

Abstract
The transcription factor T-bet (Tbx21) is critical for Th1 polarization of CD4+ T cells. Genetic deletion of Tbx21 can cause either exacerbation or attenuation of different autoimmune diseases in animal models. In the non-obese diabetic (NOD) mouse, genetic deletion of the Ifng or the Il12b (IL12p40) genes, which are both critical Th1 cytokines, does not reduce the incidence of autoimmune diabetes. These results suggest that autoimmune diabetes in the NOD may not be a Th1-driven disease. However, we report that Tbx21 deficiency in the NOD mouse completely blocks insulitis and diabetes due to defects both in the initiation of the anti-islet immune response and in the function of CD4+ effector T cells. We find defective priming of naive islet-reactive T cells by the innate immune system in Tbx21−/− animals. By contrast to naive cells, activated islet-reactive BDC2.5 TCR transgenic T cells do not require Tbx21 in recipient animals for efficient adoptive transfer of diabetes. However, when these BDC2.5 TCR transgenic effector cells lack Tbx21, they are less effective at entering the pancreas and promoting diabetes than Tbx21+/+ cells. Tbx21−/− regulatory T cells function normally in-vitro and diabetes can be restored in Tbx21−/− mice by reducing Treg numbers. Thus, the absence of diabetes in the NOD.Tbx21−/− is due to intrinsic defects in both T cells and cells of the innate immune system paired with the relative preservation of regulatory T cell function.
Keywords: T cells, diabetes, transcription factors
Introduction
Germline deletion of the Th1-lineage transcription factor T-bet (Tbx21) can alternatively exacerbate or attenuate autoimmune and inflammatory diseases in animal models. Tbx21 null animals have attenuated clinical symptoms of autoimmune experimental encephalomyelitis (1) and CD4+ Tbx21 null cells do not cause colitis when transferred into SCID or Rag-deficient mice (2). Tbx21 also plays a role in CD8+ T cell-driven disease. For example, ovalbumin-specific Tbx21-deficient CD8+ OT-I cells have intrinsic defects in tissue homing and cytotoxicity in a mouse model of myocarditis (3). Importantly, Tbx21 null animals with a transgene expressing an LCMV protein in pancreatic islets are partially protected from diabetes when infected with LCMV due to defects in the generation of antiviral CD8+ T cells (4). In collagen antibody-induced arthritis in mice, Tbx21 expression in dendritic cells was necessary to drive the disease in the absence of an adaptive immune response (5). Tbx21deficiency in B cells reduces autoantibody titers and renal immune complex deposition in Fas deficient mice (6).
By contrast to these examples of attenuated disease, several disease models show exacerbated disease in the absence of Tbx21. Tbx21-deficient animals immunized with heart myosin develop exacerbated autoimmune myocarditis compared to controls due to increased IL-17 production by effector T cells (7). Tbx21 null animals are more susceptible than wildtype animals to Th2-mediated diseases such as airway inflammation similar to human asthma (8) and bleomycin-induced skin sclerosis (9). Some strains of Tbx21-deficient mice that also lack an adaptive immune system due to Rag2 gene deficiency develop spontaneous colitis due to dysregulated cytokine production in the gut mucosa (10). These examples show that Tbx21 is important for the function of lymphocytes and non-lymphocytes in disease models and that the effects of Tbx21 deficiency on a particular disease model are difficult to predict.
This complexity in different disease models is explained in part by the many different functions of Tbx21 that have been described in lymphocytes and dendritic cells. CD8+ T cells that lack Tbx21 can produce IFN-γ in vitro (11), likely due to the expression of the Tbx21 paralogue Eomesodermin (12). However, the functions of Tbx21 and Eomesodermin overlap only partially, since Tbx21 null CD8+ T cells showed reduced IFN-γ production in mice infected with T. gondii (13). Tbx21 in dendritic cells promotes IFN-γ production and is necessary for effective in-vivo priming of antigen-specific T cells by DCs (14). Deficiency of Tbx21 in B cells skews class switching away from IgG2a (6). Conversely, upregulation of Tbx21 in cultured B cells is associated with decreased class switching to IgE and IgG1 (15). NK cells require Tbx21 for control of tumor metastasis in mice inoculated with a melanoma cell line (16). NK cells that lack Tbx21 have intrinsic cytotoxicity defects and survive poorly compared to Tbx21+/+ NK cells in vivo and in vitro (16). In sum, Tbx21 controls a wide range of Th1-related cellular phenotypes in many cell types of both the adaptive and the innate immune system.
There is evidence that polymorphisms in Th1-related genes contribute to risk of type 1 diabetes mellitus (T1DM)2 in humans. A polymorphism of Tbx21 that increases transcription from the IFN-γ promoter has been implicated as a risk gene in human T1DM in a Japanese study population (17). However, the region of human chromosome 17 that contains Tbx21 has not been implicated as a risk region for T1DM in a recent genome-wide association study (18). Separately, a polymorphism of the IL-12p40 gene has been linked to the risk of T1DM in humans (19).
Germline deletion of the Ifng gene or the IFN-γ receptor gene has been reported to delay only slightly the onset of diabetes in the NOD mouse (20–23). Since Tbx21 drives IFN-γ production in both CD4+ T cells (11) and dendritic cells (14), we sought to test whether Tbx21 was required for spontaneous autoimmune diabetes by backcrossing the Tbx21 null mouse to the NOD. Our results show that loss of Tbx21 completely blocks diabetes in NOD mice. The NOD.Tbx21−/− mice are protected from insulitis and show defects in both effector T cell function and in innate immune cell function. Regulatory T cells (Tregs) in NOD.Tbx21−/− animals function normally in in-vitro and in-vivo assays, suggesting that the balance of effector versus regulatory function of T cells is tipped toward regulation in these animals. Our results suggest that spontaneous diabetes in the NOD mouse requires a Tbx21-dependent Th1 response and that Tbx21 impacts disease pathogenesis in multiple cell types. A role for the Th1 T cell subset in diabetes in the NOD mouse was previously uncertain (22). Thus, our results highlight the importance of the Th1 effector function for diabetes in the NOD mouse.
