

Abstract
Background
The G-protein-coupled receptor (GPCR)-kinase interacting protein-1 (GIT1) is a multi-domain scaffold protein that participates in many cellular functions including receptor internalization, focal adhesion remodeling, and signaling by both GPCRs and tyrosine kinase receptors. However, there have been no in vivo studies of GIT1 function to date.
Methods and Results
To determine essential functions of GIT1 in vivo, we generated a traditional GIT1 knockout (KO) mouse. GIT1 KO mice exhibited ∼60% perinatal mortality. Pathologic examination showed that the major abnormality in GIT1-KO mice was impaired lung development characterized by markedly reduced numbers of pulmonary blood vessels and increased alveolar spaces. Since vascular endothelial growth factor (VEGF) is essential for pulmonary vascular development, we investigated the role of GIT1 in VEGF signaling in the lung and cultured endothelial cells (EC). Because activation of phospholipase-Cγ (PLCγ and ERK1/2 by angiotensin II requires GIT1, we hypothesized that GIT1 mediates VEGF-dependent pulmonary angiogenesis by modulating PLCγ and ERK1/2 activity in EC. In cultured EC, knockdown of GIT1 decreased VEGF-mediated phosphorylation of PLCγ and ERK1/2. PLCγ and ERK1/2 activity in lungs from GIT1 KO mice was reduced postnatally.
Conclusions
Our data support a critical role for GIT1 in pulmonary vascular development by regulating VEGF-induced PLCγ and ERK1/2 activation.
Keywords: G-protein-coupled receptor (GPCR)-kinase interacting protein-1, endothelial cell, PLCγ
Introduction
The G-protein-coupled receptor (GPCR)-kinase interacting protein-1 (GIT1) is a multi-domain scaffold protein that was first found due to its binding to the GPCR kinase-2 (GRK2) 1. All mammals and birds studied to date express two GIT proteins: GIT1 and GIT2. Whereas GIT1 exists almost entirely in its full-length form, GIT2 is extensively alternatively spliced in a tissue specific manner 2. Tissue distribution of GIT1 is much more restricted than GIT2 which is ubiquitous 3. Northern blotting of RNA pooled from various rat tissues with a GIT1 probe demonstrated highest levels in testis, lung and brain 1. Recently, Premont's group used mice with β-galactosidase reporters inserted into the two GIT genes to visualize GIT1 and GIT2 gene expression in mouse tissues3. Their study showed that GIT2 was expressed in most cells of the body, while GIT1 was highly restricted to brain, vessels (both endothelial cells (EC) and vascular smooth muscle cells (VSMC)), lung bronchi and liver bile ducts. There was no expression of GIT1 in parenchyma of lung or liver, nor in myocytes (heart, skeletal or smooth muscle (except vascular)).
The full-length GIT proteins have an N-terminal ARF GTPase activating protein domain, three ankyrin repeats, a Spa2-homology domain (SHD), a coiled-coil domain and a paxillin-binding site (PBS) 4. GIT1 binds to PIX, PLCγ and MEK1 through the SHD domain; and paxillin through the PBS domain. GIT1 plays a role in many cellular functions including receptor internalization, focal adhesion remodeling, and signaling pathways of both GPCRs and tyrosine kinase receptors 5,6. Recently GIT2 was shown to be essential for neutrophil function in vivo, since loss of GIT2 in neutrophils resulted in impaired GPCR-induced chemotactic direction sensing and excess superoxide production 7. However, there have been no in vivo studies of GIT1 function to date. To determine the physiologic importance of GIT1, we used gene targeting strategies to knock out GIT1 expression in mice (GIT1 KO). Here we report a dramatic pulmonary phenotype in GIT1 KO mice.
Methods And Materials
Generation of GIT1 KO Mice
GIT1 KO alleles were generated by homologous recombination in embryonic stem (ES) cells. Targeted ES cells carrying disrupted allele were injected to 129 ES cells to produce chimeric mice. Chimeras were mated with C57BL/6 females to obtain F1 mice carrying the targeted allele. For detailed procedures, please see supplemental data.
Morphometry and immunohistochemistry
Lung tissues of GIT1 WT and KO mice, embryos (E14.5) were fixed with 4% paraformaldehyde (PFA), sectioned, and processed for hematoxylin and eosin (H&E) staining or fixed for electron microscopy and as described8. Vessel density was evaluated by vWF staining (see details in supplemental data).
Pulmonary vascular imaging with X-ray and micro-CT
Lungs from GIT1 WT and KO mice (P25) and right ventricle were cannulated. A heated solution of 1% low melting agarose and 30% barium was infused. The lungs were inflation fixed with 4% PFA. The pulmonary vasculature was visualized with X-ray and Scanco vivaCT 40 (Basserdorf, Switzerland). Details of the micro-CT were published previously 9. CT data were reconstructed as 3D images. Scanco's program was used to plot vessel counts versus vessel thickness in each segment (5segments/per right lung). The total vessel numbers from all 5 segments were added.
Fluorescence microangiography
The fetal pulmonary arterial system was perfused with a solution of 1% low melting agarose containing a suspension of 0.2 μm fluorescent microspheres (1:10 (v/v)) (Molecular Probes). The lungs were then fixed in 4% PFA for 48 hours at 4°C and sections (150 μm) were cut on a vibratome (Leica VT1000S). Images were captured by optical sectioning using confocal microscope (Bio-Rad “Radiance”).
Measurement of pulmonary artery pressure (PAP) and right ventricle (RV) pressure
GIT1 WT and KO mice (2∼3 month, n=6 per group) were anesthetized with an intraperitoneal injection of ketamine (130 mg/kg) and xylazine (8.8 mg/kg) in saline (10 mL/kg). A thoracotomy was rapidly performed and the mice were ventilated with oxygen-enriched air. After median sternotomy, the right ventricle was punctured with a 27&1/2 gauge needle, and a 1.0 Fr measuring catheter (Millar Instruments, Inc. Houston Texas) was inserted into right ventricle as determined by typical RV pressure curve. The catheter was then advanced across the pulmonary valve into the pulmonary artery and pressure was recorded for 30-40 min.
