

Abstract
Purpose
To examine effects of bumetanide, a selective blocker of Na+-K+-2Cl− cotransporter (NKCC1), on hippocampal excitability and rapid kindling in immature rats.
Methods
Studies were performed in Wistar rats of three ages: postnatal day 11 (P11, neonatal), P14 (post-neonatal), and P21 (pre-adolescent). Bumetanide (0.2, 0.5, 2.5 mg/kg) was given intraperitoneally 20 minutes prior to the beginning of the studies. Hippocampal excitability was examined by measuring threshold and duration of afterdischarge, which had been elicited by electrical stimulation of ventral hippocampus. Kindling procedure consisted of 80 electrical stimulations of ventral hippocampus, delivered every 5 minutes.
Results
At P11, bumetanide (0.5 mg/kg) increased the baseline hippocampal afterdischarge threshold and shortened the afterdischarge duration. Bumetanide delayed the occurrence, and reduced the number of full motor seizures during kindling, and prevented the development of kindling-induced enhanced seizure susceptibility in a majority of animals. At P14 bumetanide (0.5 mg/kg) induced no significant antiepileptic effects, although suppression of hippocampal excitability and inhibition of kindling were observed in a subset of animals. At P21 bumetanide (0.2; 2.5 mg/kg) exerted no effects on hippocampal excitability and kindling progression.
Discussion
The obtained results provide further evidence that bumetanide may be beneficial for treating neonatal seizures, and that NKCC1 represents a potential target for antiepileptic interventions in the immature brain.
Keywords: Epilepsy, kindling, Na+-K+-2Cl− cotransporter, bumetanide
Unlike in the adulthood, in the neonatal brain, γ-amino butyric acid (GABA) acts as an excitatory neurotransmitter (Ben-Ari, 2002). Excitatory effect of GABA during early ontogenic stages are explained by the high expression of the Na+-K+-2Cl− cotransporter (NKCC1), and by the low expression of Cl− - extruding K+-Cl− cotransporter (KCC2) in the brain (Dzhala et al., 2005; Payne et al., 2003; Yamada et al., 2004; Wang et al., 2002). NKCC1 leads to elevated concentration of intracellular Cl− , while KCC2 exerts the opposite effect; thus in neurons that express NKCC1 and lack KCC2, the opening of Cl− channel by GABA leads to the outward Cl− current and to neuronal depolarization (Payne et al., 2003). These findings suggest that pharmacological blockade of NKCC1 in the neonatal brain may have antiepileptic effect. Indeed, bumetanide, an approved loop diuretic and a selective blocker of NKCC1 (Hannaert et al., 2002) exhibited acute anticonvulsant effects in the neonatal brain under conditions of both in vitro and in vivo models of epilepsy (Dzhala et al., 2005; 2008). However, it is not known, whether bumetanide would interfere with neonatal epileptogenesis, that is with the progression to the epileptic state. In part, the lack of such data is due to the fact that in a majority of rodent models of epileptogenesis, the establishment of epileptic state is outpaced by much faster brain maturation.
Rapid kindling is a model of “compressed” epileptogenesis, when epileptic state can be achieved within a matter of several hours (Lothman et al., 1985). Importantly, rapid kindling can be induced in rats as early as at postnatal day (P) 7, that is at the neonatal age (Michelson and Lothman, 1991). Such property of rapid kindling has determined its successful application in studying age-dependent effects of antiepileptic drugs (Mazarati et al., 2007; 2008).
In the present study, we compared effects of bumetanide on rapid kindling in rats of three ages: P11 (neonatal), when brain expression of NKCC1 and anticonvulsant efficacy of bumetanide are high; P14 (post-neonatal), when both brain NKCC1 expression and anticonvulsant effects of bumetanide are in decline (although still present), and P21 (pre-adolescent), when brain expression of NKCC1 is close to that in adult (i.e. very low), and bumetanide fails to exert anticonvulsant effects (Dzhala et al., 2005).