Materials and Methods
Mice
Mice with the Tbx21tm1Glm allele were backcrossed to NOD/MrkTac mice purchased from Taconic farms. NOD.BDC2.5 TCR-transgenic, NOD.Cg-Rag2tm1Fwa/JbsJ, and NOD.129S2(B6)-Cd28tm1Mak/JbsJ mice were bred and housed in a specific pathogen-free barrier facility at the University of California, San Francisco. A scan of SNPs across the genome of the NOD.Tbx21−/− mouse revealed that all chromosomes were NOD-derived except for the telomeric end of chromosome 11, which contains Tbx21 (Supplemental tables 1 and 23). Diabetes incidence was followed by periodic checks for elevated urine glucose levels using Diastix strips (Bayer Corp., Elkhart IN) and confirmed with blood glucose measurements of >250 mg/dL on at least two separate days using an Accu-Chek glucose meter (Roche). All animal experiments were approved by the University of California, San Francisco, Animal Care and Use Committee.
Histopathology
Pancreata were harvested and immediately fixed in 10% buffered formalin, embedded in paraffin, sectioned, and stained with H&E. Scoring of insulitis was done blinded to the genotype of the mouse, according to the protocol found in (24).
Cell culture and adoptive transfer
For activation assays, CD4+ T cells from NOD and NOD.Tbx21−/− and +/− mice were purified from bulk lymph node cells with a RoboSep (StemCell Technologies, Vancouver BC) and the StemCell mouse CD4+ T cell enrichment kit or with an AutoMACS (Miltenyi Bitech, Auburn CA). Typical purities were >90% CD4+. For adoptive transfer experiments, CD4+CD25+ cells were removed by incubating cells for 20 minutes in supernatant from the 7D4 hybridoma (IgM anti-CD25) followed by incubation with rabbit complement for 30 minutes at 37°C. For in vitro activation experiments, cells were resuspended in DMEM supplemented with 10% FBS, glutamine, HEPES buffer, β-mercaptoethanol, non-essential amino acids, and antibiotics. The cells were activated in plates pre-coated with 1 μg/mL anti-CD3 (clone 145-2C11) and 1 μg/mL anti-CD28 (clone PV1). For Th1 skewing, the medium was supplemented with 100 U/mL recombinant human IL-2, 20 ng/mL recombinant mouse IL-12, and 20 ug/mL anti-IL-4 (clone 11B.11). For adoptive transfer experiments with naive T cells, CD4+ cells were isolated as described above and injected into recipient mice via the tail vein. For adoptive transfer experiments with activated BDC2.5 transgenic cells, the cells were isolated as described above and activated by mixing together the mimotope peptide p31 (25) with T-depleted, irradiated splenocytes from non-diabetic NOD mice. Activation was done in supplemented DMEM as described above without skewing cytokines. Cells were collected 4 days after activation and adoptively transferred into mice via tail vein injection.
RNA isolation and realtime PCR
RNA from primary mouse T cells was extracted using the RNAeasy kit (Qiagen, Valencia CA) using the manufacturer's recommended protocol. Reverse transcription was done with a SuperScriptIII reverse transcriptase kit (Invitrogen, Carlsbad CA) using random DNA hexamers as primers, according to the manufacturer's instructions. Realtime PCR was performed on a 7500 Fast Realtime PCR System (Applied Biosystems, Foster City CA) using Taqman primer probes Mm99999054_s1 (Cxcr3) and Mm00801778_m1 (Ifng) with a Taqman primer-probe set for the eukaryotic 18S rRNA as an internal control. cDNA was diluted 10,000-fold for assay of 18S rRNA levels. Both the Cxcr3 and Ifng primer-probes were validated before use by titration against the 18S rRNA primer-probe using lymphocyte cDNA and the amplification efficiency of the individual primer-probes was calculated. Calculation of relative expression was done using the ΔΔCt method, according to the recommendations from Applied Biosystems.
Flow cytometry
Intracellular staining for FOXP3 was carried out with the eBioscience FOXP3 staining kit, according to the manufacturer's instructions. For CXCR3 surface standing, spleens were harvested, disrupted, and the splenocytes were incubated for 10 minutes in supernatant from the 2.4G2 hybridoma to block non-specific binding of IgG to splenocytes. Primary stain was done for 30 minutes with purified goat anti-mouse CXCR3, SC-9902 (Santa Cruz Biotechnology, Santa Cruz CA) at 1:5 dilution. For control stains, this primary Ab was omitted. Cells were washed and incubated with donkey anti-goat biotin F(ab′)2 (Jackson Immunoresearch, West Grove PA) at 1:5 dilution for 30 minutes. Cells were washed again and streptavidin PE (BD Biosciences, San Jose CA) was added at 1:350 dilution for 30 minutes. BDC TCR surface staining was done with a purified anti-BDC clonotype Ab produced from the original hybridoma (26). The secondary Ab was 1:2,000 diluted rat anti-mouse IgG2b-biotin (BD Biotechnologies, San Jose, CA), which was detected with 1:5,000 diluted streptavidin-PE (BD Biotechnologies, San Jose, CA). Cells were washed between all incubation steps. All cytometry data was collected on a BD Biosciences FACSCalibur or LSRII machine.
CFSE labeling
CD4+CD25− T cells were resuspended at 20×106 cells/mL in PBS. CFSE at a stock concentration of 10 mM (Invitrogen/Molecular Probes, Carlsbad CA) was diluted 1:2,000 in PBS and this dilution was mixed with an equal volume of cells. After 5 minutes, the reaction was quenched with FBS and the cells were collected for adoptive transfer.
Treg isolation and expansion
Treg cells were sorted from bulk lymph node cells of NOD and NOD.Tbx21−/− animals. Suppression assays in 96-well plates were set up as previously described (27). BDC2.5 TCR transgenic Tregs were FACS purified and expanded in-vitro as previously described before adoptive transfer of 3×106 cells into NOD.CD28−/− mice (27).
Mouse genome SNP scan
A scan of SNPs (single nucleotide polymorphisms) across the genome of the NOD.Tbx21−/− mice was done by the UT Southwestern Medical Center Microarray Core Facility using a Mouse Medium Density Linkage Panel (Illumina, San Diego, CA).
