internalization. TβR internalized through caveolin-1 lipid rafts undergoes rapid degradation, effectively decreasing TGF-β signaling. Studies have shown that caveolin-1 knockdown in vitro markedly increased collagen gene expression in normal human lung fibroblasts. Caveolin-1 was reduced in affected SSc lungs and skin and in idiopathic pulmonary fibrosis (IPF) lung tissues and fibroblasts. Increasing caveolin-1 expression markedly improved bleomycin-induced pulmonary fibrosis. Restoration of caveolin bioavailability employing penetratin, a cell-permeable peptide carrier for a bioactive caveolin-1 fragment abrogated TGF-β activation of cultured human dermal fibroblasts. Systemic administration of penetratin-caveolin-1 peptide to mice with bleomycin-induced lung fibrosis reduced fibrosis.
Summary
Caveolin-1 plays an important role in the regulation of TGF-β signaling and participates in the pathogenesis of SSc and IPF. Restoration of caveolin function employing active caveolin-1 fragments coupled to cell-permeable carrier peptides may represent a novel approach for their treatment.
Keywords: Caveolin-1, TGF-β, fibrosis, collagen, systemic sclerosis, idiopathic pulmonary fibrosis
Introduction
Fibrotic disorders, which include systemic sclerosis (SSc), idiopathic pulmonary fibrosis (IPF), cirrhosis of the liver, and the newly recognized Nephrogenic Systemic Fibrosis (NSF), are characterized by abnormal and excessive deposition of collagen and other extracellular matrix (ECM) components in various tissues [1-3]. Although their etiology is quite diverse, the presence of ECM-producing fibroblasts displaying an activated phenotype in the affected tissues is typical of fibrotic diseases. Fibroblast activation is characterized by a marked increase in the transcriptional activity of the genes encoding type I and type III collagens and fibronectin, initiation of the expression of α-smooth muscle actin (α-SMA), and the reduction of ECM degradative activities [4]. Activated fibroblasts display contractile properties resulting from the expression of stress fibers containing α-SMA, and their profibrotic activation is part of a complex set of molecular and biochemical changes that are conserved for multiple passages in vitro.
Transforming growth factor-β (TGF-β) plays a crucial role in fibroblast activation. One of the most important effects of TGF-β is its stimulation of ECM production and accumulation by increasing the expression of genes encoding various collagens and other matrix proteins and reducing the expression of genes involved in ECM degradation and turnover. Delineation and identification of the specific cellular receptors and intracellular mediators participating in the cellular response to TGF-β [1-3,5-10] and recognition that TGF-β is a major participant in the initiation and progression of tissue fibrosis have recently focused substantial attention on this growth factor as a target for the development of anti-fibrotic therapies [11-14]. Despite intensive investigations in vitro and in various animal models of fibrosis strategies to block TGF-β effects have thus far been ineffective or are still in the experimental stage.
In the last decade, however, a novel pathway capable of exerting a potent modulation of TGF-β signaling has become recognized. This pathway involves the protein caveolin-1 and caveolae, the cell membrane structures in which caveolin-1 is found. Caveolin-1-positive plasma membrane lipid rafts called caveolae co-localize TGF-β receptor with its degrading SMURF/Smad7 complex, a process which may result not only in TGF-β signal abrogation and consequent fibroblast deactivation but potentially even in the restoration of the normal balance of ECM production and degradation. Here, we review recent scientific literature that support the participation of caveolin-1 in the pathogenesis of tissue fibrosis and indicate that modulation of the TGF-β pathway by strategic management of caveolin-1 bioavailability may represent a promising and novel therapeutic approach for fibrotic conditions such as SSc.
Caveolae microdomains are crucial regulators of receptor-mediated cellular signaling
Caveolin-1 is the most important member of a family of three membrane proteins (caveolin- 1, 2 and 3) that are the major coating proteins of caveolae. Caveolae are 50- to 100-nm flask-shaped plasma membrane invaginations that since their discovery have provoked a multitude of conjectures about their function [15]. Unlike earlier views of the plasma membrane as a “fluid mosaic” [16], which posited that integral membrane proteins float and diffuse freely through a sea of homogeneous lipids, a current view is that membrane proteins are heterogeneously distributed and can be found clustered within specialized microdomains, termed lipid rafts, which are particularly rich in cholesterol and glycophosphatidylinositol-linked proteins. Caveolae represent a morphologically identifiable subset of lipid rafts which participate in the pathogenesis of numerous diseases [17-19], and extensive recent evidence has demonstrated the clustering of numerous receptors within their domains. It has also been shown that the spatial organization of cell receptors in lipid rafts and caveolae can affect the subsequent transmission of the specific signal initiated by ligand engagement of these receptors [20-23].