Methods
Experiments were performed in male Wistar rats (Charles River, Wilmington, MA), in accordance with the policies of the National Institutes of Health and the UCLA Office for the Protection of Research Subjects.
At P10, P13, or P20, under Isoflurane anesthesia, animals were implanted with a bipolar stimulating/recording electrode (Plastics 1, Roanoke, VA, diameter 0.23 mm) into ventral hippocampus (coordinates from Bregma in mm: P10- posterior 3.3, left 4.3, depth 4.4; P14- posterior 3.0; left 3.9; depth 4.2; P21- posterior 2.9; left 3.7; depth 3.8), and a tripolar recording electrode (Plastics 1, diameter 0.23 mm), into the neocortex (Mazarati et al., 2007; 2008).
Twenty four hours after surgery, (i.e. at P11, P14, or P21) animals were connected to DS8000 electrical stimulator (World Precision Instruments, Sarasota, FL), and to MP100/EEG100B acquisition system (BIOPAC, Santa Barbara, CA). Afterdischarge threshold (ADT) and afterdischarge duration (ADD) were examined by applying electrical stimuli to, and recording from the hippocampal electrode; stimulation parameters were: 10 s train, 50 ms peak interval, 1 ms pulse duration, square wave biphasic waveform, starting with 0.1 mA, with 0.05 mA increments, delivered every 10 minutes (Mazarati et al., 2007; 2008). Upon detecting ADT, animals were injected intraperitoneally with either bumetanide (Sigma, St Louis, MO) at doses of 0.2 mg/kg, 0.5 mg/kg, or 2.5 mg/kg (the latter dose was given to P21 only) or saline as control treatment. The lowest dose of bumetanide was chosen based on earlier studies (Dzhala et al., 2005). Twenty minutes later, afterdischarge properties were examined again, and the animals were subjected to rapid kindling: eighty stimulations delivered every 5 minutes at the parameters described above, but at 0.05 mA above ADT, as determined after bumetanide or saline injection (Mazarati et al, 2007; 2008). During kindling, electrical activity was recorded from the cortical electrode; animals' behavior was recorded using digital camera. An additional ½ or the original dose of bumetanide was injected in middle of the kindling procedure (i.e. after the 40th stimulation). Twenty four hours after the last kindling stimulation, hippocampal afterdischarge properties were examined again.
Upon completion of the studies, animals were euthanized with Pentobarbital (100 mg/kg); proper position of the electrode in the hippocampus was verified in fresh-frozen cryostat-cut coronal sections. The implants did not induce significant tissue damage beyond electrode tracks, similar to that reported earlier (Mazarati et al., 2007).
The following parameters were analyzed: before kindling- ADT and ADD; during kindling- number of stimulations required to develop first stage 4 (rearing) - 5 (falling) behavioral seizure (Racine, 1972), total number of stage 4-5 seizures, and median duration of electrographic seizures; after kindling- ADT, ADD and behavioral seizure score in response to the threshold stimulation. Statistical analysis employed paired t-test or Mann-Whitney test were appropriate. In addition to the group analysis, we examined individual responses to bumetanide. For this, we introduced the following arbitrary criteria for the treatment efficacy. For baseline afterdischarge properties - the increase of ADT equal to or more than 2 standard deviations from mean values before treatment in the same group; for kindling progression - the reduction of the number of stage 4-5 seizures by 2 standard deviations or more from mean values in age-matched controls; for post-kindling - ADT within pre-kindling Mean ± 2 standard deviations. These data were analyzed using Fisher's exact test. Each group included 8 animals at the beginning of the study; one P21 rat was not used for the post-kindling analysis due to the loss of the electrode cap.
Results
At 0.2 mg/kg, bumetanide induced no effects on either of the examined parameters, and in either of the age groups (data not shown). Data described below were obtained for the dose of 0.5 mg/kg (and 2.5 mg/kg in some of P21 rats).