Cell culture
Human umbilical vein endothelial cells (HUVEC) were isolated and grown as described previously 10. Rat pulmonary microvascular endothelial cells (RPMEC) were a generous gift from Dr. R. James White and growth in 10% FBS DMEM. GIT1 siRNA (AAGCTGCCAAGAAGAAGCTAC) and control non-silencing siRNA(AATTCTCCGACACGTGTCACT) were transfected as previously described by our laboratory6. Matri-gel was purchased from Chemicon, and the tube formation assays were performed as instructed by the manufacturer.
3H-thymidine incorporation
To assess the effect of GIT1 on RPMEC proliferation, the rate of DNA synthesis was measured as described11.
Immunoblotting
Western blotting was performed as described 6. Results were normalized by arbitrarily setting the densitometry of control samples to 1.0.
Statistical Analysis
All values are expressed as mean ± SE, and represent data from three to six independent experiments. Survival was assessed by the Kaplan-Meier method and compared between the 2 genotypes using a log rank test. Comparisons of parameters between 2 groups were analyzed with unpaired t-test. Comparisons of different parameters between the 2 genotypes were analyzed with 2-way ANOVA and comparisons of different parameters between the 2 genotypes or groups at different time points were analyzed with a two-way repeated measures ANOVA. Statistical significance was evaluated with StatView (StatView 5.0, SAS Institute Inc, Cary, NC). A value of P<0.05 was considered statistically significant.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Generation of GIT1 KO Mice
To study the physiologic relevance of GIT1, targeting vectors were created to knock out the gene by homologous recombination replacing exons 2-7 with a neomycin resistance cassette (Fig. 1A). The deleted region encodes the ARF-GAP domain and ankyrin repeats. Targeting of GIT1 was confirmed by RT-PCR and western blot. RT-PCR analysis of RNA from lung tissue of GIT1WT and KO mice at postnatal day 4 (P4) revealed that GIT1 transcripts were absent in homozygous mice. Transcripts encoding GIT2 were expressed at normal levels in KO mice (Fig. 1B). There was no GIT1 protein expression in any tissue from GIT1 KO mice (Fig. 1C).
Figure 1. Generation of GIT1 KO mice.
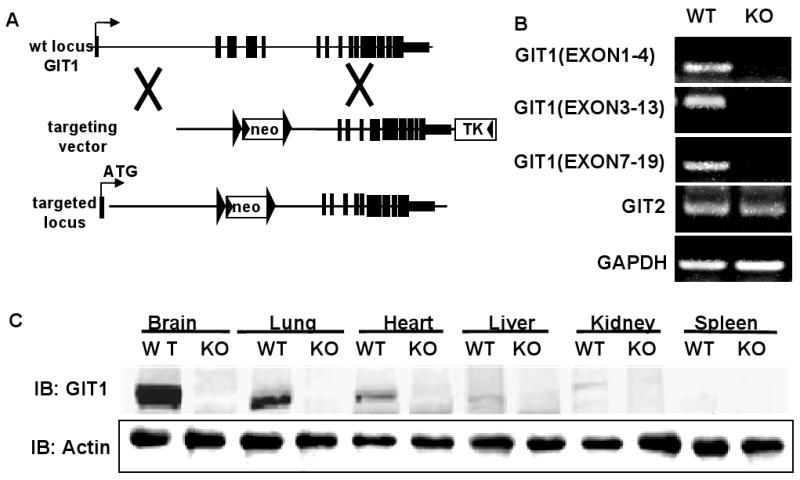
A. Strategy to generate GIT1 KO mice by homologous recombination. Exons 2-7 were replaced with the targeting vector containing a neomycin-resistance cassette. B. Analysis of GIT1 and GIT2 transcripts by RT-PCR. C. GIT1 protein expression in different tissues of WT and KO mice.
High mortality of GIT1 KO mice
GIT1 KO mice were born at the expected Mendelian ratio and were fertile. However many GIT1 KO mice died soon after birth. To characterize the time course for death, pregnant GIT1 KO mice and WT mice were observed daily at term (8 litters for each group) and during the 10 days after birth. Litter sizes of GIT1 KO animals varied from six to eight pups at birth. Kaplan-Meier curves showed that 60% (Fig. 2A, log-rank test, P value=0.011) of GIT1 KO offspring succumbed within 10 days of birth, whereas minimal neonatal mortality (4%) was evident in pups of WT litters.
Figure 2. Survival, gross anatomy and histopathology in GIT1 WT and KO mice.
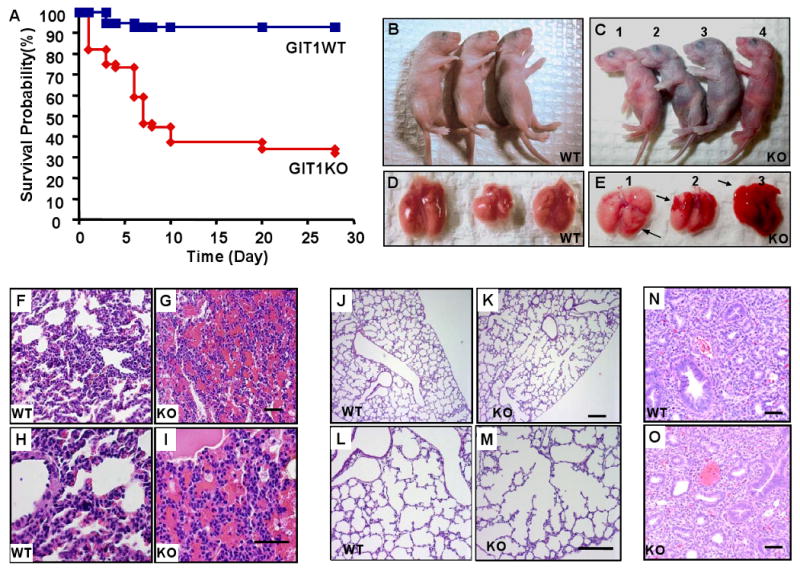
A. Kaplan-Meier survival analysis. B-C. GIT1 WT and KO neonates (P4): WT neonates (B) and two KO neonates appeared normal (C, #1 and 4) while others (C, #2 and 3) showed varying degrees of cyanosis and respiratory distress. D-E. Fetal lungs (P4) from WT mice (D) had a normal gross appearance, whereas lungs from KO mice (E) showed scattered hemorrhages. F-I. Hematoxylin and eosin (H&E) staining of lung sections from P5 GIT1 WT and KO mice. There was obvious hemorrhage in parenchyma of GIT1 KO mice (G, I) compared to WT mice (F, H). J-M. GIT1 WT and KO mice lungs were perfused as described in methods, then the tissues were embedded and stained with H&E. GIT1 WT mice show normal, well-developed saccular and alveolar airway structures (J, L), whereas GIT1 KO showed abnormally large airspaces (K; M). N-O H&E staining of lung sections from embryos (E14.5) of GIT1 WT and KO mice. There were no obvious differences in histological appearance between WT and KO embryonic lungs.