Caveolin-1 regulated TGF-β signaling
Recent studies have shown that caveolin-1 plays a very important role in the regulation of TGF-β signaling owing to its participation in TβR internalization. TβR are internalized both by caveolin-1 associated lipid rafts and by early endosome antigen 1 (EEA-1) non-lipid raft pathways. It has been shown, furthermore, that non-lipid raft associated internalization increases TGF-β signaling, whereas, caveolin-associated internalization increases TβR degradation thus, effectively decreasing or abolishing TGF-β signaling [24,25]. Specifically, it was demonstrated that SARA and SMURF, upstream regulators of either TGF-β signaling or TβR degradation, respectively, were localized in distinct subcellular compartments [26,27]. The complex SARA/Smad2/3 which initiates TβR-1 signaling was found localized in a non-lipid raft, EEA-1 positive compartment, whereas, the SMURF/Smad7 complex which is responsible for initiating proteasome degradation of TβR was found localized in caveolin-1 positive lipid rafts. The localization of the TβR in the EEA-1 positive compartment was responsible for downstream Smad activation through recruitment and phosphorylation of Smad-2/3 and subsequent nuclear translocation of the Smad2/3-Smad4 complex, whereas, the localization of the TβR-I (or II) and receptor complex in caveolae lipid rafts caused recruitment of SMURF/Smad7 and subsequent receptor ubiquitination and rapid turnover.
This novel mechanism of regulation of the TβR function and activity follows ligand engagement and is regulated by the fluidity of the membrane and the membrane density of the distinct caveolin-1 and non-caveolin compartments. Thus, absence of one compartment or imbalance in the densities of the two compartments may affect the level of TGF-β pathway activity given the same amount of ligand binding. Furthermore, since this process occurs at the level of internalization of the TβR immediately following ligand engagement, it likely represents an important mechanism of regulation of TGF-β signaling as illustrated in Figure 1. One unique feature of this pathway is that it provides a cogent and plausible mechanism for the perpetuation of tissue fibrosis following an initial triggering event. The triggering event results in a decrease in caveolin-1 gene expression; this decrease then shifts TGF-β internalization through the EEA-1 pathway, leading to accentuation and perpetuation of the TGF-β-induced fibrotic effects and simultaneously causing further downregulation of caveolin-1 gene expression. Thus, the current evidence indicates that caveolin-1 is a crucial regulator of TGF-β intracellular signaling and TβR endosomal degradation and, therefore, may play a key role in the pathogenesis of disorders characterized by exaggerated tissue fibrosis.
Figure 1.

Schematic diagram for the involvement of caveolae in TGF-β signal transduction and for the mechanisms whereby caveolin-1 downregulation in SSc would result in tissue fibrosis and further caveolin-1 downregulation (adapted from ref. 25).
Early evidence of the role of caveolin-1 in pathologic tissue fibrosis
The earliest evidence linking caveolin-1 to a fibrotic phenotype was provided by the study of Kasper et al. [28] more than a decade ago. These authors demonstrated that caveolin expression was markedly downregulated in alveolar epithelium of rats and mini-pigs following radiation-induced lung injury. The profound downregulation of caveolin preceded the onset of radiation-induced lung fibrosis. Furthermore, the amount of immunoreactive caveolin correlated with the severity of lung fibrosis, with normal amounts present in non-affected tissue in contrast with a marked decrease in areas of fibrosis. The role of caveolin-1 in the development of tissue fibrosis was confirmed in two independent studies which simultaneously reported the generation of mice lacking caveolin-1 by gene targeting [29,30]. Drab et al. [29] showed that these mice displayed profound histopathological and electron microscopical alterations in the lung alveolar septae, with replacement of the normal alveolar bilayer lining by a disorganized, multilayered structure containing abundant connective tissue. Razani et al. [30] described thickened alveolar septae, marked hypercellularity, increased reticulin deposition and thickening of the basement membrane.