Effects of bumetanide on baseline afterdischarge properties
Bumetanide (0.5 mg/kg) induced statistically significant increase of ADT and shortening of ADD in P11 (Fig. 1A), but not in groups of P14 (Fig. 1B) and P21 (Fig. 1C) rats. Analysis of individual responses revealed that in 7 out of 8 P11 animals, the increase of ADT after bumetanide was equal to or larger than 2 standard deviations from mean pre-treatment value (Fig. 1D); this was statistically different from the age-matched controls (0 out of 8, p<0.05). At P14, 3 out of 8 subjects followed the above-mentioned trend (Fig. 1D), although the number was not statistically different from that in control group (0 out of 8, p>0.05). None of P21 animals showed substantial increase of ADT after bumetanide (0.5 mg/kg) injection (Fig. 1D). A separate group of P21 rats (n=8) was injected with the drug at a dose of 2.5 mg/kg; however, even at this higher dose, bumetanide did not alter afterdischarge properties in any of the animals (ADT=0.82±0.14 mA before bumetanide, 0.77±0.12 mA after bumetanide; ADD=21.8±4.7 s before bumetanide, 24.2±3.7 s after bumetanide, p>0.05; no animals showed the increase of ADT equal to or more than 2 standard deviations of mean pre-treatment value).
Fig. 1. Effects of bumetanide on baseline afterdischarge properties.
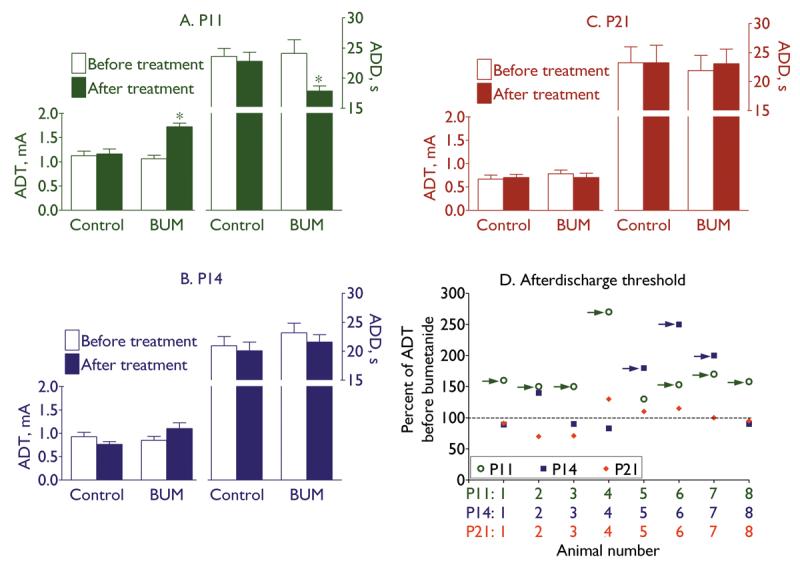
Treatment with bumetanide (BUM, 0.5 mg/kg) increased afterdischarge threshold (ADT) and shortened afterdischarge duration (ADD) in P11 rats (A), but did not modify afterdischarge properties in groups of P14 (B) and P21 (C) animals. On A-C data are presented as Mean±SEM; asterisk on A- p<0.05 vs. “Before treatment” (paired t-test). D: Plot of ADT in response to bumetanide in individual animals of three ages. For each animal, the effect of bumetanide is expressed as percent of ADT before bumetanide treatment (Y-axis). Color-coded numbers under the X-axis correspond to respective data points for each animal and age. Arrows point to those subjects, in which the increase of ADT after bumetanide injection was equal to or larger than 2 standard deviations from mean values before treatment.