Abnormal lung morphology in GIT1 KO mice
Some newborn GIT1 KO pups exhibited severe respiratory distress and were obviously cyanotic (Fig. 2C, #2 and #3) compared to WT pups (Fig. 2B). The lung tissues of these pups showed gross pleural hemorrhages (Fig. 2E) compared to WT lungs (Fig. 2D). No obvious hemorrhages were observed in the lungs of adult GIT1 KO mice that survived past 1 month, although lung morphology was abnormal as shown by increased air spaces (3 month old, supplemental Fig.1). Gross morphology of all other organs was completely normal. Histopathological analyses of tissues that express GIT1 including brain and liver were also normal (not shown). Histolopathological examination of the lungs of WT mice showed normal saccular expansion at days 4 -7 (Fig. 2F & H), while there was evidence of marked hemorrhage in parenchyma of GIT1 KO mice (Fig. 2G & I). In the perfused lungs from P7 mice, WT mice showed a well developed alveolar structure (Fig. 2J & L), while abnormally large airspaces were present in KO mice (Fig. 2K & M). In contrast, there were no differences in the histological appearance of WT and KO lungs at earlier periods of gestation (E14.5, Fig. 2N&O). To gain insight into mechanisms responsible for impaired pulmonary development and hemorrhage, we studied ultrastructural details of alveoli and vessels. Transmission electron microscopy revealed that the tight junctions of arteries, capillaries and venules were intact in KO mice (Supplemental Fig. 2). There were no significant differences in hematologic parameters including bleeding times between GIT1 WT and KO (Supplemental Table 1, two-way ANOVA).
GIT1 protein expression in neonate lung tissue of GIT1 WT mice
Premont's group showed that GIT1 expression was restricted to cells lining blood vessels and bronchi in adult mouse lung tissue 3. However, GIT1 protein expression in neonatal lung was not investigated. Immunostaining of GIT1 in neonatal lung (P5) revealed that GIT1 was predominantly colocalized with platelet endothelial cell adhesion molecule-1 (PECAM-1, an endothelial cell marker) in vessels (supplemental Fig. 3 A-C), especially capillaries (supplemental Fig. 3 D-F), suggesting an important role for GIT1 in EC function. GIT1 was also expressed in smooth muscle and epithelial cells of bronchi (Supplemental Fig. 3A-C). There was no significant difference in smooth muscle cell α-actin protein expression in lungs of GIT1 WT and KO mice (P7, P=0.36, unpaired t test, n=3, supplemental Fig. 4), suggesting that differences in smooth muscle cell function did not explain the GIT1 KO phenotype.
Defective pulmonary development in GIT1 KO mice
To better define the abnormalities in pulmonary vasculature we performed X-ray and 3-dimensional analysis of the pulmonary arterial tree using micro-CT and fluorescence microangiography. WT lungs demonstrated uniform filling of the distal pulmonary arterial tree, with homogeneous perfusion of the microvasculature (Fig. 3A, C, E). In contrast, lungs from GIT1 KO mice showed dramatic pruning of arteriolar branches and regions of marked capillary hypoperfusion (Fig. 3B, D, F). The numbers of vessels in the right lung were counted based on the vessel size. This analysis showed that the greatest difference in KO lungs was a significant decrease in vessels of the smallest size (0-50 μm), although there was a trend in vessels of 50-200 μm (Fig. 3G). To characterize further the alterations in pulmonary microvessels we assayed the presence of EC. Cells expressing the EC marker, vWF, could be demonstrated by immunostaining in lung sections from both P7 WT (Fig. 4A) and KO mice (Fig. 4B). However, the number of vWF-positive cells was reduced significantly in lungs of GIT1 KO mice (Fig. 4C, unpaired t test). Moreover, expression of the VEGF receptor 2 (VEGFR2), a key receptor for EC migration and proliferation, time dependently increased in lungs of GIT WT mice (maximum at P5), to a much greater extent than in the KO mice (Fig. 4D & Fig. 4E, two-way repeated measures ANOVA).
Figure 3. Imaging of lungs shows reduced microvasculature in GIT1 KO mice.
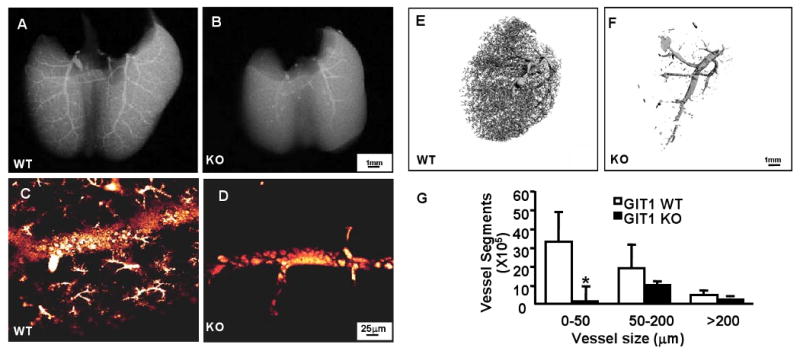
X-ray (A-B), fluorescein microangiography (C-D) micro-CT (E-F) were performed as described in the methods. WT mice displayed numerous small pulmonary arterioles extending into the capillary circulation (A, C, E), whereas there were strikingly fewer arterioles and capillaries in lungs from GIT1 KO mice (B, D, F) G. Quantification of micro-CT data according to the size of the vessels (mean ±SE; n =3,*P<0.05, compared with WT).