Caveolin-1 downregulation in SSc and pulmonary fibrosis
Subsequent studies extended the early observations described above to human diseases such as IPF and SSc. Tourkina et al. [31] demonstrated that caveolin-1 knockdown in vitro resulted in a 5-fold increase in normal human lung fibroblast collagen gene expression whereas increased caveolin-1 expression induced by PKC-α caused a reduction in collagen production. The same study showed that fibroblasts from affected skin or lung from SSc patients contained less caveolin-1. More recently, additional evidence for caveolin-1 participation in pulmonary fibrosis was presented. Wang et al. [32] observed marked reduction of caveolin-1 expression in lung tissues and fibroblasts from IPF patients compared with controls. Furthermore, induction of caveolin-1 expression with an adenovirus-caveolin-1 expression vector ameliorated bleomycin-induced pulmonary fibrosis. These authors also showed that TGF-β1 decreased caveolin-1 expression in human lung fibroblasts and that caveolin-1 expression suppressed TGF-β1-induced stimulation of ECM production in these cells. Odajima et al. [33••] examined the expression of caveolin-1 during the development of bronchiolization, a key process in fibrosing lung, in mice with bleomycin-induced pulmonary fibrosis and in lungs from patients with various forms of interstitial pneumonias. The results showed a marked decrease of caveolin-1 mRNA levels in the lungs from the bleomycin-treated mice as well as in bronchiolar epithelial cells isolated by laser capture microdissection. Also, significant reduction in caveolin-1 protein and mRNA levels were found in affected tissues from patients with pulmonary fibrosis. Thus, the results of these studies collectively supported a pivotal role for caveolin-1 in the pathogenesis of pulmonary fibrosis.
Recently our group demonstrated that caveolin downregulation is also an important feature of SSc [34••]. In these studies we found a substantial reduction in caveolin-1 immunofluorescence in affected SSc lungs and skin in contrast with normal caveolin-1 expression in histopathologically non-affected areas (Figure 2). Furthermore, TGF-β treatment of normal dermal fibroblasts induced a potent downregulation of caveolin-1 expression. These findings suggested that the observed decrease in caveolin-1 expression in SSc affected tissues may have been caused by TGF-β or other cytokines released by inflammatory cells present in the tissue and could in turn be responsible for further amplification of TGF-β signaling, a crucial event in the pathogenesis of tissue fibrosis in SSc. Specifically, as depicted in Figure 1, decreased caveolin-1 expression in SSc would be responsible for an amplification loop of TGF-β signaling; the increase in TGF-β signaling causing, in turn, a further downregulation of caveolin expression at both the mRNA and protein levels, triggering a repetitive cycle of signal amplification that ultimately leads to tissue fibrosis.
Figure 2.
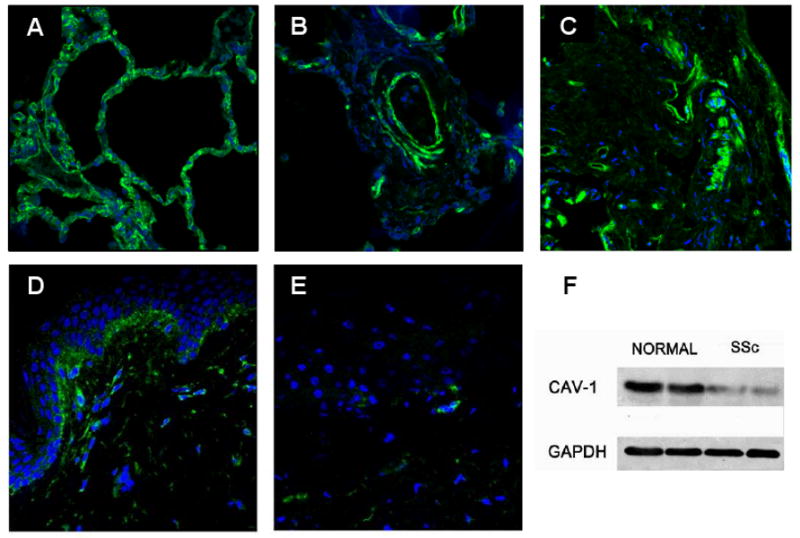
Reduction of caveolin-1 expression in SSc lung and skin. A. Confocal image of normal lung immunostained for caveolin-1. Note the homogeneous green membrane staining for caveolin-1 on the surface of alveolar wall cells. B and C. Confocal image of SSc lung stained for caveolin-1. Note that in contrast with A, there is low expression of caveolin-1 in most cells of the vessel wall and interstitium. D. Normal skin. Note the expression of caveolin-1 surrounding almost every cell in the dermis. E. SSc skin. Note the low expression of caveolin-1 in most cells. F. Cell lysates from two SSc dermal fibroblast cell lines and two normal dermal fibroblast cell lines analyzed by Western blot for caveolin-1. Reproduced from ref. 34 with permission form Arthritis Rheum.