Results
Tbx21−/− mice are protected from diabetes
The Tbx21−/− mouse was backcrossed to NOD for 10 generations. A scan of SNPs throughout the genome of the NOD.Tbx21−/− mouse revealed that all chromosomes were NOD-derived except for part of chromosome 11, which contains the null allele of Tbx21 (Supplemental tables 1 and 2). To determine diabetes incidence in these animals, female offspring of NOD.Tbx21+/− parents were followed for diabetes incidence for 30 weeks. Both NOD.Tbx21−/− and NOD.Tbx21+/− animals were completely protected from disease (Figure 1A). The NOD.Tbx21+/+ littermates became diabetic with normal kinetics for our colony, with 56% of female mice diabetic by 30 weeks of age. Histological analysis of pancreata showed that 12-week old NOD.Tbx21−/− females are protected from insulitis (Figure 1B). Heterozygous mice show partial protection from insulitis, indicating that gene dosage of Tbx21 affects progression to insulitis.
FIGURE 1.

Tbx21 deficiency protects NOD mice from insulitis and diabetes. A, Female NOD.Tbx21−/− mice and their +/− and +/+ littermates were followed for diabetes for 30 weeks. B, Mice from the above cross were sacrificed for histopathological analysis of the pancreas at 12 weeks of age. Grade 0, no insulitis; grade 1, perivascular/periductal mononuclear cell infiltration outside of islet perimeter; grade 2, mononuclear cell penetration of up to 25% of islet; grade 3, mononuclear cell penetration of up to 75% of islet; grade 4, islet destruction with less than 20% of islet mass remaining. Results are from at least 100 islets from 4 animals per genotype.
Priming of diabetogenic T cells is defective in Tbx21−/− mice
Since defects in APC function have been described in Tbx21−/− mice (14), we tested initial priming of diabetogenic T cells in Tbx21−/− hosts. Naive CD4+CD25− BDC2.5 TCR transgenic cells, which have a TCR specific for an islet antigen in the context of I-Ag7 (28), were labeled with CFSE and adoptively transferred into NOD or NOD.Tbx21−/− recipients. After three days, the pancreatic lymph nodes of the recipient mice were harvested and analyzed by flow cytometry. The percentage of Tbx21+/+ BDC2.5 TCR transgenic T cells that went into cycle in the pancreatic lymph nodes of the recipient animals was reduced by about half in the Tbx21−/− recipients (Figure 2A). Deficiency of Tbx21 in the T cells did not affect proliferation when the recipients were NOD.Tbx21+/+. Interestingly, Tbx21 deficient cells proliferated better in NOD.Tbx21−/− hosts compared to NOD.Tbx21+/+ cells in NOD.Tbx21−/− hosts (p<0.01), suggesting that NOD.Tbx21+/+ T cells may have a greater requirement for Tbx21 in antigen-presenting cells than NOD.Tbx21−/− T cells. In sum, the expression of Tbx21 in the recipient animals was critical for optimal priming of diabetogenic T cells.
FIGURE 2.

Tbx21 is necessary in both T cells and APCs in an adoptive transfer model of diabetes. A, CD4+CD25− BDC2.5 TCR transgenic T cells from Tbx21−/− and Tbx21+/+ mice were labeled with CFSE. The cells were collected, recounted, and 1×106 were adoptively transferred into NOD or NOD.Tbx21−/− recipients. After 72 hours, pancreatic lymph nodes from the recipient mice were harvested and bulk lymph node cells were stained for CD4 and the BDC2.5 TCR. Each data point is an individual mouse, and the line is the mean of all mice in a group. ** indicates a p-value of <0.01 using a 2-tailed unpaired T test. n.s. indicates that p>0.05. B, 1×106 or 5×103 freshly isolated CD4+CD25− BDC2.5 transgenic T cells from Tbx21+/+ mice were adoptively transferred into NOD.Rag2−/−.Tbx21−/− or NOD.Rag2−/−.Tbx21+/+ mice. The mice were followed for diabetes incidence. Results are from two or three independent experiments. C, 1×106 or 5×103 freshly isolated CD4+CD25− BDC2.5 transgenic T cells from Tbx21−/− mice were adoptively transferred into NOD.Rag2−/−.Tbx21−/− or NOD.Rag2−/−.Tbx21+/+ mice. The mice were followed for diabetes incidence. Results are from three independent experiments.
To test for a functional role of Tbx21 in APCs that prime autoreactive diabetogenic T cells, low numbers of naive CD4+CD25−Tbx21+/+ BDC2.5 TCR transgenic cells were transferred into Rag2−/− animals. At a dose of 5×103 BDC2.5 transgenic cells per mouse, all of the Tbx21+/+ recipients but only 3 of 12 Tbx21−/− recipients had become diabetic by 90 days after adoptive transfer. At a high dose of 1×106 cells per animal, all recipients became diabetic between 11 and 21 days after adoptive transfer, regardless of the Tbx21 genotype of the recipient animal (Figure 2B). These results suggest that there is a defect in T cell priming in Tbx21−/− mice after adoptive transfer of a low number of autoreactive T cells. However, this defect can be overcome with the transfer of higher cell numbers. A statistically significant difference (p=0.03) was found between the percentage of CD4+ cells in the pancreatic lymph nodes of Tbx21+/+ and Tbx21−/− recipient mice 11 days after adoptive transfer. In the Tbx21+/+ recipients, 20% (range 14.8% to 23%, n=3) of cells were CD4+, compared to 11.1% (range 10.5% to 12.1%, n=3) of cells in the Tbx21−/− recipients. Together, these results suggest that Tbx21 is necessary in the innate immune system for efficient priming and proliferation of diabetogenic T cells.