Figure 4. Decreased EC number and vessel density in GIT1 KO mice.
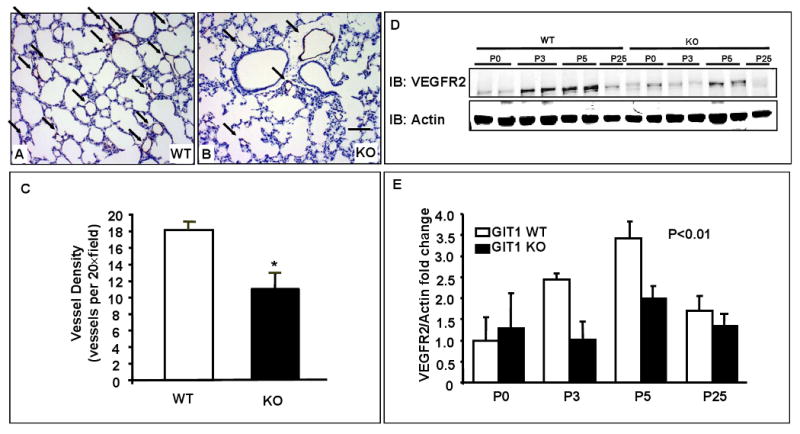
A-B. vWF staining of lungs of GIT1 WT (A) and KO (B) at P7. Black arrows indicate vWF positive vessels (brown staining). Bar=20 μm C. Quantification of vWF staining. 5 fields were chosen from each sample (n=3 per group). D. VEGFR2 expression in lungs of GIT1 WT and KO mice at different ages. E. Quantitation of the relative expression of VEGFR2 normalized to actin (WT P0 = 1.0). *P< 0.01 compared with WT groups (mean ±SE; n =4).
To determine the role of GIT1 in formation of alveoli we studied both VEGFR2 expression and surfactant protein C (SP-C, a pneumocyte marker) expression. When GIT1 expression was decreased with siRNA, there was no change in the expression of VEGFR2 in HUVEC (Fig. 5A), demonstrating that decreased expression of VEGFR2 in lung was likely due to decreased numbers of EC. Because alveoli development may be regulated by pulmonary vasculature formation12, SP-C positive cells were also measured by immunostaining. As anticipated, SP-C positive cells in GIT1 KO mice decreased about 30% compared with WT (supplemental Fig. 5, unpaired t-test), whereas the ratio of SP-C positive cells and total cell number was similar in two groups. These data suggest that deficiency of GIT1 did not affect differentiation of pneumocytes.
Figure 5. GIT1 is required for activation of MEK1, ERK1/2 and PLCγ signaling pathways in EC and lung.
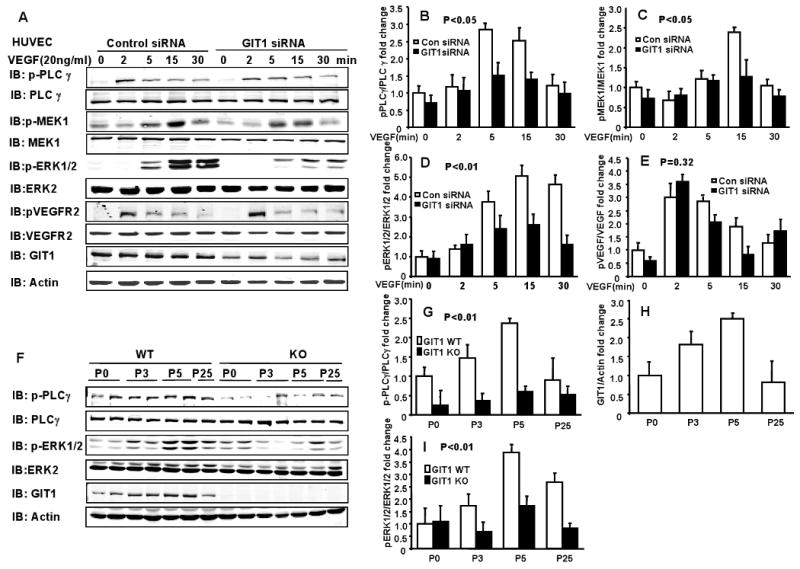
A. GIT1 and control siRNA were transfected for 48 h into HUVECs. After serum starvation for 4 h, the cells were stimulated with 20 ng/ml VEGF for the indicated times and phosphorylation of the indicated proteins was determined. B-E. Right panels are quantitation of relative increase of PLCγ, MEK1, ERK1/2 and VEGFR2 phosphorylation compared to control siRNA group without VEGF stimulation. P< 0.05 compared with control siRNA groups (mean ±SE; n =3). F. Phosphorylation of PLCγ and ERK1/2 as well as expression of GIT1 in lungs of GIT1 WT and KO mice were detected by immunoblot. Total PLCγ, ERK1/2 and actin were assayed as loading controls. G-I. Quantitation of relative changes of PLCγ and ERK1/2 phosphorylation, as well as GIT1 expression (normalized to actin in WT P0 group). P< 0.01 compared with WT (mean ±SE; n =4).
GIT1 KO mice have normal pulmonary artery pressures (PAP)
Since some newborn GIT1 KO pups exhibited severe respiratory distress and were obviously cyanotic (Fig. 2C, #2 and #3), it is possible that adult GIT1 KO mice will develop pulmonary hypertension. Interestingly, we found that GIT1 KO and WT mice had similar PAPs (9.49±0.76 mmHg versus 11.25±1.17 mmHg, P=0.16, two-way ANOVA, n=6 per group), as well as right ventricle pressures (RVP, 19.34±1.78mmHg verse 20.29±0.10 mmHg, P=0.41, n=6 per group, supplemental Table 2). There were also no significant differences in heart rate (note that heart rate was recorded under anesthesia), or right ventricle dp/dt values (P=0.22, n=6). Thus pulmonary vascular tone appears to be similar in GIT1 KO and WT mice that survive to adulthood.