Effects of bumetanide on kindling progression
At P11, bumetanide (0.5 mg/kg) significantly delayed the occurrence, and decreased the number of stage 4-5 seizures, as compared with controls (Fig. 2A); no such effect was observed in groups of P14 (Fig. 2B) and P21 (Fig. 2C) rats. In 5 out of 8 P11 rats, bumetanide decreased the number of stage 4-5 seizures by 2 standard deviations or more from mean value in control group (Fig. 2D; p<0.05 vs. Control, where such a decrease was not observed in any animals). At P14, the same three animals which had shown significant increase of baseline ADT in response to bumetanide, developed fewer stage 4-5 seizures (p>0.05 vs. Control, Fig. 2D). None of P21 rats showed the decrease of stage 4-5 seizure count after bumetanide (0.5 mg/kg) treatment (Fig. 2D). Furthermore, administration of bumetanide at a dose of 2.5 mg/kg, did not modify kindling progression in any of P21 animals (n=8; number of stimulations to first stage 4-5 seizure=18.8±2.7; median EEG seizure duration=64.5±4.5; number of stage 4-5 seizures=29.4±1.9, and for all animals was within Mean±2 standard deviations of respective value in Control group; p>0.05 vs. Control).
Fig. 2. Effects of bumetanide on kindling progression.

In P11 animals (A), pretreatment with bumetanide (0.5 mg/kg) delayed the occurrence, and reduced the number of stage 4 seizures; neither of the analyzed indices was modified in groups of P14 (B) and P21 (C) rats. On A-C data are presented as Mean±SEM; asterisk on A- p<0.05 vs. Control (Mann-Whitney test). D: Plot of stage 4-5 seizure count in individual animals of three ages. For each animal, the effect of bumetanide is expressed as percent of the mean number of stage 4-5 seizures in control groups (Y-axis). Color-coded numbers under the X-axis correspond to respective data points for each animal and age. Arrows point to those subjects, in which bumetanide pretreatment reduced number of stage 4-5 seizures by 2 standard deviations or more from mean values in age-matched control groups.
Effects of pretreatment with bumetanide on kindling-induced enhanced hippocampal excitability
Twenty four hours after the last kindling stimulation, control animals of all three ages showed significant decrease of ADT and prolongation of ADD, as compared with pre-kindling values (Fig. 3A-C). Furthermore, animals developed behavioral seizures in response to the threshold stimulation. Minimal/maximal/median seizure score was 2/4/3 at P11 and P14, and 3/4/4 at P21. Pre-treatment with bumetanide did not prevent kindling-induced increase of hippocampal excitability and seizure responses in groups of P14 and P21 animals (Fig. 3B,C); however, in P11 rats pre-treated with bumetanide, ADT and ADD were statistically similar to their pre-kindling values, and minimal/maximal/median seizure score was 0/2/1 (Fig. 3A). In 7 out of 8 bumetanide-treated P11 animals, ADT was within pre-kindling Mean ± 2 standard deviations (Fig. 3D), which was statistically different from age-matched controls (0 out of 8, p<0.05). At P14, similar effect of bumetanide pre-treatment was observed in 2 out of 8 subjects (p>0.05 vs. age-matched controls). Those were same animals which responded to bumetanide treatment during kindling procedure (Fig. 3D). Pre-treatment with bumetanide at a dose of 2.5 mg/kg, did not attenuate kindling-induced increase of hippocampal excitability in any of P21 rats (n=8, ADT= 45±2.1% of pre-kindling value; ADD= 203±9% of pre-kindling value; average seizure score=3.5±5, p>0.05 vs control).
Fig. 3. Effects of pretreatment with bumetanide on the enhanced seizure susceptibility 24 hours after kindling.