Effector T cells require Tbx21 for efficient disease transfer
Different numbers of naive CD4+CD25− Tbx21−/− BDC2.5 transgenic T cells were transferred into Rag2−/− recipients to test for the capacity of the Tbx21-deficient cells to cause diabetes. When 1×106 Tbx21−/− cells were transferred, all Rag2−/−.Tbx21+/+ and Rag2−/−.Tbx21−/− animals became diabetic with rapid kinetics (Figure 2C). However, when only 5×103 cells were transferred, the disease incidence was decreased to 8% (Tbx21−/− cells into Rag2−/−.Tbx21+/+ hosts, n=12) and 20% (Tbx21−/− cells into Rag2−/−.Tbx21−/− hosts, n=11), by 50 days after adoptive transfer. By comparison, when 5×103 naive CD4+CD25−Tbx21+/+ BDC2.5 transgenic T cells were transferred into Rag−/−.Tbx21+/+ animals, 100% of mice became diabetic within 40 days of adoptive transfer (Figure 2B). These results show a clear defect in the ability of Tbx21 deficient T cells to cause diabetes when the number of diabetogenic cells is limited in this system.
In the experiments described above, differences in initial priming and homeostatic expansion could account for the observed defects in Tbx21-deficient T cells. We sought to test for a cell-intrinsic defect in the Tbx21−/− T cells unrelated to these processes. Lymph nodes were harvested from BDC2.5 TCR transgenic NOD or NOD.Tbx21−/− mice and CD4+ cells were purified by magnetic bead selection. These cells were then cultured with T-depleted irradiated splenocytes from NOD mice in the presence of the peptide mimetope Acp31. After four days, no difference in T cell proliferation (Figure 3A) or CD25 expression (data not shown) were detected between the −/− and +/+ transgenic cells. Adoptive transfer of 1×106 of these activated cells into NOD or NOD.Tbx21−/− animals revealed that activated Tbx21−/− T cells induce diabetes at lower rates and with slower kinetics than activated Tbx21+/+ cells (Figure 3B). These results show that Tbx21 is required in the activated transgenic T cells but not in the recipient animals for robust diabetes transfer. These results could be explained by a defect in tissue trafficking in the activated Tbx21−/− cells or by some other intrinsic defect in effector function. To distinguish between these possibilities, we harvested pancreata from pre-diabetic animals from the above experiment at day 5 post-transfer for histological scoring of islet infiltration. This analysis showed that the animals transferred with activated Tbx21−/− cells had infiltration in fewer than 50% of their islets (Figure 3C). By contrast, animals transferred with activated Tbx21+/+ BDC2.5 T cells showed complete destruction of more than 75% of their islets, with few islets free of mononuclear infiltration.
FIGURE 3.

Defective adoptive transfer of diabetes with activated Tbx21−/− BDC2.5 TCR transgenic T cells. A, CD4+CD25− BDC2.5 transgenic T cells from Tbx21−/− and Tbx21+/+ mice were activated in-vitro using irradiated T-depleted NOD splenocytes and the Acp31 mimeotope peptide. Cells in culture were counted before initial activation and again immediately before adoptive transfer. B, 1×106 of these activated BDC2.5 TCR transgenic cells were transferred into age-matched non-diabetic NOD or NOD.Tbx21−/− animals, which were followed for diabetes. Survival of Tbx21−/− and Tbx21+/+ recipients transferred with Tbx21+/+ BDC2.5 T cells is not significantly different (log rank test, p=0.10). C, Alternatively, some of the adoptively-transferred mice were sacrificed at day 5 post-transfer for histopathological analysis of pancreatic islet inflammation. Results are from two independent experiments.
In a study of autoimmune myocarditis induced by activated Tbx21−/− transgenic T cells, it was found that these cells become sequestered in the heart draining lymph node after adoptive transfer, likely due to a defect in CXCR3 expression (3). However, TCR-specific staining for activated BDC2.5 TCR+ T cells revealed no significant differences in the percentage of transgenic T cells among the total CD4+ population of the pancreatic lymph nodes of recipients of activated BDC2.5 T cells 5 days after adoptive transfer (Figure 4A). To test if differences CXCR3 expression could play a role in the efficacy of BDC2.5 T cell transfers, Cxcr3 mRNA was measured by quantitative PCR in the activated BDC2.5 TCR+ T cells used for the adoptive transfer experiments described above. Ifng expression was decreased 100-fold in Tbx21-deficient T cells. In contrast, Cxcr3 mRNA expression in Tbx21−/− cultures was decreased to 25% of the levels in Tbx21+/+ cultures (Figure 4B). Thus, the activated Tbx21−/− T cells appeared to enter the pancreas in lower numbers than Tbx21+/+ T cells although they were not sequestered in the pancreatic lymph nodes despite lower levels of CXCR3 expression. Tbx21+/− CD4+ T cells activated under Th1 skewing conditions showed an intermediate phenotype, with Ifng mRNA and protein expression decreased by half and Cxcr3 mRNA expression levels between 80 and 96% of Tbx21+/+ (Supplemental figure 1A and B). These results are consistent with previous reports showing that a single copy of Tbx21 is not sufficient to drive normal immune cell effector function (8, 29). The decrease in Cxcr3 expression in Tbx21−/− cells in the experiments described above was modest as compared to the observed 30-fold decrease in Cxcr3 mRNA previously reported in CD4+ cells activated in a Th0 culture (30) and even larger decreases reported in other settings (3). In fact, surface staining revealed only a modestly lower level of CXCR3 protein expression on freshly isolated Tbx21−/− CD4+ splenocytes as compared to Tbx21+/+ controls (Figure 4C). Although this difference was statistically significant in CD4+ splenocytes, there was no difference in CXCR3 protein expression between Tbx21+/+ and −/− CD8+ splenocytes (Figure 4D).
FIGURE 4.