Caveolin-null mice and tissue fibrosis
Early studies on caveolin-1 null mice [29,30] described thickening of lung alveolar septa caused by proliferation of endothelial cells and accumulation of reticulin and other matrix proteins, however, they did not describe whether the caveolin-1-null animals had evidence of fibrosis in other tissues. We performed an extensive histopathologic analysis and measured the hydroxyproline content of skin and lungs of caveolin-1 knockout mice [34••]. The results indicated that as early as 12 weeks of age, caveolin-1-null mice displayed a marked increase in collagen deposition in skin and lungs compared with their normal littermates (Figure 3). These histopathological changes were accompanied by a threefold increase in lung and skin collagen content. In vitro studies performed with caveolin-1 null fibroblasts indicated that the observed tissue fibrosis was associated with a profibrotic phenotype of tissue fibroblasts strikingly similar to that of SSc fibroblasts. These data indicate that the lung involvement in caveolin-1-null mice is secondary to a combination of pulmonary artery hypertension resulting from the known hyper-proliferative phenotype of caveolin-1-null vascular smooth muscle cells and their increased sensitivity to endothelin-1 [35,36] combined with parenchymal lung fibrosis caused by the profibrotic gene expression pattern of caveolin-1 null fibroblasts.
Figure 3.
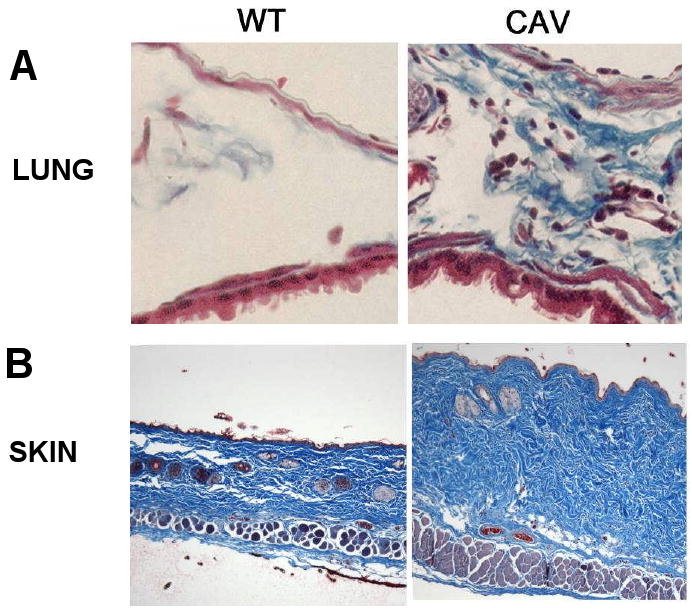
Increased collagen in lungs and skin of caveolin-1 knockout mice. A. Masson's Trichrome staining for collagen in interstitium of wild type (WT) and caveolin-1 null lung (CAV). B. Masson's Trichrome staining for collagen in the skin of wild type (WT) and caveolin-1 null skin (CAV). Reproduced from ref. 34 with permission from Arthritis Rheum.
The role of caveolin-1 in lung fibrosis has been indirectly confirmed by a study in which specific endothelial overexpression of caveolin-1 was induced in caveolin-1 knockout mice. Selective re-expression of caveolin-1 in endothelial cells partially reverted the phenotype of pulmonary artery hypertension, whereas, in contrast, the extent of lung parenchymal fibrosis was not affected by the endothelial cell-specific rescue of caveolin-1 expression [37••].