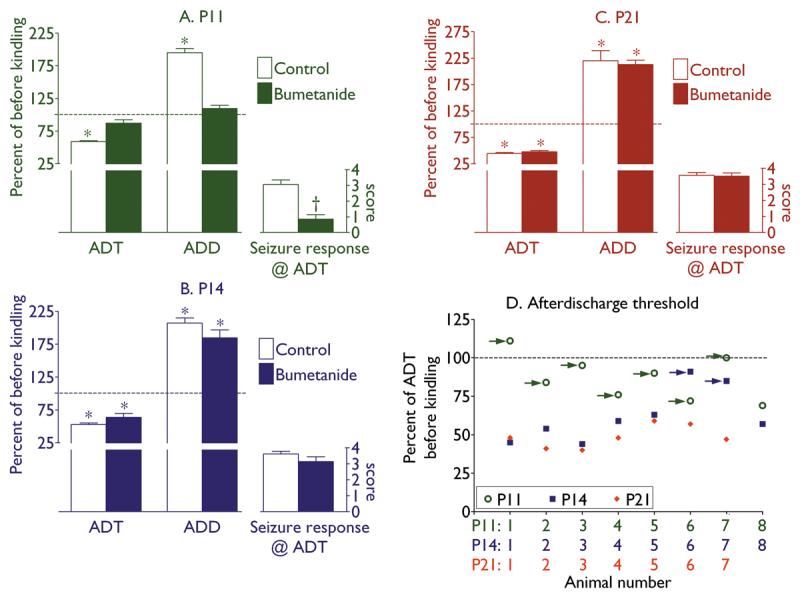
In P11 animals (A), pretreatment with bumetanide prevented kindling-induced decrease of afterdischarge threshold (ADT), increase of afterdischarge duration (ADD), and limited seizure response to threshold stimulation to a maximum of stage 2 seizures. Both P14 (B) and P21 (C) animals, exhibited significant decrease of ADT, increase of ADD, as compared with pre-kindling values, and developed up to stage 4 seizures in response to threshold stimulation. On A-C data are presented as Mean±SEM; asterisks on A-C- p<0.05 vs. respective values before kindling (paired t-test); dagger on A- p<0.05 vs. Control (Mann-Whitney test). D: Plot of ADT after kindling in individual animals of three ages. For each animal, the effect of bumetanide is expressed as percent of ADT before kindling and bumetanide injection (Y-axis). Color-coded numbers under the X-axis correspond to respective data points for each animal and age. Arrows point to those subjects in which ADT was within pre-kindling Mean ± 2 standard deviations.
Discussion
Our experiments showed that bumetanide induced age-dependent antiepileptic effects under conditions of rapid kindling. In neonatal rats (P11), bumetanide decreased hippocampal excitability, which was evident as the increase of ADT and shortening of ADD. During kindling, the drug delayed the occurrence and reduced the number of full motor seizures. Finally bumetanide prevented the establishment of the post-kindling enhanced seizure susceptibility. Thus, while bumetanide did not completely block rapid kindling epileptogenesis, it exerted significant positive disease- modifying effect. None of the described effects were observed at P21. Interestingly antiepileptic effects of bumetanide were found in a subset of P14 rats, which conceivably reflected residual expression of NKCC1 in some animals at this age. In contrast, lack of effects of bumetanide in a small subset of P11 animals can be attributed to an earlier than commonly occurring ontogenic decrease of NKCC1 expression. In the rat cortex, maximal NKCC1 expression has been reported between P3 and P14, with the peak during first 10 postnatal days, and with the dramatic decrease starting from P21 (Dzhala et al., 2005). Bumetanide effectively suppressed high K+ - induced epileptiform activity in hippocampal slices from P6-P9, but not from P15-P23 rats, while at P10-P12 the drug was partially effective (Dzhala et al, 2005). Furthermore, at P9-P12, bumetanide attenuated kainate-induced seizures in vivo (Ibid). Thus, in our studies, bumetanide followed general age-dependent pattern of antiepileptic effects which had been observed in other seizure models.