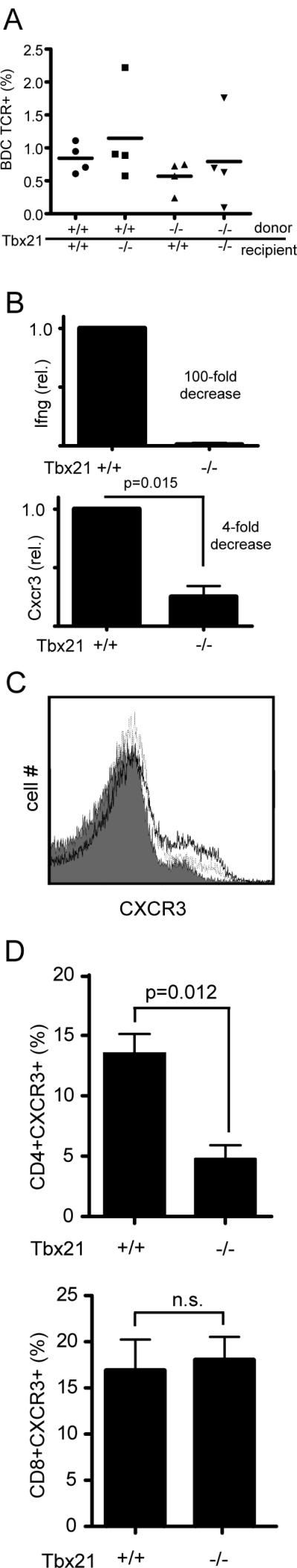
Tbx21 deficient BDC2.5 TCR transgenic T cells are defective in IFN-γ production and Cxcr3 expression. A, Pancreatic lymph nodes were harvested from mice mentioned in Figure 3C at day 5 after adoptive transfer of activated BDC2.5 TCR transgenic T cells. Lymph node cells were stained for the BDC2.5 TCR. Percentages indicated are out of total CD4+ cells. B, CD4+CD25− BDC2.5 TCR transgenic T cells from Tbx21−/− and Tbx21+/+ mice were activated in-vitro using irradiated T-depleted NOD splenocytes and the Acp31 mimeotope peptide. Total cellular RNA was collected 4 days after activation and assayed by realtime PCR for levels of Ifng and Cxcr3 mRNA transcript. Expression of each gene in Tbx21+/+ samples was indexed to 1. Data was pooled from three independent experiments and analyzed with a one-sample T test. C, Freshly isolated CD4+ splenocytes were stained using a polyclonal Ab for cell surface expression of CXCR3 protein and measured by flow cytometry. Gray tracing is no primary Ab; dotted line is Tbx21−/− splenocytes; and dark line is Tbx21+/+ splenocytes. Data is representative of three experiments. D, Quantitative analysis of CXCR3+ cells in freshly isolated splenocytes in the total CD4+ population (top panel) and the CD8+ population (bottom panel). Data was pooled from three independent experiments and analyzed with a 2-tailed upaired T test. Error bars represent the standard error of the mean.
Regulatory T cells contribute to protection of Tbx21-deficient mice from diabetes
CD4+FOXP3+ Treg function is critical for controlling the balance between effector function and regulation in diabetes in the NOD mouse (31). There was no difference in the percentage of FOXP3+ cells among total CD4+ cells in the spleen or pancreatic lymph nodes of 6-week old pre-diabetic NOD and NOD.Tbx21−/− mice (Figure 5A). As shown in Figure 5B, Tbx21−/− Tregs were able to suppress Tbx21+/+ effector cell proliferation with the same efficiency as Tbx21+/+ Tregs. Given the defects in effector function described above, we sought to test whether the dominant regulatory role of FOXP3+ cells was responsible for preventing diabetes in the NOD.Tbx21−/− animal. To test this hypothesis, the NOD.Tbx21−/− mice were crossed onto a NOD.CD28−/− background. The NOD.CD28−/−mouse exhibits rapid, synchronized diabetes onset at about 12 weeks of age due to a defect in Treg survival and function (31). In contrast to the NOD.Tbx21−/− mice, the NOD.Tbx21−/−.CD28−/− animals showed diabetes onset starting from 9 weeks of age. Overall incidence of diabetes in this genotype increased until about 20 weeks of age before reaching a plateau at 72% (Figure 6A). This data suggests that Tbx21−/− Tregs are necessary in-vivo to prevent diabetes. To more rigorously test this hypothesis, NOD.CD28−/− mice were adoptively transferred with in-vitro expanded NOD.BDC2.5 Tregs that were either Tbx21−/− or +/+. The recipient mice were then followed for spontaneous diabetes incidence (Figure 6B). Protection of CD28−/− mice from diabetes with Tbx21−/− Tregs was statistically significant (log rank test, p<0.05). There was no statistical difference between the groups treated with Tbx21+/+ and Tbx21−/− Tregs (log rank test, p=0.16). Lastly, we bred NOD.Tbx21−/− mice that expressed the BDC2.5 transgene and were also Rag2 deficient. These animals showed rapid diabetes onset at or soon after weaning (Figure 6C), similar to published data on this genotype in the context of Tbx21 sufficiency (32). By contrast, Rag+/+ animals were protected from early onset of diabetes (log rank test, p<0.01). These results point to a critical role for regulatory T cells in controlling diabetes in Tbx21-deficient animals, since expression of endogenously rearranged TCRα chains is necessary for the generation of BDC2.5 Tregs.
FIGURE 5.

Tbx21-deficient mice have normal numbers of functional Tregs. A, Splenocytes and pancreatic lymph node cells from NOD and NOD.Tbx21−/− mice were stained for CD4 and FOXP3. Each data point represents an individual mouse. Groups were compared with an unpaired two-tailed T test. n.s. is not statistically significant. B, CD4+CD25+CD62L+ regulatory T cells were purified by FACS and titrated in a suppression assay with CD4+CD25− responder cells. Responders were activated with soluble anti-CD3 and irradiated syngeneic splenocytes. A 1:1 ratio of Tregs to responders has 5×104 of each cell type. Data is representative of three independent experiments. Error bars are standard error of the mean.
FIGURE 6.

Tregs are required to prevent diabetes in Tbx21−/− mice. A, NOD.Tbx21−/−.CD28−/− mice were followed for spontaneous diabetes incidence. B, NOD.CD28−/− mice were treated at 7 weeks of age with 3×105 in-vitro expanded Tregs from either BDC2.5 Tbx21+/+ or BDC2.5 Tbx21−/− mice, or with saline as a control. The mice were then followed for diabetes incidence. The survival curve of mice that received Tbx21−/− Tregs is significantly different from the survival curve of the mice that did not receive any Tregs (log rank test, p<0.01). C, NOD.BDC2.5.Rag2−/−.Tbx21−/− mice were followed for diabetes incidence.