Restoration of caveolin-1 levels as a therapeutic approach for fibrotic diseases
The data summarized here provide robust evidence supporting the involvement of caveolin-1 downregulation in the pathogenesis of tissue fibrosis. The next logical step, as suggested in a recently published editorial [38••] is to develop therapeutic tools that may increase caveolin-1 bioavailability in the target cells. One of the most promising and novel approaches to accomplish this goal is the utilization of cell permeable carriers capable of shuttling small peptides and proteins inside cells [39]. One such carrier is penetratin, a 16-amino acid-long fragment of the third helix of the homeodomain of the Antennapedia homeoprotein [40]. Penetratin is internalized by cells in a specific, non-receptor-mediated manner, and is able to translocate through the cell membrane [40]. On the other hand, Razani et al. [41] demonstrated previously that the interaction between caveolin-1 and the TβR was mediated by a small region within the caveolin-1 protein identified as the caveolin scaffolding domain (CSD) which specifically recognizes and binds to a short amino acid sequence termed the caveolin binding motif present in TβR and other serine-threonine kinases. Numerous studies have provided ample experimental evidence that the caveolin-1 kinase inhibitor function is mediated by the CSD [42,43] and in some studies caveolin-1 function has been restored following successful delivery of the CSD to cells employing a penetratin-CSD fusion cell permeable peptide.
In our studies [34••] we employed the CSD-penetratin fusion peptide to explore the effects of caveolin-1 function restoration on the profibrotic phenotype of SSc fibroblasts and on the effects of exogenous TGF-β stimulation. We found that the CSD-penetratin fusion peptide was not cytotoxic, inhibited the increased collagen production and α-SMA expression in SSc dermal fibroblasts, and suppressed TGF-β stimulation of collagen and α-SMA expression. Furthermore, the peptide inhibited TGF-β induced Smad2/3 nuclear translocation. Similar studies were recently published by Tourkina et al. [44••], who, employing the same peptide on SSc lung fibroblasts in vitro demonstrated an inhibitory effect of important non-Smad intracellular signaling TGF-β pathways including MEK, ERK, JNK, and Akt. Of further interest was their demonstration that systemic administration of CSD-penetratin peptide to mice with bleomycin-induced lung fibrosis blocked epithelial cell apoptosis and inflammatory cell infiltration and reduced fibrotic changes, thus supporting the potential efficacy of this treatment for SSc and pulmonary fibrosis.
Conclusion
Numerous studies published during the last decade have provided strong evidence that caveolin-1, the main protein component of caveolae, participates in the pathogenesis of fibrotic diseases through regulation of TβR degradation and activation. A profound reduction in caveolin-1 expression has been demonstrated in affected tissues from IPF and SSc patients, and the important role of the protein in TGF-β signaling and stimulation of ECM production has been conclusively demonstrated. Restoration of caveolin-1 function employing novel cell-permeable peptides coupled to active caveolin-1 fragments abrogated the profibrotic effects of TGF-β and, therefore, may represent a novel treatment for SSc and other fibrotic disorders.
Acknowledgments
S.A.J. and F.D.G. were supported by NIH/NIAMS grant RO1-AR-019616 and by the generous support of the John Murray Foundation.
M.P.L. and his laboratory were supported by grants from the NIH/NCI (R01-CA-80250; R01-CA-098779; R01-CA-120876), the American Association for Cancer Research (AACR), and the Department of Defense-Breast Cancer Research Program (Synergistic Idea Award).
We thank Susan V. Castro, Ph.D., for her assistance in the preparation of the manuscript.