The dose of bumetanide required for the inhibiting of kindling (0.5 mg/kg) was somewhat higher than that reported earlier (0.2 mg/kg, Dzhala et al., 2005). This difference may be due to different seizure models (kindling vs. kainic acid-induced seizures), as well as to different methodological approaches used for quantifying both seizures and effects of the drug. An important putative ramification of the higher antiepileptic dose as indentified in our studies, might be the involvement of another bumetanide target – inhibition of KCC2 (Payne et al., 2002). However, bumetanide is a highly selective NKCC1 blocker, with 500-fold preference for NKCC1 over KCC2 (Payne et al., 2003). Therefore, it is not highly likely that a 2.5-fold increase of the required dose of bumetanide would block KCC2. Moreover, potential inhibition of KCC2 should have had facilitatory effects on seizures, due to the blockade of Cl− extrusion and consequent GABA-induced depolarization (Payne et al., 2003); however, we did not observe such an effect of bumetanide at a dose of 0.5 mg/kg at any age, or even at a dose of 2.5 mg/kg in P21 animals, that is at the age when KCC2 expression dominates over that of NKCC1 (Dzhala et al., 2005).
Another possible scenario of mitigation of seizures by bumetanide may involve diuretic response and subsequent increase of the size of extracellular space, which may dampen epileptiform activity (Kilb et al., 2006; Sykova, 2004). However, the dose of bumetanide that inhibited kindling was significantly lower than the dose which increases diuresis: thus at a dose of 1 mg/kg, bumetanide did not affect diuresis in the rat (Halladay et al., 1978). A dose reportedly required to induce diuretic response in this species was 10 mg/kg (Olsen, 1981), while 15 mg/kg was used to mitigate brain edema after traumatic brain injury (Lu et al., 2008). Thus, while we cannot provide direct evidence favoring the assumption that inhibition of NKCC1 is primarily responsible for antiepileptic effects of bumetanide in the rapid kindling model (and agreeably other mechanisms cannot be excluded with the absolute certainty), such an assumption seems plausible, based on different binding affinities of bumetanide towards NKCC1 and KCC2; on opposite expected outcomes of the inhibition of these two co-transporters; and on the anticonvulsant effect at a dose below the one known to induce diuretic response.
Age-dependent decline of anticonvulsant efficacy of bumetanide may be attributed to the ontogenic decrease of its transport into the brain, or to its increased rate of elimination (Eades and Christensen, 1998). We did not measure brain concentration of bumetanide; however, lack of its anticonvulsant effect in P21 rats even at a significantly high dose (five-fold higher than the effective anticonvulsant dose in P11 rats), suggests that the failure of bumetanide to inhibit kindling in older animals cannot be attributed exclusively to differences in tissue distribution. At the same time, even if pharmacokinetic aspect does play a role in determining age-specific effects of the drug, such finding by itself would be useful for designing clinical trials of loop diuretics as antiepileptic drugs.
It should be noted, that the reported effects of bumetanide were not uniformly anticonvulsant. Thus, according to Kilb et al (2007) bumetanide facilitated seizure activity induced in vitro by kainic acid or zero Mg2+; the authors proposed that anticonvulsant effects of bumetanide were specific for those protocols which enhance extracellular K+, or interfere with Cl− transport. Zhu et al (2008) observed that both NKCC1 gene knock-out in mice and bumetanide application in mouse hippocampal slices enhanced the excitability of CA3 pyramidal neurons in vitro, possibly due to the reduced rate of clearance of extracellular K+, which in turn is known to rise during neuronal activation. Rapid kindling, being an in vivo complex phenomenon, may not be suitable for dissecting cellular mechanisms involved in anticonvulsant effects of bumetanide; nevertheless, our findings were consistent with previously reported antiepileptic potential of bumetanide (Dzhala et al., 2005).
In conclusion, the presented data provide further support for the inclusion of bumetanide in the treatment of neonatal seizures, and possibly for the role of NKCC1 as a potential target for antiepileptogenic intervention in the immature brain. Clinical availability of bumetanide further increases the appeal for its introduction into the clinical epilepsy practice. In addition, the study expands the utility of rapid kindling as a model for screening of antiepileptic drugs tailored for specific ages.
Acknowledgments
This work was supported by research grants NS059505 (AM) NS046516 (RS) from NIH/NINDS, and by DAPA Foundation (RS).
The authors confirm that they have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Footnotes
Conflict of interest. None of the authors has any conflict of interest.