GIT1 knockdown inhibited VEGF-induced activation of PLCγ in EC
Because the GIT1 KO mouse phenocopies the VEGF120 mouse 13, we hypothesized that VEGF signaling would be significantly inhibited by loss of GIT1. We and others have shown important roles for GIT1 in signaling mediated by tyrosine kinase receptors, especially activation of MEK1-ERK1/2, PLCγ and FAK 5. Because the PLCγ KO mice also has a vascular phenotype 14, we further hypothesized that GIT1 is required for activation of PLCγ induced by VEGF. Importantly, VEGF-dependent activation of the PLCγ pathway in EC contributes to ERK1/2 activation (via Raf-MEK1) and to DNA synthesis 15,16. To gain insight into the functional significance of GIT1 in VEGF-mediated signaling events in EC, we measured the effect of GIT1 knockdown on VEGF-mediated PLCγ, MEK1 and ERK1/2 activation. HUVEC were transfected with GIT1 siRNA for 48 h and then stimulated with VEGF (Fig. 5A). Treatment of HUVEC with GIT1 siRNA significantly reduced endogenous GIT1 expression, whereas control siRNA had no effect (Fig. 5A). Decreasing GIT1 expression significantly inhibited VEGF activation of PLCγ (Fig. 5A&B), MEK1 (Fig. 5A&C) and ERK1/2 (Fig. 5A&D). There was no significant effect of GIT1 knockdown on VEGFR2 activation as measured by phosphorylation of tyrosine 1175 which is important for activation of PLCγ (Fig. 5A&E, two-way repeated measures ANOVA). These data show that GIT1 is essential for activation of PLCγ MEK1 and ERK1/2 by VEGF.
Phosphorylation of PLCγ and ERK1/2 were decreased in lungs of GIT1 KO mice
To confirm the role of GIT1 in VEGF signaling in vivo, activation of PLCγ and ERK1/2 was investigated in the lungs of GIT1 WT and KO mice. Both PLCγ and ERK1/2 phosphorylation increased in lungs isolated from GIT1 WT mice with peak at P5 (Fig. 5F-I). In contrast, in GIT1 KO mice phosphorylation of PLCγ and ERK1/2 was markedly decreased compared with WT mice (Fig. 5F-I, two-way repeated ANOVA). GIT1 expression also increased from P0 to P5 (Fig. 5G&I). These data suggest that GIT1-dependent activation of PLCγ and ERK1/2 may play an important role during the postnatal period of lung development.
GIT1 is required for serum-induced DNA synthesis in EC in vitro and proliferation in vivo
VEGF-dependent activation of the MEK1- ERK1/2 pathway contributes to DNA synthesis and cell proliferation in EC16. To determine whether GIT1 is required for EC proliferation, we examined the effect of GIT1 knockdown on serum-induced DNA synthesis in microvascular EC. As shown in Fig. 6A, GIT1 siRNA significantly inhibited DNA synthesis (two-way ANOVA). To confirm the role of GIT1 in EC proliferation in vivo, we assayed expression of proliferating cell nuclear antigen (PCNA) in lung. PCNA was highly expressed in lungs of GIT1 WT mice during P3-5 and decreased at P25 (Fig. 6B), while PCNA expression was dramatically decreased in GIT1 KO (0.4-fold±0.2, p<0.05 compared to WT group, unpaired t test, n=4). These data suggest that GIT1 is essential for postnatal EC proliferation.
Figure 6. Effect of GIT1 on EC proliferation in vitro and in vivo.
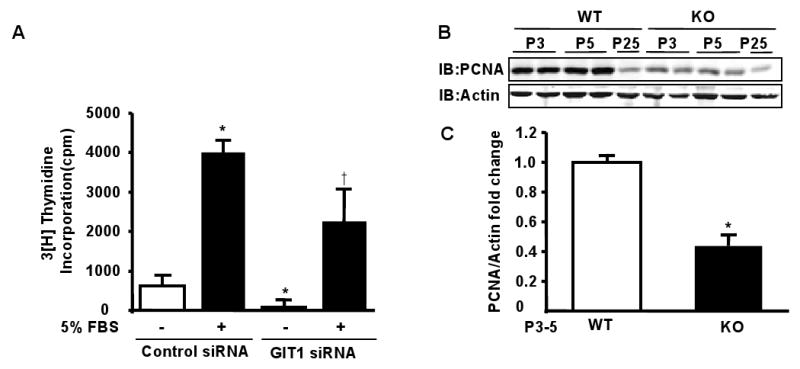
A. Microvascular EC were transfected with GIT1 siRNA. After serum starvation for 24 h, the cells were stimulated with 5% FBS and DNA synthesis was evaluated by 3H-thymidine incorporation. Note that FBS was used as microvascular EC respond weakly to VEGF. B. Expression of PCNA in lungs of GIT1WT and KO mice were detected by immunoblot. Total actin was assayed as loading control. C. Quantitation of relative changes of PCNA expression (normalized to actin in WT P3-5 group). *P<0.01compared with WT groups of same time point (mean ±SE; n =4).
GIT1 is required for EC tube formation induced by VEGF
VEGF stimulated EC tube formation in vitro is a well established assay for angiogenesis. To evaluate the role of GIT1 in angiogenesis, the effect of GIT1 knockdown on tube formation was studied. VEGF increased tube formation by 4-fold, which was significantly reduced by GIT1 knockdown (Fig. 7, two-way ANOVA).
Figure 7. Effect of GIT1 on EC tube formation.
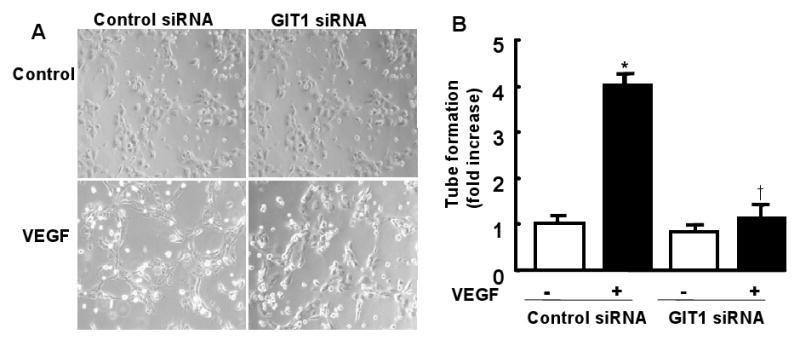
HUVEC were transfected with control siRNA or GIT1 siRNA. After serum starvation for 24 h, the cells were seeded on 6-well plates at 1×105 cells/well precoated with Matrigel and maintained in serum free medium, or with VEGF (50 ng/ml). Tube formation was visualized after 6 h (A) and analyzed using ImageJ (B).