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
•• of outstanding interest
- 1.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of Systemic Sclerosis. Ann Int Med. 2004;140:37–50. [PubMed] [Google Scholar]
- 4.Jelaska A, Korn JH. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 2000;43:2230–2239. doi: 10.1002/1529-0131(200010)43:10<2230::AID-ANR10>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Blobe GC, Schiemann EP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 6.Schmierer B, Hill CS. TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. Review. [DOI] [PubMed] [Google Scholar]
- 7.Massagué J, Gomis RR. The logic of TGFβ signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. Review. [DOI] [PubMed] [Google Scholar]
- 8.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. Review. [DOI] [PubMed] [Google Scholar]
- 9.Verrecchia F, Mauviel A, Farge D. Transforming growth factor-β signaling through the Smad proteins: Role in systemic sclerosis. Autoimmun Rev. 2006;5:563–569. doi: 10.1016/j.autrev.2006.06.001. Review. [DOI] [PubMed] [Google Scholar]
- 10.Leask A, Abraham DJ. TGF-β signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. Review. [DOI] [PubMed] [Google Scholar]
- 11.Lafyatis R. Targeting fibrosis in systemic sclerosis. Endocr Metab Immune Disord Drug Targets. 2006;6:395–400. doi: 10.2174/187153006779025766. Review. [DOI] [PubMed] [Google Scholar]
- 12.Denton CP, Black CM, Abraham DJ. Mechanisms and consequences of fibrosis in systemic sclerosis. Nat Clin Pract Rheumatol. 2006;2:134–144. doi: 10.1038/ncprheum0115. Review. [DOI] [PubMed] [Google Scholar]
- 13.Leask A. Scar wars: Is TGFβ the phantom menace in scleroderma? Arthritis Res Ther. 2006;8:213. doi: 10.1186/ar1976. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbloom J, Jimenez SA. Molecular ablation of TGF-β signaling pathways by tyrosine kinase inhibition: The coming of a promising new era in the treatment of tissue fibrosis. Arthritis Rheum. 2008 August; doi: 10.1002/art.23634. In Press. Invited Editorial. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Razani B, Lisanti MP. Caveolin-deficient mice: Insights into caveolar function human disease. J Clin Invest. 2001;108:1553–1561. doi: 10.1172/JCI14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 17.Cohen AW, Hnasko R, Schubert W, et al. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. Review. [DOI] [PubMed] [Google Scholar]
- 18.Michel V, Bakovic M. Lipid rafts in health and disease. Biol Cell. 2007;99:129–140. doi: 10.1042/BC20060051. Review. [DOI] [PubMed] [Google Scholar]
- 19.Quest AF, Leyton L, Párraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem Cell Biol. 2004;82:129–144. doi: 10.1139/o03-071. Review. [DOI] [PubMed] [Google Scholar]
- 20.Kabouridis PS. Lipid rafts in T cell receptor signalling. Mol Membr Biol. 2006;23:49–57. doi: 10.1080/09687860500453673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hothersall E, McSharry C, Thomson NC. Potential therapeutic role for statins in respiratory disease. Thorax. 2006;61:729–734. doi: 10.1136/thx.2005.057976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–391. doi: 10.1146/annurev.pharmtox.48.121506.124841. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwencke C, Braun-Dullaeus RC, Wunderlich C, et al. Caveolae and caveolin in transmembrane signaling: Implications for human disease. Cardiovasc Res. 2006;70:42–49. doi: 10.1016/j.cardiores.2005.11.029. Review. [DOI] [PubMed] [Google Scholar]
- 24.Zhang XL, Topley N, Ito T, et al. Interleukin-6 regulation of transforming growth factor (TGF)-beta receptor compartmentalization and turnover enhances TGF-beta1 signaling. J Biol Chem. 2005;280:12239–45. doi: 10.1074/jbc.M413284200. [DOI] [PubMed] [Google Scholar]
- 25.Di Guglielmo GM, Le Roy C, Goodfellow AF, et al. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–21. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 26.Hayes S, Chawla A, Corvera S. TGF beta receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol. 2002;158:1239–1249. doi: 10.1083/jcb.200204088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Runyan CE, Schnaper HW, Poncelet AC. The role of internalization in transforming growth factor beta1-induced Smad2 association with Smad anchor for receptor activation (SARA) and Smad2-dependent signaling in human mesangial cells. J Biol Chem. 2005;280:8300–8308. doi: 10.1074/jbc.M407939200. [DOI] [PubMed] [Google Scholar]