References
- Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222–235. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Eades SK, Christensen ML. The clinical pharmacology of loop diuretics in the pediatric patient. Pediatr Nephrol. 1998;12:603–616. doi: 10.1007/s004670050514. [DOI] [PubMed] [Google Scholar]
- Hannaert P, Alvarez-Guerra M, Pirot D, Nazaret C, Garay RP. Rat NKCC2/NKCC1 cotransporter selectivity for loop diuretic drugs. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:193–199. doi: 10.1007/s00210-001-0521-y. [DOI] [PubMed] [Google Scholar]
- Halladay SC, Carter DE, Sipes IG. A relationship between the metabolism of bumetanide and its diuretic activity in the rat. Drug Metab Dispos. 1978;6:45–49. [PubMed] [Google Scholar]
- Kilb W, Dierkes PW, Sykova E, Vargova L, Luhmann HJ. Hypoosmolar conditions reduce extracellular volume fraction and enhance epileptiform activity in the CA3 region of the immature rat hippocampus. J Neurosci Res. 2006;84:119–129. doi: 10.1002/jnr.20871. [DOI] [PubMed] [Google Scholar]
- Kilb W, Sinning A, Luhmann HJ. Model-specific effects of bumetanide on epileptiform activity in the in-vitro intact hippocampus of the newborn mouse. Neuropharmacology. 2007;53:524–533. doi: 10.1016/j.neuropharm.2007.06.015. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Hatlelid JM, Zorumski CF, Conry JA, Moon PF, Perlin JB. Kindling with rapidly recurring hippocampal seizures. Brain Res. 1985;360:83–91. doi: 10.1016/0006-8993(85)91223-5. [DOI] [PubMed] [Google Scholar]
- Lu KT, Cheng NC, Wu CY, Yang YL. NKCC1-mediated traumatic brain injury-induced brain edema and neuron death via Raf/MEK/MAPK cascade. Crit Care Med. 2008;36:917–922. doi: 10.1097/CCM.0B013E31816590C4. [DOI] [PubMed] [Google Scholar]
- Mazarati A, Shin D, Auvin S, Sankar R. Age-dependent effects of topiramate on the acquisition and the retention of rapid kindling. Epilepsia. 2007;48:765–773. doi: 10.1111/j.1528-1167.2007.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati A, Wu J, Shin D, Kwon YS, Sankar R. Antiepileptogenic and antiictogenic effects of retigabine under conditions of rapid kindling: An ontogenic study. Epilepsia. 2008;49:1777–1786. doi: 10.1111/j.1528-1167.2008.01674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson HB, Lothman EW. An ontogenetic study of kindling using rapidly recurring hippocampal seizures. Brain Res Dev Brain Res. 1991;61:79–85. doi: 10.1016/0165-3806(91)90116-z. [DOI] [PubMed] [Google Scholar]
- Olsen UB. Investigation of the role of kidney kallikrein on bumetanide induced diuresis in rats. Acta Pharmacol Toxicol (Copenh) 1981;49:321–326. doi: 10.1111/j.1600-0773.1981.tb00914.x. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Sykova E. Extrasynaptic volume transmission and diffusion parameters of the extracellular space. Neuroscience. 2004;129:861–876. doi: 10.1016/j.neuroscience.2004.06.077. [DOI] [PubMed] [Google Scholar]
- Wang C, Shimizu-Okabe C, Watanabe K, Okabe A, Matsuzaki H, Ogawa T, Mori N, Fukuda A, Sato K. Developmental changes in KCC1, KCC2, and NKCC1 mRNA expressions in the rat brain. Brain Res Dev Brain Res. 2002;139:59–66. doi: 10.1016/s0165-3806(02)00536-9. [DOI] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ. Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurons is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Polley N, Mathews GC, Delpire E. NKCC1 and KCC2 prevent hyperexcitability in the mouse hippocampus. Epilepsy Res. 2008;79:201–212. doi: 10.1016/j.eplepsyres.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]