

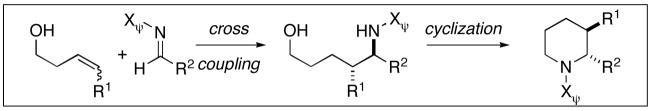
The development of reactions that facilitate the syntheses of nitrogen-containing small molecules continues to be an intense area of research in organic chemistry. Whereas many powerful methods exist for the preparation of such targets, recent accomplishments in reductive coupling chemistry have provided new strategies of untapped potential for the convergent synthesis of secondary carbinolamines. To date, methods have been described for bimolecular alkyne–imine coupling that provide a convenient route to substituted allylic amines.1 The corresponding cross coupling reaction between internal olefins and imines represents a significantly more complex and potentially more powerful bond construction.2 In addition to overcoming the lower reactivity of substituted olefins in bimolecular carbometalation reactions, a synthetically useful cross coupling process must address the additional complexity that results from the need to simultaneously control site selectivity and diastereoselection in the carbon–carbon bond forming event (2+3→4–7, Figure 1). The development of a synthetic method to accomplish the cross coupling of internal olefins and imines in a regio- and stereoselective manner would represent a significant advance, and provide a fundamentally new strategy for the synthesis of stereodefined secondary carbinolamines.
Figure 1.

Bimolecular coupling of imines and substituted olefins via formation of azametallacyclopentanes.
Here we describe a method for cross coupling of aromatic imines with terminal, 1,1-disubstituted, (E)- and (Z)-disubstituted olefins. Additionally, we describe the use of this coupling reaction in single- and double asymmetric synthesis, as well as define a novel route for convergent stereoselective synthesis of substituted piperidines.
Previously, we have described titanium alkoxide-mediated reactions for alkyne–alkyne,3 alkyne–alkene,4 and alkyne–imine5 cross coupling. These methods have provided a means to overcome the sluggish reactivity of highly substituted π-systems in reductive coupling chemistry, and defined a means to control regioselection in bimolecular carbometalation that is independent of the differential steric environment around an unsymmetrically substituted alkyne or alkene. As depicted in Figure 2, the present study explores alkoxide-directed, titanium-mediated, reductive cross coupling between homoallylic alkoxides 8 and imines 9 as a method for the synthesis of acyclic aminoalcohols (i.e. 10 or 12) and substituted heterocycles (i.e. 11 or 13). In contrast to established reductive cross coupling reactions between alkynes and heterosubstituted π-systems (alkyne–aldehyde, alkyne–imine), this alkene–imine cross coupling process is significantly more complex as the carbon–carbon bond formation proceeds with the simultaneous generation of two stereogenic sp3 carbon centers.
Figure 2.

Cross coupling of substituted olefins and imines – a potential pathway for the synthesis of aminoalcohols and substituted heterocycles.
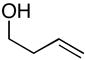
To examine the potential of this proposed reaction pathway for regio- and diastereoselective C–C bond formation, a study was initiated to explore the reactivity of azametallacyclopropanes (generated in situ from aromatic imines) with homoallylic alkoxides of general structure 8. As depicted in entry 1 of Table 1, deprotonation of homoallylic alcohol 14 (n-BuLi, Et2O) and addition to a preformed azametallacyclopropane derived from imine 15 (Ti(Oi-Pr)4, c-C5H9MgCl, Et2O, −70 to −40 °C), followed by protonation of the presumed bicyclic azametallacyclopentane resulted in formation of the 1,5-amino alcohol 16 in 76% yield (rr ≥ 95:5). The success of this coupling reaction not only defines a convergent route to aminoalcohols, but also provides a convenient means to access substituted piperidines. Treatment of 1,5-aminoalcohol 16 with PPh3 and CCl4 (reflux) resulted in the formation of the substituted piperidine 17 in 85% yield.6
Table 1.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | homoallylic alcohol | imine | 1,5-aminoalcohola | yield (%) | rr | dr | piperidineb (yield) |
| 1 |
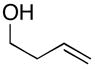 14 |
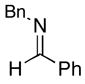 15 |
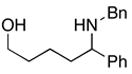 16 |
76 | ≥95.5 | n/a |
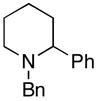 17 (85%) |
| 2 |
18 |
15 |
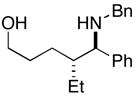 19 |
73 | ≥95.5 | ≥95.5 |
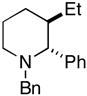 20 (81%) |
| 3 |
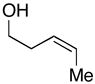 21 |
15 |
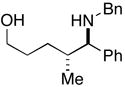 22 |
63 | ≥95.5 | ≥95.5 |
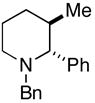 23 (77%) |
| 4 |
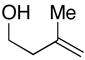 24 |
15 |
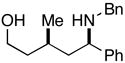 25 |
67 | ≥95.5 | 4:1 |
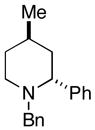 26 (76%) |
Reaction conditions for cross coupling: imine (1 eq), Ti(Oi-Pr)4 (1.5 eq), c-C5H9MgCl (3.0 eq), Et2O (−78 to −40 °C), then add alkoxide (1.5 eq) (−40 to −20 °C, 0 °C, or rt -see Supporting Information for details).