Discussion
The major finding of this paper is the presence in GIT1 KO mice of profound abnormalities in lung morphogenesis including decreased pulmonary vascular density, parenchymal hemorrhage and increased alveolar spaces. Organ development and function in the GIT1 KO mouse was otherwise normal, suggesting that there is a unique requirement for GIT1 in the lung. Interestingly, Premont's lab showed that GIT1 expression in the lung was confined to EC and bronchi, while GIT2 was ubiquitous3. While we observed no compensation by GIT2 in the lung, the ubiquitous expression of this protein may be sufficient to compensate for GIT1 deficiency in most tissues. The pulmonary findings in the GIT1 KO mice phenocopy the VEGF120 mice13, suggesting that GIT1 is a critical mediator of VEGF signaling in the lung. Support for this concept is provided by impaired signaling downstream of VEGF including reduced activation of PLCγ and ERK1/2 in the lungs of GIT1 KO. We show further that this pathway is critical for EC tube formation and proliferation. These data support a novel mechanism for VEGF signaling in the lung mediated by a VEGF-GIT1-PLCγ-MEK1-ERK1/2 pathway. Importantly, this phenotype resembles the human diseases bronchopulmonary dysplasia (BPD) and alveolar capillary dysplasia (ACD), in which impaired vascular development retards pulmonary development chronically.
The abnormalities in pulmonary vascular development in GIT1 KO mice were most striking in the decreased numbers of distal arteriolar branches and capillaries with diameters of 0-50 μm as shown by X-ray, micro-CT, and fluorescein angiography. Correspondingly, total EC number was obviously reduced as demonstrated by decreased expression of the EC markers, vWF and VEGFR2, in lungs of GIT1 KO mice. However, embryonic lung morphogenesis (E14.5) of GIT1 KO was normal suggesting that the primary problem in the GIT1 KO is a failure of pulmonary angiogenesis rather than epithelial morphogenesis. In addition we found that tight junctions in arteries and veins were normal in GIT1 KO based on electron microscopy. Bleeding time and platelet number were also normal in GIT1 KO mice. Thus the impressive pulmonary hemorrhage we observed was likely not due to abnormalities in vascular permeability or hemostasis. Therefore the essential requirement for GIT1 in pulmonary vascular development likely reflects unique developmental and perinatal events in the pulmonary vasculature (and possibly alveoli as discussed below). Future studies using EC-specific knockout of GIT1 will be necessary to define the roles of GIT1 more precisely.
Because of reduced pulmonary vascularity with chronic hypoxemia, we hypothesized that adult GIT1 KO mice may develop pulmonary hypertension. Surprisingly, adult GIT1 KO mice showed PAP and RV pressures similar to WT mice. Furthermore, components of the pulmonary vasoconstrictor pathway - endothelin-1 (ET-1)17,18 and the endothelin-1 receptor A were unchanged in GIT1 KO mice (Supplemental Fig. 6). These results suggest that compensatory mechanisms exist in the GIT1 KO mice that survive to adulthood. It will be interesting in future studies to characterize these compensatory pathways.
The finding that the GIT1 KO has many morphologic similarities to the VEGF120 mouse suggests that GIT1 is a novel mediator for VEGF signaling in the lung13. There is close coordination of airway and vessel growth that is essential for normal lung development, especially during the saccular and alveolar stages of development. Angiogenesis has been shown to be necessary for alveolarization during lung development, and inhibiting pulmonary vascular growth impedes alveolar growth12 19. Thus we propose that inhibiting pulmonary vascular growth during a critical period of postnatal lung growth impairs alveolarization, suggesting that endothelial-epithelial cross talk, especially via VEGF signaling, is critical for normal lung growth following birth. This concept is supported by decreased SP-C positive cell numbers and decreased expression of VEGF mRNA in GIT1 KO mice (Supplemental Fig. 7,). We believe that a requirement for GIT1 in pneumocyte development is unlikely since these cells express primarily GIT23. In addition to VEGF, fibroblast growth factor-10 (FGF-10) has been shown to be essential for lung morphogenesis. We believe it is unlikely that FGF-10 is involved in the GIT1 KO pulmonary phenotype since mRNA expression of FGF-10 was normal in GIT1 KO mice (Supplemental Fig. 6).
Both VEGF164 and VEGF18820 have been shown to be essential to development of the lung vasculature. The disruption of even a single copy of the VEGF gene results in embryonic lethality due to the failure of endothelial differentiation and blood vessel formation20. To overcome this limitation, Galambos et al13 engineered knock-in transgenic mice expressing only VEGF120, which led to a selective impairment in the development of the distal pulmonary arteriolar branches, strongly resembling the vascular defects of GIT1-deficient animals13. VEGF contributes to blood vessel formation in the developing lung 21,22, including the arteriolar branches and capillary formation. Postnatal lung development (alveolar stage) is primarily dependent upon angiogenesis, which correlates with increased expression of GIT1 (Fig. 5F, I) and VEGF13,22-24 in the lung. Based on these findings we suggest that GIT1 function is required for VEGF signaling. VEGF signaling involves activation of c-Src, PLC-γ, and ERK1/2, which are important in angiogenesis6,25. Our previous data showed that GIT1 is a substrate of c-Src and functions as a scaffold protein for PLCγ and MEK16,26,27. Thus we believe that GIT1 is required for PLCγ-MEK1-ERK1/2 activation by VEGF and for lung microvasculature development. In cultured EC, knockdown of GIT1 by siRNA obviously decreased the activation of PLC-γ, MEK1 and ERK1/2 (Fig. 5) which play important roles in EC proliferation and migration 6,25. This role of GIT1 was confirmed by the findings of significantly decreased EC proliferation and tube formation after GIT1 knockdown (Figs. 6, 7).