Discussion
In this paper, we show that the Tbx21 transcription factor, which is a critical regulator of Th1 responses, is necessary for diabetes in the NOD mouse. We have found defects in both the innate and adaptive immune systems in the NOD.Tbx21−/− mouse. Cells of the innate immune system in Tbx21−/− mice are defective in driving activation and cycling of adoptively transferred naive BDC2.5 TCR transgenic T cells. This cycling defect was paralleled by a defect in diabetes transfer by naive islet-reactive cells into Rag2−/−.Tbx21−/−mice. The observed failure of small numbers of anti-islet Ag-specific transgenic T cells to cause disease could be overcome with high numbers of cells. This result suggests that efficient in-vivo proliferation, which is absent in Tbx21−/− recipients, is critical to generate large enough numbers of islet-reactive cells to cause disease. The defects in initial T cell priming and proliferation helps to explain why the NOD.Tbx21−/− animals fail to develop insulitis, which is normally completely penetrant in the NOD mouse (34). Further work with a conditional allele of Tbx21 will more clearly elucidate the role of this transcription factor in cells of the innate immune system.
In addition to defects in the innate immune system, there are also profound defects in diabetogenic CD4+ T cells that lack Tbx21. Activated NOD.BDC2.5 TCR transgenic T cells that lack Tbx21 fail to enter the pancreas and cause islet destruction similar to Tbx21+/+ cells. The Tbx21−/− cells were not sequestered in the target organ draining lymph nodes, a result reported in another adoptive transfer model using transgenic Tbx21−/− cells (3). Expression of Cxcr3 mRNA is decreased in Tbx21−/− BDC2.5 TCR transgenic T cells activated in vitro compared to Tbx21+/+ cells. However, this decrease is much more modest (25% of WT) than that described by others in Tbx21−/− cells (3, 30). By contrast, IFN-γ expression is completely dependent upon Tbx21 expression. This modest difference in CXCR3 expression implies that the trafficking defects in Tbx21−/− T cells may be due to other factors besides decreased CXCR3 expression. We speculate that Tbx21 is important in the innate immune system in the early initiating stages of autoimmune diabetes and that the T cell defects become relevant at later stages of pathogenesis after the generation of activated effector cells.
The absence of diabetes in the NOD.Tbx21−/− mouse is unexpected. Although there is some evidence that Th1 cytokines such as IL-12 help to promote diabetes in the NOD (35, 36), both the IL-12 knockout (37) and the IFN-γ knockout (20) NOD mice show robust progression to diabetes. These results have led to uncertainty about the importance of IFN-γ-producing Th1 T cells in the pathogenesis of diabetes in the NOD. Importantly, a recent report argues that IFN-γ can protect against diabetes by decreasing a pathogenic anti-islet Th17 response (38). These results raise the prospect that diabetes in the NOD is a Th17-driven disease. This hypothesis is unlikely for two reasons. First, Stat4−/− mice on the NOD background are mostly but not completely protected from diabetes (39, 40). Stat4 is critical for IL-12 receptor signaling (41) and its functions in driving gene expression during Th1 differentiation only partially overlap with those of Tbx21 (42). Second, we have found that the phenotype of the NOD.Tbx21−/− mouse is even stronger than the NOD.Stat4−/− since none of the Tbx21−/− NOD mice develop diabetes or exhibit insulitis. This profound block in diabetes and insulitis shows that the effects of Tbx21 in diabetes extended beyond driving IFN-γ production. Indeed, many other reports have shown that Tbx21 is necessary not only for the production of IFN-γ or IL-12, but also for a wide range of phenotypes including lymphocyte trafficking (30), cytotoxic activity of T cells (43), and priming of T cells by dendritic cells (14).
The 129S6/SvEvTac-derived genetic region surrounding the null Tbx21 allele in the NOD.Tbx21−/− mouse extends for up to 15 Mb, raising the possibility that 129-derived genes other than Tbx21 could contribute to the protection of the NOD.Tbx21−/− mouse from diabetes. Importantly, chromosome 11 contains Idd4 (44–46), a region initially derived from B10.H-2g7 mice that partially protects NOD mice from developing diabetes. In these studies, mice heterozygous for the B10-derived portion of chromosome 11 had rates of diabetes of about half of the homozygous controls and similar amounts of insulitis (45). Several other non-NOD regions spread along the length of mouse chromosome 11 have been associated with protection from diabetes, but none of the NOD.Idd4 congenic mice in the published literature show a complete absence of diabetes as in the Tbx21+/− and Tbx21−/− mice (47–49). The region of chromosome 11 defined as Idd4 by the Jackson Laboratory is completely NOD-derived in the NOD.Tbx21−/− mouse (Supplemental table 1). Thus, although we cannot exclude the possibility that some part of the observed phenotype of the NOD.Tbx21−/− mouse is due to 129-derived genes linked to Tbx21, the genetic ablation of Tbx21 is very likely to be critical for a large part of the observed phenotype of the mouse.
Regulatory T cells play a role in controlling diabetes in the NOD.Tbx21−/− mouse. Deficiency of CD28 in the NOD mouse has been shown to decrease the number of Tregs and increase the kinetics and penetrance of diabetes (50). Strikingly, the NOD.Tbx21−/−.CD28−/− mouse shows robust progression to diabetes, implying that Tbx21−/− Tregs are necessary to prevent disease in this animal. To directly test whether Tbx21−/− Tregs are capable of preventing diabetes in-vivo, we adoptively transferred in-vitro expanded BDC2.5 Tregs that were either Tbx21+/+ or −/− into NOD.CD28−/− mice. The Tbx21−/− Tregs are competent to prevent or delay disease in these animals compared to untreated controls. However, 2 of 5 CD28−/− mice in the group treated with Tbx21−/− Tregs became diabetic during the course of the experiment, compared to 0 of 5 in the Tbx21+/+ Treg-treated group. These results suggest that although Tbx21−/− Tregs are functional, they may be less effective than Tbx21+/+ Tregs at similar doses. However, the survival curve of the mice treated with Tbx21−/− Tregs was not statistically different from those treated with Tbx21+/+ Tregs. Further, NOD.Rag2−/−.Tbx21−/−.BDC2.5 TCR transgenic mice, which lack endogenous regulatory T cells, show rapid progression to diabetes between 4 and 8 weeks of age. Taken together, these data demonstrate that regulatory T cells contribute to the control of diabetes in Tbx21−/− animals and that Tbx21−/− effector cells are still competent to cause diabetes if regulatory T cells are removed. Thus, the balance between regulation and autoimmunity in the NOD is shifted toward tolerance by the deficiency in Tbx21, but the system can still be pushed into autoimmunity. Additional studies using tissue-specific genetic deletion of Tbx21 will be needed to define precisely which Tbx21-expressing cell types are the most important for spontaneous diabetes in the NOD.