Reaction conditions for cyclization: PPh3, imidazole, CCl4, reflux.
More highly substituted olefins were well tolerated in this cross coupling reaction. As illustrated in entry 2, coupling of the (E)-disubstituted homoallylic alcohol 18 with imine 15 provided the 4,5-anti-1,5-aminoalcohol 19 in 73% yield (dr ≥ 95:5; rr ≥ 95:5), and on cyclization, the 2,3-trans-disubstituted piperidine 20 in 81% yield. Coupling of the related (Z)-disubstituted homoallylic alcohol 21 with imine 15 provided the 4,5-anti-1,5-aminoalcohol 22 in 63% yield (dr ≥ 95:5; rr ≥ 95:5), and on cyclization, the 2,3-trans-disubstituted piperidine 23 in 77% yield (entry 3).
As depicted in Figure 3, the stereoconvergence observed in these coupling reactions can be rationalized by assuming preferential reaction by way of transition state structures that avoid 1,2-steric interactions in a developing cis-fused bicyclo[3.3.0] ring system (B and C).7, 8
Figure 3.

A model for stereoselection in imine–alkene cross coupling.7, 8
The homoallylic alcohol 24, bearing a 1,1-disubstituted olefin, was also an effective coupling partner with imine 15 providing the 1,3-syn-1,5-aminoalcohol product 25 in 67% yield (dr = 4:1; rr ≥ 95:5),9 and on cyclization, the corresponding 2,4-trans-disubstituted piperidine 26 in 76% yield. The stereoselectivity in this case is consistent with preferential progression through a transition state geometry that has the phenyl substituent occupying a position outside the developing cis-fused bicyclo[3.3.0] ring system (F in Figure 3).7, 8
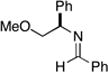
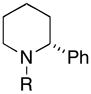
Next, coupling reactions of achiral homoallylic alcohols with chiral imines were examined. As depicted in Table 2, coupling of homoallylic alcohols with chiral imine 28 proceeded in a generally efficient manner (61–83%).10 Highest selectivities were observed in coupling reactions of the terminal- (14), and (Z)-disubstituted (27) olefins, and provided products with dr ≥ 20:1 (entries 1 and 2). Cyclization of these 1,5-aminoalcohols (29 and 31) provides the corresponding substituted piperidines 30 and 32 with good efficiency (77 and 87% yield). Coupling of the (E)-disubstituted olefin 18 with chiral imine 28 also led to aminoalcohol 31, yet this product was obtained with lower levels of stereoselection (25:4:1) as that observed with the isomeric olefin 27.
Table 2.
 | ||||||
|---|---|---|---|---|---|---|
| entry | homoallylic alcohol | imine | 1,5-aminoalcohola | yield (%) | dr | piperidineb (yield) |
| 1 |
 14 |
 28 |
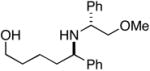 29 |
83 | 24:1 |
 30 (77%) |
| 2 |
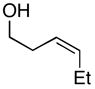 27 |
28 |
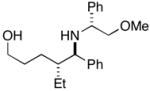 31 |
75 | 20:1 |
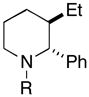 32 (87%) |
| 3 |
18 |
28 | 31 | 61 | 25:4:1 | |
Reaction conditions for cross coupling: imine (1 eq), Ti(Oi-Pr)4 (1.5 eq), c-C5H9MgCl (3.0 eq), Et2O (−60 to −30 °C), then add alkoxide (1.5 eq) (−40 to 0 °C or rt).
Reaction conditions for cyclization: For 30 - 2-NsCl, Et3N, DMAP, CH2Cl2, rt. For 32-MsCl, Et3N, CH2Cl2, rt. R = CH(Ph)CH2OMe.