Among the components of the VEGF-GIT1-PLCγ-MEK1-ERK1/2 signaling pathway we believe that activation of PLCγ is most critical to the pulmonary vasculature. Neither the MEK1 or ERK1/2 KO mice have a significant vascular phenotype, while PLCγ KO mice die at embryonic day 8-9 due to deficiencies in EC function14. PLCγ phosphorylation, a measure of activity, exhibited a time course in GIT1 WT lungs that matched GIT1 and VEGF expression, suggesting a role in postnatal lung maturation (Fig. 5). Importantly, phosphorylation of VEGFR2 at tyrosine 1175 which is required for activation of PLCγ was not decreased by deletion of GIT1 (Fig. 5)16. Thus the difference between GIT1 and PLCγ KO in terms of time of fetal death likely reflects a requirement for PLCγ protein expression (rather than activity) and/or some compensation by GIT2 during development.
Clinical significance
The diminished numbers of EC and lack of capillary in-growth into the alveolar septae found in the GIT1 KO lungs represent characteristic features of BPD and ACD. BPD is a chronic lung disease that develops in premature infants treated with ventilation and supplemental oxygen28. As a consequence of impaired alveolar capillary in-growth, normal air-blood barriers fail to form29,30. BPD has been associated with decreased expression of VEGF and VEGFR228, suggesting mechanisms similar to GIT1 KO. ACD occurs in infants with persistent pulmonary hypertension refractory to vasodilator treatment. ACD also is characterized by deficiency of alveolar capillary in-growth and failure to form normal air-blood barriers between EC and type I epithelial cells. While the gene defect is unknown, a vascular etiology appears likely since children with ACD often display misalignment of pulmonary veins, with both venous and arterial vessels sharing a common adventitial sheath31. It is possible that abnormal GIT1 expression and function might contribute to impaired alveolar vascularization in BPD and ACD patients. In conclusion, the present report provides novel evidence of a previously unrecognized contribution of GIT1 to pulmonary vascular development and maturation. Thus, pharmacological or genetic manipulation of the GIT1 signaling pathway (especially PLCγ) in the perinatal period may represent a therapeutic strategy to improve respiratory function in premature infants and in clinical conditions associated with abnormalities in lung morphogenesis.
Supplementary Material
Acknowledgments
We acknowledge technical assistance by Dr. Michael Zuscik and Laura Yanoso for the micro-CT assay and advice on histopathology by Dr. Haodong Xu.
Sources of Funding: This work was supported by grants from the NIH to B.C.B. (HL63462), and to C.Y. (HL77789). This work was also supported by Scientist Development grant from American Heart Associate to Jinjiang Pang (0835626D).
Footnotes
Disclosures: The authors declare no conflicts.
Clinical Perspective: The mechanisms by which blood vessels contribute to organ development remain unknown. Here we show that deficiency of G-protein-coupled receptor (GPCR)-kinase interacting protein-1 (GIT1), which mediates critical aspects of vascular endothelial growth factor receptor 2 (VEGFR2) signal transduction such as activation of phospholipase-Cγ and extracellular signal regulated kinase 1/2, impairs lung development. The diminished numbers of endothelial cells (EC) and lack of capillary in-growth into the alveolar septae found in the lungs of the GIT1 knockout mice represent characteristic features of the human diseases bronchopulmonary dysplasia (BPD) and alveolar capillary dysplasia (ACD). BPD is a chronic lung disease that develops in premature infants treated with ventilation and supplemental oxygen. BPD has been associated with decreased expression of VEGF and VEGFR2, suggesting mechanisms similar to GIT1 deficiency. ACD also is characterized by decreased alveolar capillary in-growth and failure to form normal air-blood barriers between EC and type I epithelial cells. A vascular etiology appears likely since children with ACD often display misalignment of pulmonary veins with both venous and arterial vessels sharing a common adventitial sheath. It is possible that abnormal GIT1 expression and function might contribute to impaired alveolar vascularization in BPD and ACD patients. Thus, pharmacological or genetic manipulation of the GIT1 signaling pathway may represent a therapeutic strategy to improve respiratory function in premature infants and individuals with abnormal lung development.
References
- 1.Premont RT, Claing A, Vitale N, Freeman JL, Pitcher JA, Patton WA, Moss J, Vaughan M, Lefkowitz RJ. beta2-Adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc Natl Acad Sci U S A. 1998;95:14082–7. doi: 10.1073/pnas.95.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Premont RT, Claing A, Vitale N, Perry SJ, Lefkowitz RJ. The GIT family of ADP-ribosylation factor GTPase-activating proteins. Functional diversity of GIT2 through alternative splicing. J Biol Chem. 2000;275:22373–80. doi: 10.1074/jbc.275.29.22373. [DOI] [PubMed] [Google Scholar]
- 3.Schmalzigaug R, Phee H, Davidson CE, Weiss A, Premont RT. Differential Expression of the ARF GAP Genes GIT1 and GIT2 in Mouse Tissues. J Histochem Cytochem. 2007 doi: 10.1369/jhc.7A7207.2007. [DOI] [PubMed] [Google Scholar]
- 4.Brown MC, West KA, Turner CE. Paxillin-dependent Paxillin Kinase Linker and p21-Activated Kinase Localization to Focal Adhesions Involves a Multistep Activation Pathway. Mol Biol Cell. 2002;13:1550–65. doi: 10.1091/mbc.02-02-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoefen RJ, Berk BC. The multifunctional GIT family of proteins. J Cell Sci. 2006;119:1469–75. doi: 10.1242/jcs.02925. [DOI] [PubMed] [Google Scholar]