Supplementary Material
Acknowledgements
We thank members of the Bluestone laboratory for helpful advice. We thank X. Zhang and the UCSF Helen Diller Family Comprehensive Cancer Center Mouse Pathology Core for assistance with preparation of H&E slides. We thank Paul Wegfahrt and Nicolas Martinier for assistance with mouse handling and Shuwei Jiang for assistance with sorting Tregs. We thank Dimitri de Kouchkovsky for assistance with genotyping mice.
This work was supported by National Institutes of Health grants R01 AI46643 and AI56388, P30 DK63720 and CA112663. J.H.E. received funding from the NIH Medical Scientist Training Program (GM007618).
Footnotes
Abbreviations used in this paper: NOD, non-obese diabetic; T1DM, Type 1 diabetes mellitus; Treg, regulatory T cell; SNP, single nucleotide polymorphism.
The online version of this article contains supplemental material.
References
- 1.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, Bhan A, Autschbach F, Sullivan BM, Szabo SJ, Glimcher LH, Blumberg RS. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taqueti VR, Grabie N, Colvin R, Pang H, Jarolim P, Luster AD, Glimcher LH, Lichtman AH. T-bet controls pathogenicity of CTLs in the heart by separable effects on migration and effector activity. J Immunol. 2006;177:5890–5901. doi: 10.4049/jimmunol.177.9.5890. [DOI] [PubMed] [Google Scholar]
- 4.Juedes AE, Rodrigo E, Togher L, Glimcher LH, von Herrath MG. T-bet controls autoaggressive CD8 lymphocyte responses in type 1 diabetes. J Exp Med. 2004;199:1153–1162. doi: 10.1084/jem.20031873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Fathman JW, Lugo-Villarino G, Scimone L, von Andrian U, Dorfman DM, Glimcher LH. Transcription factor T-bet regulates inflammatory arthritis through its function in dendritic cells. J Clin Invest. 2006;116:414–421. doi: 10.1172/JCI26631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A. 2002;99:5545–5550. doi: 10.1073/pnas.082114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, Penninger JM, Eriksson U. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–2019. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, Ackerman K, Haley K, Galle PR, Szabo SJ, Drazen JM, De Sanctis GT, Glimcher LH. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295:336–338. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 9.Aliprantis AO, Wang J, Fathman JW, Lemaire R, Dorfman DM, Lafyatis R, Glimcher LH. Transcription factor T-bet regulates skin sclerosis through its function in innate immunity and via IL-13. Proc Natl Acad Sci U S A. 2007;104:2827–2830. doi: 10.1073/pnas.0700021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 12.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 13.Mayer KD, Mohrs K, Reiley W, Wittmer S, Kohlmeier JE, Pearl JE, Cooper AM, Johnson LL, Woodland DL, Mohrs M. Cutting edge: T-bet and IL-27R are critical for in vivo IFN-gamma production by CD8 T cells during infection. J Immunol. 2008;180:693–697. doi: 10.4049/jimmunol.180.2.693. [DOI] [PubMed] [Google Scholar]
- 14.Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, Glimcher LH. T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci U S A. 2003;100:7749–7754. doi: 10.1073/pnas.1332767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu N, Ohnishi N, Ni L, Akira S, Bacon KB. CpG directly induces T-bet expression and inhibits IgG1 and IgE switching in B cells. Nat Immunol. 2003;4:687–693. doi: 10.1038/ni941. [DOI] [PubMed] [Google Scholar]
- 16.Werneck MB, Lugo-Villarino G, Hwang ES, Cantor H, Glimcher LH. T-bet plays a key role in NK-mediated control of melanoma metastatic disease. J Immunol. 2008;180:8004–8010. doi: 10.4049/jimmunol.180.12.8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki Y, Ihara K, Matsuura N, Kohno H, Nagafuchi S, Kuromaru R, Kusuhara K, Takeya R, Hoey T, Sumimoto H, Hara T. Identification of a novel type 1 diabetes susceptibility gene, T-bet. Hum Genet. 2004;115:177–184. doi: 10.1007/s00439-004-1146-2. [DOI] [PubMed] [Google Scholar]
- 18.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JH, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G, Maisuria M, Meadows W, Smink LJ, Healy B, Burren OS, Lam AA, Ovington NR, Allen J, Adlem E, Leung HT, Wallace C, Howson JM, Guja C, Ionescu-Tirgoviste C, Simmonds MJ, Heward JM, Gough SC, Dunger DB, Wicker LS, Clayton DG. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morahan G, Huang D, Ymer SI, Cancilla MR, Stephen K, Dabadghao P, Werther G, Tait BD, Harrison LC, Colman PG. Linkage disequilibrium of a type 1 diabetes susceptibility locus with a regulatory IL12B allele. Nat Genet. 2001;27:218–221. doi: 10.1038/84872. [DOI] [PubMed] [Google Scholar]
- 20.Hultgren B, Huang X, Dybdal N, Stewart TA. Genetic absence of gamma-interferon delays but does not prevent diabetes in NOD mice. Diabetes. 1996;45:812–817. doi: 10.2337/diab.45.6.812. [DOI] [PubMed] [Google Scholar]
- 21.Serreze DV, Post CM, Chapman HD, Johnson EA, Lu B, Rothman PB. Interferon-gamma receptor signaling is dispensable in the development of autoimmune type 1 diabetes in NOD mice. Diabetes. 2000;49:2007–2011. doi: 10.2337/diabetes.49.12.2007. [DOI] [PubMed] [Google Scholar]
- 22.Serreze DV, Chapman HD, Post CM, Johnson EA, Suarez-Pinzon WL, Rabinovitch A. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J Immunol. 2001;166:1352–1359. doi: 10.4049/jimmunol.166.2.1352. [DOI] [PubMed] [Google Scholar]