- 28.Kasper M, Reimann T, Hempel U, et al. Loss of caveolin expression in type I pneumocytes as an indicator of subcellular alterations during lung fibrogenesis. Histochem Cell Biol. 1998;109:41–48. doi: 10.1007/s004180050200. [DOI] [PubMed] [Google Scholar]
- 29.Drab M, Verkade P, Elger M, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 30.Razani B, Zhang XL, Bitzer M, et al. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem. 2001;276:6727–6738. doi: 10.1074/jbc.M008340200. [DOI] [PubMed] [Google Scholar]
- 31.Tourkina E, Gooz P, Pannu J, et al. Opposing effects of protein kinase Cα and protein kinase Cε on collagen expression by human lung fibroblasts are mediated via MEK/ERK and caveolin-1 signaling. J Biol Chem. 2005;280:13879–13887. doi: 10.1074/jbc.M412551200. [DOI] [PubMed] [Google Scholar]
- 32.Wang XM, Zhang Y, Kim HP, et al. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med. 2006;203:2895–2906. doi: 10.1084/jem.20061536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33••.Odajima N, Betsuyaku T, Nasuhara Y, et al. Loss of caveolin-1 in bronchiolization in lung fibrosis. J Histochem Cytochem. 2007;55:899–909. doi: 10.1369/jhc.7A7203.2007. [DOI] [PubMed] [Google Scholar]; This study demonstrates reduction of caveolin-1 in patients with various forms of fibrosing pneumonias and in lungs of mice with bleomycin-induced pulmonary fibrosis
- 34••.Del Galdo F, Sotgia F, de Almeida C, et al. Decreased expression of caveolin-1 in systemic sclerosis: Crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. 2008 September; doi: 10.1002/art.23791. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated a marked reduction of caveolin-1 in affected skin and lungs from patients with systemic sclerosis. Cutaneous and pulmonary fibrosis was documented in caveolin-1 mice and in vitro studies showed that restoration of caveolin-1 function in caveolin-1-null fibroblasts abrogated TGF-β signaling
- 35.Zhao YY, Liu Y, Stan RV, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hassan GS, Williams TM, Frank PG, et al. Caveolin-1-deficient aortic smooth muscle cells show cell autonomous abnormalities in proliferation, migration, and endothelin-based signal transduction. Am J Physiol Heart Circ Physiol. 2006;290:H2393–2401. doi: 10.1152/ajpheart.01161.2005. [DOI] [PubMed] [Google Scholar]
- 37••.Murata T, Lin MI, Huang Y, et al. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med. 2007;204:2373–2382. doi: 10.1084/jem.20062340. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors accomplished specific expression of caveolin-1 in endothelial cells of caveolin-1-null mice and demonstrated a partial reversal of pulmonary hypertension in these mice, although no effects on the pulmonary fibrosis phenotype were observed
- 38••.Verma S, Slutsky AS. Idiopathic pulmonary fibrosis--new insights. N Engl J Med. 2007;356:1370–1372. doi: 10.1056/NEJMcibr070490. [DOI] [PubMed] [Google Scholar]; This paper is a very illustrative review focusing on novel insights into the pathogenesis of pulmonary fibrosis and of novel therapeutic approaches
- 39.Deshayes S, Morris MC, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci. 2005;62:1839–1849. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derossi D, Chassaing G, Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8:84–87. Review. [PubMed] [Google Scholar]
- 41.Razani B, Lisanti MP. Caveolin-deficient mice: insights into caveolar function human disease. J Clin Invest. 2001;108:1553–61. doi: 10.1172/JCI14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu SW, Howat SL, Renzoni EA, et al. Endothelin-1 induces expression of matrix-associated genes in lung fibroblasts through MEK/ERK. J Biol Chem. 2004;279:23098–23103. doi: 10.1074/jbc.M311430200. [DOI] [PubMed] [Google Scholar]
- 43.Chang CC, Lin MT, Lin BR, et al. Effect of connective tissue growth factor on hypoxia-inducible factor 1 β degradation and tumor angiogenesis. J Natl Cancer Inst. 2006;98:984–995. doi: 10.1093/jnci/djj242. [DOI] [PubMed] [Google Scholar]
- 44••.Tourkina E, Richard M, Gööz P, et al. Antifibrotic properties of caveolin-1 scaffolding domain in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2008;294:L843–861. doi: 10.1152/ajplung.00295.2007. [DOI] [PubMed] [Google Scholar]; This study demonstrated that restoration of caveolin-1 function in vivo in mice with bleomycin-induced pulmonary fibrosis employing a cell-permeable peptide coupled to a caveolin-1 fragment reduced the fibrotic changes and inhibited important intracellular fibrotic signaling pathways