Cross coupling reactions were also examined to determine the extent to which stereochemical matching and/or mismatching might occur in double diastereodifferentiating cases.11 As illustrated in Table 3, coupling of the (Z)-homoallylic alcohol 33 with chiral imine ent - 28 furnished the 1,4-anti-1,5-syn-1,5-aminoalcohol 34 in 76% yield (dr ≥ 50:1), cyclization of which provided the 2,3-trans-2,5-trans-tetrasubstituted piperidine 35 (entry 1a). Coupling of the same homoallylic alcohol (33) with 28 proceeded with low conversion (30%) and provided a mixture of diastereomeric products (dr = 4:1:1; entry 2a). Although the efficiency of this coupling reaction can be increased on warming (entry 2b), the dr is affected. The disparate reactivity and selectivity observed defines a clear matched (entry 1) and mismatched (entry 2) double asymmetric relationship in these coupling reactions.
Table 3.
 | ||||||
|---|---|---|---|---|---|---|
| entry | homoallylic alcohol | imine | 1,5-aminoalcohola | yield (%) | dr | piperidineb (yield) |
| 1 |
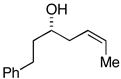 33 |
ent-28 |
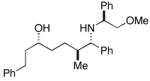 34c |
76 | ≥50:1 |
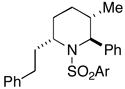 35 (79%) |
| 2a |
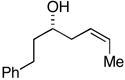 33 |
28 |
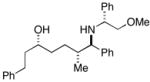 36c |
30 | 4:1:1 | |
| 2b | 85 | 6:3:1 | ||||
| 3 |
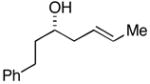 38 |
28 |
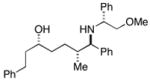 36c |
88 | 35:4:1 |
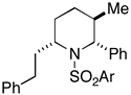 37(78%) |
| 4 |
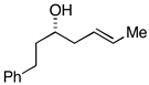 38 |
ent-28 |
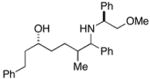 39 |
76 | 19:12:4:1 | |
Reaction conditions for cross coupling: imine (1 eq), Ti(Oi-Pr)4 (1.5 eq), c-C5H9MgCl (3.0 eq), Et2O (−60 to −30 °C), then add alkoxide (1.5 eq) (−30 to 0 °C (entries 1, 2a, 3 and 4) or rt (entry 2b)).
Reaction conditions for cyclization: 1) H2 (1 atm), Pd(OH)2, AcOH/MeOH (1:10 v/v), rt, 2) 2-NsCl, Et3N, DMAP, CH2Cl2, 0 °C to rt, 3) PPh3, DIAD, THF, rt. Ar = 2-nitrophenyl.
Major diastereomer shown.
Cyclization of 36 to 37 was performed with the enantiomer of 36 (for data on ent-37, see Supporitng Information).
The (E)-disubstituted homoallylic alcohol 38 displayed inverse double asymmetric behavior as compared with (Z)-disubstituted homoallylic alcohol 33. As illustrated in entries 3 and 4, coupling of 38 with 28 represents a matched double asymmetric process (88% yield, dr = 35:4:1), whereas coupling of 38 with ent - 28 represents a mismatched double asymmetric reaction (dr = 19:12:4:1).
A preliminary empirical model to explain these double asymmetric reactions is proposed in Figure 4. The model is based on: (1) diastereoselective coordination of the pendent methyl ether to the titanium center,10 (2) intramolecular delivery of the olefin to the azametallacyclopropane,3–5 (3) minimization of 1,2-interactions in the developing five-membered ring, and (4) positioning of the alkyl group (R) outside of the developing cis-fused ring system. The sum of these factors may contribute to defining matched double asymmetric reactions for the synthesis of either 1,4-anti-1,5-syn (34) or 1,4-syn - 1, 5 - anti (36) aminoalcohols via transition state geometries G and H.7,8 Based on this empirical model, the mismatched double asymmetric reactions (Table 3, entries 2 and 4) may suffer from the requirement of placing the alkyl substituent (R) on the inside of the developing cis-fused ring system (I, J; Figure 4).
Figure 4.

A model for stereoselection in the double asymmetric cross coupling reactions of disubstituted olefins with chiral imines.
These double asymmetric cross coupling reactions proceed with a great deal of variability in stereoselection that is dependent on the stereochemical relationship of the secondary alcohol, imine and olefin. Interestingly, matched double asymmetric reactions can provide stereoselective access to either the 1,4-anti-1,5-syn-1,5-aminoalcohol or 1,4-syn-1,5-anti-1,5-aminoalcohol products, and the corresponding trisubstituted piperidines 35 and 37. The variability in the diastereomeric ratio of products observed in each of these matched double asymmetric coupling reactions is consistent with a stereoreinforcing12 relationship based on olefin geometry: (S,Z)-homoallylic alcohol + ent-28 is matched and benefits from a stereoreinforcing relationship with the (Z)-olefin, whereas (S,E)-homoallylic alcohol + 28 is also a matched double asymmetric reaction but there is a non-stereoreinforcing relationship with the (E)-olefin.