- 23.Kanagawa O, Xu G, Tevaarwerk A, Vaupel BA. Protection of nonobese diabetic mice from diabetes by gene(s) closely linked to IFN-gamma receptor loci. J Immunol. 2000;164:3919–3923. doi: 10.4049/jimmunol.164.7.3919. [DOI] [PubMed] [Google Scholar]
- 24.Leiter EH. Current Protocols in Immunology. John Wiley and Sons; 2003. The NOD Mouse: A Model for Insulin-Dependent Diabetes Mellitus. [DOI] [PubMed] [Google Scholar]
- 25.Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol. 2001;166:908–917. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- 26.Kanagawa O, Militech A, Vaupel BA. Regulation of diabetes development by regulatory T cells in pancreatic islet antigen-specific TCR transgenic nonobese diabetic mice. J Immunol. 2002;168:6159–6164. doi: 10.4049/jimmunol.168.12.6159. [DOI] [PubMed] [Google Scholar]
- 27.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 29.Ishizaki K, Yamada A, Yoh K, Nakano T, Shimohata H, Maeda A, Fujioka Y, Morito N, Kawachi Y, Shibuya K, Otsuka F, Shibuya A, Takahashi S. Th1 and type 1 cytotoxic T cells dominate responses in T-bet overexpression transgenic mice that develop contact dermatitis. J Immunol. 2007;178:605–612. doi: 10.4049/jimmunol.178.1.605. [DOI] [PubMed] [Google Scholar]
- 30.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–3439. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest. 2004;114:979–987. doi: 10.1172/JCI20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.You S, Slehoffer G, Barriot S, Bach JF, Chatenoud L. Unique role of CD4+CD62L+ regulatory T cells in the control of autoimmune diabetes in T cell receptor transgenic mice. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14580–14585. doi: 10.1073/pnas.0404870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson Laboratory Verification of markers at known diabetes susceptibility/resistance (Idd) loci. 2009 http://type1diabetes.jax.org/gqc_resistance_suceptibility_loci.html.
- 34.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 35.Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med. 1995;181:817–821. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Beller DI. Distinct pathways for NF-kappa B regulation are associated with aberrant macrophage IL-12 production in lupus- and diabetes-prone mouse strains. J Immunol. 2003;170:4489–4496. doi: 10.4049/jimmunol.170.9.4489. [DOI] [PubMed] [Google Scholar]
- 37.Trembleau S, Penna G, Gregori S, Chapman HD, Serreze DV, Magram J, Adorini L. Pancreas-infiltrating Th1 cells and diabetes develop in IL-12-deficient nonobese diabetic mice. J Immunol. 1999;163:2960–2968. [PubMed] [Google Scholar]
- 38.Jain R, Tartar DM, Gregg RK, Divekar RD, Bell JJ, Lee HH, Yu P, Ellis JS, Hoeman CM, Franklin CL, Zaghouani H. Innocuous IFNgamma induced by adjuvant-free antigen restores normoglycemia in NOD mice through inhibition of IL-17 production. J Exp Med. 2008;205:207–218. doi: 10.1084/jem.20071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyton RJ, Davies S, Marden C, Fantino C, Reynolds C, Portugal K, Dewchand H, Altmann DM. Stat4-null non-obese diabetic mice: protection from diabetes and experimental allergic encephalomyelitis, but with concomitant epitope spread. Int Immunol. 2005;17:1157–1165. doi: 10.1093/intimm/dxh293. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Chen M, Ellett JD, Fialkow LB, Carter JD, McDuffie M, Nadler JL. Autoimmune diabetes is blocked in Stat4-deficient mice. J Autoimmun. 2004;22:191–200. doi: 10.1016/j.jaut.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 41.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202:139–156. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- 42.Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, Kaplan MH. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. 2008;29:679–690. doi: 10.1016/j.immuni.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci U S A. 2003;100:15818–15823. doi: 10.1073/pnas.2636938100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maier LM, Wicker LS. Genetic susceptibility to type 1 diabetes. Curr Opin Immunol. 2005;17:601–608. doi: 10.1016/j.coi.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh S, Palmer SM, Rodrigues NR, Cordell HJ, Hearne CM, Cornall RJ, Prins JB, McShane P, Lathrop GM, Peterson LB, et al. Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nat Genet. 1993;4:404–409. doi: 10.1038/ng0893-404. [DOI] [PubMed] [Google Scholar]
- 46.Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, Hearne CM, Knight AM, Love JM, McAleer MA, Prins JB, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature. 1991;351:542–547. doi: 10.1038/351542a0. [DOI] [PubMed] [Google Scholar]
- 47.McDuffie M. Derivation of diabetes-resistant congenic lines from the nonobese diabetic mouse. Clin Immunol. 2000;96:119–130. doi: 10.1006/clim.2000.4893. [DOI] [PubMed] [Google Scholar]
- 48.Ivakine EA, Fox CJ, Paterson AD, Mortin-Toth SM, Canty A, Walton DS, Aleksa K, Ito S, Danska JS. Sex-specific effect of insulin-dependent diabetes 4 on regulation of diabetes pathogenesis in the nonobese diabetic mouse. J Immunol. 2005;174:7129–7140. doi: 10.4049/jimmunol.174.11.7129. [DOI] [PubMed] [Google Scholar]
- 49.Grattan M, Mi QS, Meagher C, Delovitch TL. Congenic mapping of the diabetogenic locus Idd4 to a 5.2-cM region of chromosome 11 in NOD mice: identification of two potential candidate subloci. Diabetes. 2002;51:215–223. doi: 10.2337/diabetes.51.1.215. [DOI] [PubMed] [Google Scholar]
- 50.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.