- 6.Yin G, Haendeler J, Yan C, Berk BC. GIT1 functions as a scaffold for MEK1-extracellular signal-regulated kinase 1 and 2 activation by angiotensin II and epidermal growth factor. Mol Cell Biol. 2004;24:875–885. doi: 10.1128/MCB.24.2.875-885.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazaki Y, Hashimoto S, Tsujimura T, Morishige M, Hashimoto A, Aritake K, Yamada A, Nam JM, Kiyonari H, Nakao K, Sabe H. Neutrophil direction sensing and superoxide production linked by the GTPase-activating protein GIT2. Nat Immunol. 2006;7:724–31. doi: 10.1038/ni1349. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P, Moons L, Dewerchin M, Mackman N, Luther T, Breier G, Ploplis V, Muller M, Nagy A, Plow E, Gerard R, Edgington T, Risau W, Collen D. Insights in vessel development and vascular disorders using targeted inactivation and transfer of vascular endothelial growth factor, the tissue factor receptor, and the plasminogen system. Ann N Y Acad Sci. 1997;811:191–206. doi: 10.1111/j.1749-6632.1997.tb52002.x. [DOI] [PubMed] [Google Scholar]
- 9.Jorgensen SM, Demirkaya O, Ritman EL. Three-dimensional imaging of vasculature and parenchyma in intact rodent organs with X-ray micro-CT. Am J Physiol. 1998;275:H1103–14. doi: 10.1152/ajpheart.1998.275.3.H1103. [DOI] [PubMed] [Google Scholar]
- 10.Jin ZG, Wong C, Wu J, Berk BC. Flow shear stress stimulates Gab1 tyrosine phosphorylation to mediate protein kinase B and endothelial nitric-oxide synthase activation in endothelial cells. J Biol Chem. 2005;280:12305–9. doi: 10.1074/jbc.M500294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arkin IT, Xu H, Jensen MO, Arbely E, Bennett ER, Bowers KJ, Chow E, Dror RO, Eastwood MP, Flitman-Tene R, Gregersen BA, Klepeis JL, Kolossvary I, Shan Y, Shaw DE. Mechanism of Na+/H+ antiporting. Science. 2007;317:799–803. doi: 10.1126/science.1142824. [DOI] [PubMed] [Google Scholar]
- 12.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol. 2002;283:L555–62. doi: 10.1152/ajplung.00408.2001. [DOI] [PubMed] [Google Scholar]
- 13.Galambos C, Ng YS, Ali A, Noguchi A, Lovejoy S, D'Amore PA, DeMello DE. Defective pulmonary development in the absence of heparin-binding vascular endothelial growth factor isoforms. Am J Respir Cell Mol Biol. 2002;27:194–203. doi: 10.1165/ajrcmb.27.2.4703. [DOI] [PubMed] [Google Scholar]
- 14.Liao HJ, Kume T, McKay C, Xu MJ, Ihle JN, Carpenter G. Absence of erythrogenesis and vasculogenesis in Plcg1-deficient mice. J Biol Chem. 2002;277:9335–41. doi: 10.1074/jbc.M109955200. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A- dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. Embo J. 2001;20:2768–78. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci U S A. 2005;102:1076–81. doi: 10.1073/pnas.0404984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durmowicz AG, Stenmark KR. Mechanisms of structural remodeling in chronic pulmonary hypertension. Pediatr Rev. 1999;20:e91–e102. [PubMed] [Google Scholar]
- 18.Stenmark KR, Mecham RP. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu Rev Physiol. 1997;59:89–144. doi: 10.1146/annurev.physiol.59.1.89. [DOI] [PubMed] [Google Scholar]
- 19.Stenmark KR, Balasubramaniam V. Angiogenic therapy for bronchopulmonary dysplasia: rationale and promise. Circulation. 2005;112:2383–5. doi: 10.1161/CIRCULATIONAHA.105.574061. [DOI] [PubMed] [Google Scholar]
- 20.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 21.deMello DE, Sawyer D, Galvin N, Reid LM. Early fetal development of lung vasculature. Am J Respir Cell Mol Biol. 1997;16:568–81. doi: 10.1165/ajrcmb.16.5.9160839. [DOI] [PubMed] [Google Scholar]
- 22.Hislop AA. Airway and blood vessel interaction during lung development. J Anat. 2002;201:325–34. doi: 10.1046/j.1469-7580.2002.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hislop A. Developmental biology of the pulmonary circulation. Paediatr Respir Rev. 2005;6:35–43. doi: 10.1016/j.prrv.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Frey U, Hislop A, Silverman M. Branching properties of the pulmonary arterial tree during pre- and postnatal development. Respir Physiol Neurobiol. 2004;139:179–89. doi: 10.1016/j.resp.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, Inai Y, Silver M, Isner JM. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. Embo J. 1999;18:3964–72. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haendeler J, Yin G, Hojo Y, Saito Y, Melaragno M, Yan C, Sharma VK, Heller M, Aebersold R, Berk BC. GIT1 mediates Src-dependent activation of phospholipase Cgamma by angiotensin II and epidermal growth factor. J Biol Chem. 2003;278:49936–44. doi: 10.1074/jbc.M307317200. [DOI] [PubMed] [Google Scholar]
- 27.Yin G, Zheng Q, Yan C, Berk BC. GIT1 Is a Scaffold for ERK1/2 Activation in Focal Adhesions. J Biol Chem. 2005;280:27705–12. doi: 10.1074/jbc.M502271200. [DOI] [PubMed] [Google Scholar]
- 28.Maniscalco WM, Watkins RH, Pryhuber GS, Bhatt A, Shea C, Huyck H. Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Physiol Lung Cell Mol Physiol. 2002;282:L811–23. doi: 10.1152/ajplung.00325.2001. [DOI] [PubMed] [Google Scholar]
- 29.Chess PR, D'Angio CT, Pryhuber GS, Maniscalco WM. Pathogenesis of bronchopulmonary dysplasia. Semin Perinatol. 2006;30:171–8. doi: 10.1053/j.semperi.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 30.D'Angio CT, Maniscalco WM. Bronchopulmonary dysplasia in preterm infants: pathophysiology and management strategies. Paediatr Drugs. 2004;6:303–30. doi: 10.2165/00148581-200406050-00004. [DOI] [PubMed] [Google Scholar]
- 31.Rabah R, Poulik JM. Congenital alveolar capillary dysplasia with misalignment of pulmonary veins associated with hypoplastic left heart syndrome. Pediatr Dev Pathol. 2001;4:167–74. doi: 10.1007/s100240010125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.