Overall, we report a titanium alkoxide-mediated reaction for regio- and stereoselective cross coupling of substituted olefins and imines. The reported process provides convenient access to substituted 1,5-aminoalcohols and piperidines. Our studies focusing on simple diastereoselection have shown that this reaction is stereoconvergent with respect to olefin stereochemistry – both (E) and (Z) disubstituted homoallylic alcohols provide anti-products in coupling reactions with achiral imines. We have studied this reaction in both single and double asymmetric modes, have defined matched and mismatched double asymmetric relationships, and have shown a reversal of stereochemical matching/mismatching as a function of the olefin geometry of the chiral homoallylic alcohol. Research focused on further understanding the nature of stereoselection in this cross coupling reaction, as well as application to the synthesis of complex heterocycles is underway.
Supplementary Material
Experimental procedures and tabulated spectroscopic data for new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We gratefully acknowledge financial support of this work by the American Cancer Society (RSG-06-117-01), the American Chemical Society (PRF-45334-G1), the Arnold and Mabel Beckman Foundation, Boehringer Ingelheim, Eli Lilly & Co., the National Institutes of Health –NIGMS (GM80266) and Yale University.
References
- 1.For cross coupling of imines and alkynes, see: (promoted by Zr): Buchwald SL, Watson BT, Wannamaker MW, Dewan JC. J Am Chem Soc. 1989;111:4486–4494.Jensen M, Livinghouse T. J Am Chem Soc. 1989;111:4495–4496.Brossman RB, Davis WM, Buchwald SL. J Am Chem Soc. 1991;113:2321–2322. promoted by Ti.Gao Y, Harada K, Sato F. Tetrahedron Lett. 1995;36:5913–5916. catalyzed by Ni.Patel SJ, Jamison TF. Angew Chem Int Ed. 2003;42:1364–1367. doi: 10.1002/anie.200390349.Patel SJ, Jamison TF. Angew Chem Int Ed. 2004;43:3941–3944. doi: 10.1002/anie.200460044. coupling of conjugated alkynes with N-sulfinyl-iminoacetates catalyzed by Rh)Kong JR, Cho CW, Krische MJ. J Am Chem Soc. 2005;127:11269–11276. doi: 10.1021/ja051104i.
- 2.For cross coupling of zirconium-imine complexes and terminal olefins, see 1(a).
- 3.Ryan J, Micalizio GC. J Am Chem Soc. 2006;128:2764–2764. doi: 10.1021/ja057352w. [DOI] [PubMed] [Google Scholar]
- 4.Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440–1443. doi: 10.1002/anie.200603515. [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin M, Takahashi M, Micalizio GC. Angew Chem Int Ed. 2007;46 doi: 10.1002/anie.200605060. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For an example of a related PPh3, CCl4-promoted cyclodehydration, see: Song YC, Okamoto S, Sato F. Tetrahedron Lett. 2002;43:8635–8637.
- 7.The stereochemistry of the C–Ti bond in the proposed bicyclic azametallacyclopentanes is not known.
- 8.The coordination number of titanium in the transition states for these carbometalation reactions is not known. The stereoselection observed can be accounted for by assuming a transition state structure for the generation of a cis-fused bicyclo[3.3.0]-metallacycle. Proposition of an azametallacyclopentane intermediate is in accord with well-known reactivity patterns of azametallacyclopropanes.
- 9.For a nickel-catalyzed coupling of substituted 1,3-dienes to imines for the synthesis of related 1,3-syn-functionalized products (i.e. 25), see: Kimura M, Miyachi A, Kojima K, Tanaka S, Tamaru Y. J Am Chem Soc. 2004;126:14360–14361. doi: 10.1021/ja0450354.
- 10.For examples of stereoselective alkyne–imine cross coupling reactions with chiral imine 28, see: Fukuhara K, Okamoto S, Sato F. Org Lett. 2003;5:2145–2148. doi: 10.1021/ol034599u. (b) ref. 5
- 11.Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem Int Ed Engl. 1985;24:1–30. For an example of triple asymmetric synthesis, see Duplantier AJ, Nantz MH, Roberts JC, Short RP, Somfai P, Masamune S. Tetrahedron Lett. 1989;30:7357–7360.
- 12.Evans DA, Dart MJ, Duffy JL, Yang MG. J Am Chem Soc. 1996;118:4322–4343. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and tabulated spectroscopic data for new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.